

Abstract
Background/Aim: Schlafen 12 (SLFN12) expression correlates with survival in triple negative breast cancer (TNBC). SLFN12 slows TNBC proliferation and induces TNBC differentiation, but whether SLFN12 affects the tumoral response to chemotherapy or radiation is unknown. Materials and Methods: We over-expressed SLFN12 in MDA-MB-231 cells using two different lentiviral vectors. We assessed viable cell numbers via crystal violet assay after treatment with carboplatin, paclitaxel, olaparib, zoledronic acid, camptothecin, or cesium irradiation. CHK1 and CHK2 phosphorylation was assessed by western blot and the effects of inhibiting CHK1/CHK2 by AZD7762 were examined. Key findings were confirmed in Hs578t and BT549 TNBC cells after adenoviral SLFN12 over-expression. Results: SLFN12 over-expression increased TNBC sensitivity to radiation, carboplatin, paclitaxel, zoledronic acid, and camptothecin, but not to olaparib. SLFN12 over-expression decreased CHK1 and CHK2 phosphorylation after treatment with the DNA damaging agent camptothecin (CPT). The CHK1/CHK2 inhibitor diminished the significant cytotoxicity difference between over-expression and baseline SLFN12 levels in response to carboplatin. Conclusion: SLFN12 increases TNBC sensitivity to DNA-damaging agents at least in part by reducing CHK1/2 phosphorylation. This may contribute to improved survival in patients whose TNBC over-expresses SLFN12. Therefore, SLFN12 levels may be used to customize or predict radiotherapy and chemotherapy effects in TNBC.
- TNBC
- SLFN12
- chemotherapy
- radiotherapy
- CHK1
- CHK2
Breast cancer is the second most common cancer in women in the United States, afflicting 13% of women over their lifetime (1). Breast cancer can be categorized phenotypically by receptor expression and biology. Surgical excision remains the sine qua non of treatment, but many cases of breast cancer can be further adjunctively treated specifically by hormonal agents like tamoxifen or by targeting HER-2/neu. In contrast, triple negative breast cancer (TNBC) is not only uniquely aggressive (2) but is also unresponsive to endocrine or targeted therapy because it lacks estrogen and progesterone receptors and does not express HER-2/neu (3, 4). Therefore, TNBC can only be treated by less specific and more toxic interventions such as cytotoxic chemotherapy or radiation. However, TNBC is relatively chemoresistant and radioresistant as well (3-5), leading to a much worse prognosis than other breast cancers (3, 4, 6). TNBCs are rich in CD44+CD24− cells, breast cancer stem cells (BCSCs) that are themselves relatively chemoresistant and radiation-resistant, and this abundance of cancer stem cells reinforces the aggressive nature of these tumors (4, 7, 8). Therefore, the search for alternatives to improve the prognosis for TNBC patients is crucial.
The Schlafens (SLFN) are a family of genes encoding a novel set of proteins that are differently expressed in various species. These proteins are classified according to their structure and size to short, intermediate, and long (4, 9-12). The long human Schlafen proteins (SLFN5, SLFN11, and SLFN13) have been reported to play a role in the reduction of invasiveness and proliferation of human cancers (12, 13) through transcription modulation, containing nuclear targeting sequences and negatively controlling the expression of several genes that influence malignant cell motility. SLFN11 has also been reported to sensitize cancer cells to DNA-damaging agents (14, 15).
In contrast to the long Schlafens, intermediate Schlafens lack a nuclear targeting sequence and therefore must act differently in the cytosol. Less is known about the role of intermediate SLFNs in cancer. SLFN12 is the only intermediate Schlafen protein expressed in humans (4, 9). SLFN12 induces differentiation in both enterocytes (11) and prostate cancer cells (12), and its over-expression inhibits prostate cancer cell proliferation (12). Patients with TNBC that express relatively higher levels of SLFN12 have longer survival than patients whose tumors express less SLFN12 (4). Similar findings have been reported in lung adenocarcinoma, although this is not true for lung squamous cell carcinoma (10). However, whether such survival differences reflect only differences in tumor biology or might also reflect differences in the tumor cell’s response to chemotherapy and radiotherapy has not been clear.
We hypothesized that over-expression of SLFN12 would sensitize TNBC cells to specific chemotherapy and radiotherapy treatments. To test this hypothesis, we used MDA-MB-231 cells, which are a common model for TNBC (16). An MDA-MB-231 subclone that stably over-expresses SLFN12 (denoted LV) was compared to another subclone that had been transfected with an empty vector virus and therefore, expressed only baseline levels of SLFN12, denoted EV (4). These two cell lines were treated with different chemotherapeutic drugs or subjected to cesium irradiation and cell viability was assessed. To further evaluate a potential mechanism for our observations, we also examined the kinetics of CHK1 and CHK2 phosphorylation in LV and EV using western blot. We further confirmed our results in another clone of SLFN12-over-expressing MDA-MB-231 cells with a different construct, Flag-tagged SLFN12 (denoted LV-Flag) compared to control cells that over-express Flag-tag alone (denoted EV-Flag), and then in Hs578t and BT549 cells after transient adenoviral SLFN12 over-expression.
Materials and Methods
Antibodies. We obtained Phospho-Chk1 (Ser317) (D12H3) XP® Rabbit mAb #12302, Phospho-Chk2 (Thr68) (C13C1) Rabbit mAb #2197, Chk1 (2G1D5) Mouse mAb #2360 and Chk2 (1C12) Mouse mAb #3440 from Cell Signaling Technology (Danvers, MA, USA). IRDye® 680RD Donkey anti-Rabbit IgG Secondary Antibody and IRDye® 800CW Donkey anti-Mouse IgG Secondary Antibody were used as secondary antibodies and obtained from LI-COR (Lincoln, NE, USA).
Drugs. Carboplatin (NSC 241240) Catalog #S1215, Paclitaxel (NSC 125973) Catalog No#S1150, Zoledronic acid (ZOL 446) Catalog No#S1314, Olaparib (AZD2281) Catalog No#S1060 and AZD7762-CHK1/CHK2 inhibitor were from Selleck Chemicals (Houston, TX, USA). CPT (C-9911) was from Sigma-Aldrich (St. Louis, MO, USA).
Cells and reagents. Crystal Violet C-6158 was from Sigma-Aldrich. MDA-MB-231, Bt549, and Hs578t cells were originally obtained from the American Tissue Culture Collection (ATCC, Manassas, VA, USA). MDA-MB-231 cells were subjected to lentiviral infection with either a vector encoding for SLFN12 or an empty lentiviral vector before selecting stably expressing lines as previously described (5). MDA-MB-231 cells over-expressing SLFN12 were denoted as LV and those expressing only baseline levels of SLFN12 after infection with the empty vector control were denoted EV. Cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) cat #: 25-500 (Genesee Scientific, El Cajon, CA, USA) supplemented with 10% fetal bovine serum (FBS) (Genesee Scientific).
A different SLFN12 over-expressing MDA-MB-231 clone was generated using lentiviral vectors that express either Flag-tagged SLFN12 (LV-Flag) or the Flag-tag alone as a control (EV-Flag), which were constructed by SignaGen (Rockville, MD, USA). MDA-MB-231 cells were seeded into a six-well plate at 50,000 cells per well in DMEM medium supplemented with 10% FBS. On the following day, the lentiviral vector was prepared in different infecting particle dilutions (1, 10, 20, 40 viral particles per cell) in 1 ml Opti-MEM media (#11058021, ThermoFisher Scientific, Waltham, MA, USA) supplemented with 5% FBS, 1% GlutaMAX (#35050061, ThermoFisher Scientific) and 10 μg/ml polybrene (#sc-134220, Santa Cruz Biotechnology, Dallas, TX, USA), and then the mixture was added to the cells. Cells were allowed to grow for 24 h, after which another 2 ml of fresh Opti-MEM media supplemented with 5% FBS and 1% GlutaMAX was added. After an additional 24 h of incubation, Opti-MEM medium was replaced with DMEM medium supplemented with 10% FBS, and cells were cultured for an additional 48 h at 37°C and 5% CO2. The selection process was then started using selection medium (DMEM medium supplemented with 10% FBS and 2.5 mg/ml Puromycin (#A1113803, ThermoFisher Scientific) The selection process was performed for one week, after which the surviving cell clones were detached with 0.25% trypsin, diluted with selection medium, re-seeded into 24-well plates and allowed to grow in the selection medium until cells reached 80% confluence. The surviving clones were then re-seeded into 6-well plates and allowed to grow in the selection medium until the cells again reached 80% confluence. Cells were detached with 0.25% trypsin and seeded into a T25-flask in DMEM medium supplemented with 10% FBS. Cells were passaged three times, after which RNA was extracted to confirm successful stable over-expression of SLFN12.
Transient adenoviral transfection studies. Adenoviral vector expressing SLFN12 was constructed by Applied Biological Materials (Richmond, BC, Canada) using a pAdeno vector, CMV promoter, and human SLFN12 insert (accession # NM_018042). For transient transduction, Hs578t and BT549 cells were seeded in 12-well plates at 100,000 cells per well and incubated at 37°C and 5% CO2 in DMEM media supplemented with 10% FBS. On the following day, cells were transduced with adenoviral vector containing SLFN12 (AdSLFN12) or empty control vector (AdCMV) at 4000 viral particles per cell. Carboplatin (43 μM) was added simultaneously with the adenoviral transduction, while 20 nM paclitaxel was added 48-h after adenoviral transduction. Cells were allowed to grow for 72 h after viral transduction, and then cells were processed for crystal violet staining.
RNA and qPCR. Cells were seeded into 6 well plates at 200,000 cells per well, allowed to grow for 72 h, and then lysed in 100 μl of RLT lysis buffer which has a proprietary composition (Qiagen, Hilden, Germany). RNA was isolated from cells using the RNeasy Mini Kit, Qiashredders, and the QiaCube instrument per manufacturer’s protocols (Qiagen). cDNA was synthesized from the extracted RNA using a SMARTScribe Reverse Transcription kit (Takara Clontech, Mountain View, CA, USA) according to the manufacturer’s protocol. cDNA samples were analyzed by qPCR using the BioRad CFX96 Touch Real-Time PCR Detection System (Hercules, CA, USA) and the PrimeTime Gene Expression Master Mix from Integrated DNA Technology (IDT, Coralville, IA, USA). qPCR cycle conditions were 1 cycle of 2 min at 95°C, 50 cycles of 10 s at 95°C and 45 s at the annealing temperature of 55°C. Expression levels were determined from the threshold cycle (Ct) values using the method of 2–∆∆Ct using RPLP0 or HPRT as the reference control genes.
Primers were obtained from IDT and their sequences were the following:
HPRT, forward primer: TTG TTG TAG GAT ATG CCC TTG A, reverse GCG ATG TCA ATA GGA CTC CAG, probe/5HEX/AGC CTA AGA/ZEN/TGA GAG TTC AAG TTG AGT TTG G/3IABkFQ/.
SLFN12, forward primer: GGG AGC AGG TAA TGA CGT ATT TAT T, reverse primer: CAG TTG ACC AGG AAG GAA TGG, probe: FAM/ATC CAG TTC/ZEN/ATG GTG GAG GCT GAA/3IABkFQ.
Cytotoxicity assay. Cell viability was assayed using a crystal-violet-based assay. Cells were seeded at 100,000 cells per well in 12-well plates. Both SLFN12 over-expressing cells (LV) and cells with only native SLFN12 expression (EV) were seeded into 6 wells of the same 12-well plate. Cells were allowed to adhere at 37°C in 5% CO2 for 48 h. Three wells of cells for each cell type were treated with the experimental condition (chemotherapy drug) and three remained untreated as controls. The cells were then incubated for 24-72 h, followed by counting using a crystal violet-based assay as previously described by Feoktistova (17). Briefly, the cells were washed quickly in distilled water to remove non-adherent cells before adding a solution of crystal violet in methanol for 30 min at room temperature that stains and fixes the cells. The fixed cells were then washed three additional times in distilled water to remove the crystal violet that did not stain the cells before air drying for 24 h. Cells were solubilized with methanol and optical density was measured with the Spark® Multimode Microplate Reader by Tecan (Männedorf, Switzerland) at 570 nm. The optical density of the drug-treated cells in each well was normalized to the mean of the control cells of the same cell type and multiplied by 100 to calculate the percentage of cell viability, which was subtracted from 100 to calculate the cytotoxicity percentage.
Cesium irradiation. SLFN12 over-expressing cells (LV) and control cells (EV) were collected into 15 ml tubes. Different 15 ml tubes from each cell type were cesium irradiated with 0-12 Gy using Gammacell 3000 Elan by Theratronics, formerly known as MDS Nordion (Ontario, Canada). After irradiation, the cells were seeded at 100,000 cells per well into 12-well plates, seeding LV and EV in parallel into the same plate in each case. Cells were allowed to adhere and incubated at 37°C and 5% CO2 for 24 h. Cell viability was then assessed by crystal violet assay as described above. The optical density of each well of cesium-irradiated cells was normalized to the mean of control cells (0 GY) of the same cell type and multiplied by 100 to calculate the percentage of cell viability which was subtracted from 100 to calculate the cytotoxicity percentage.
Western blot analysis of phosphorylation of CHK1 and CHK2. LV and EV were seeded at 300,000 cell/well into 6-well plates, incubated at 37°C and 5% CO2 for 48 h, treated with CPT (3 μM) for 1 h, and then washed twice with phosphate buffered saline. The cells were then lysed in radioimmunoprecipitation assay (RIPA) lysis buffer supplemented with Halt protease inhibitors (ThermoFisher Scientific). Lysate protein was sonicated, centrifuged at 12,500 rpm (17,469×g) for 15 min at 4°C and quantified using a bicinchoninic acid protein assay (ThermoFisher Scientific, #87786). Then, 40 μg of protein per lane was resolved by 10%-SDS-PAGE and transferred to 0.2 μm nitrocellulose membranes using Tris-Glycine transfer buffer supplemented with 20% methanol. Membranes were blocked with Odyssey blocking buffer (LI-COR) for 1 h at room temperature followed by overnight incubation at 4°C with the appropriate primary and secondary antibodies for p-CHK1, p-CHK2, CHK1, and CHK2 as above. Membranes were washed four times in Tris-buffered saline with 0.1% Tween (TBS-T) buffer for 5 min each and incubated for 1 h at room temperature with appropriate secondary antibodies: Donkey anti-mouse CW800 and Donkey anti-rabbit RD680 (LI-COR) followed by four 5-min washes in TBS-T buffer. Membranes were imaged using a LI-COR CLX machine. Data were analyzed using Image Studio v5.2 (LI-COR).
CHK1/CHK2 inhibitor cytotoxicity assay. LV and EV were seeded at 20,000 cells per well into 24-well plates, with both cell types being seeded in parallel into 12 wells of each plate. These were incubated at 37°C and 5% CO2 for 48 h. Cells in 4 wells for each cell type were left untreated as controls, cells in 4 wells were treated with 43 μM carboplatin only, and the last 4 wells of each cell type were treated with both carboplatin 43 μM and AZD7762 (CHK1/CHK2 inhibitor) (1.25 μM) for 72 h. Crystal violet assay was then performed as described above. The optical density of carboplatin (43 μM) only treated cells was normalized to the mean of the control cells of the same cell type and multiplied by 100 to calculate the percentage of cell viability which was subtracted from 100 to calculate the cytotoxicity percentage. The optical density of carboplatin (43 μM) and AZD7762 (1.25 μM) treated cells was also normalized to the mean of the control cells of the same type and using the calculation mentioned above, the cytotoxicity percentage was calculated.
Statistical analysis. Data are expressed as mean±SEM and analyzed by GraphPad prism v9 and Excel via student t-test or one-way ANOVA unless stated otherwise. Both crystal violet uptake assays and western blots were analyzed within the linear range of the assay.
Results
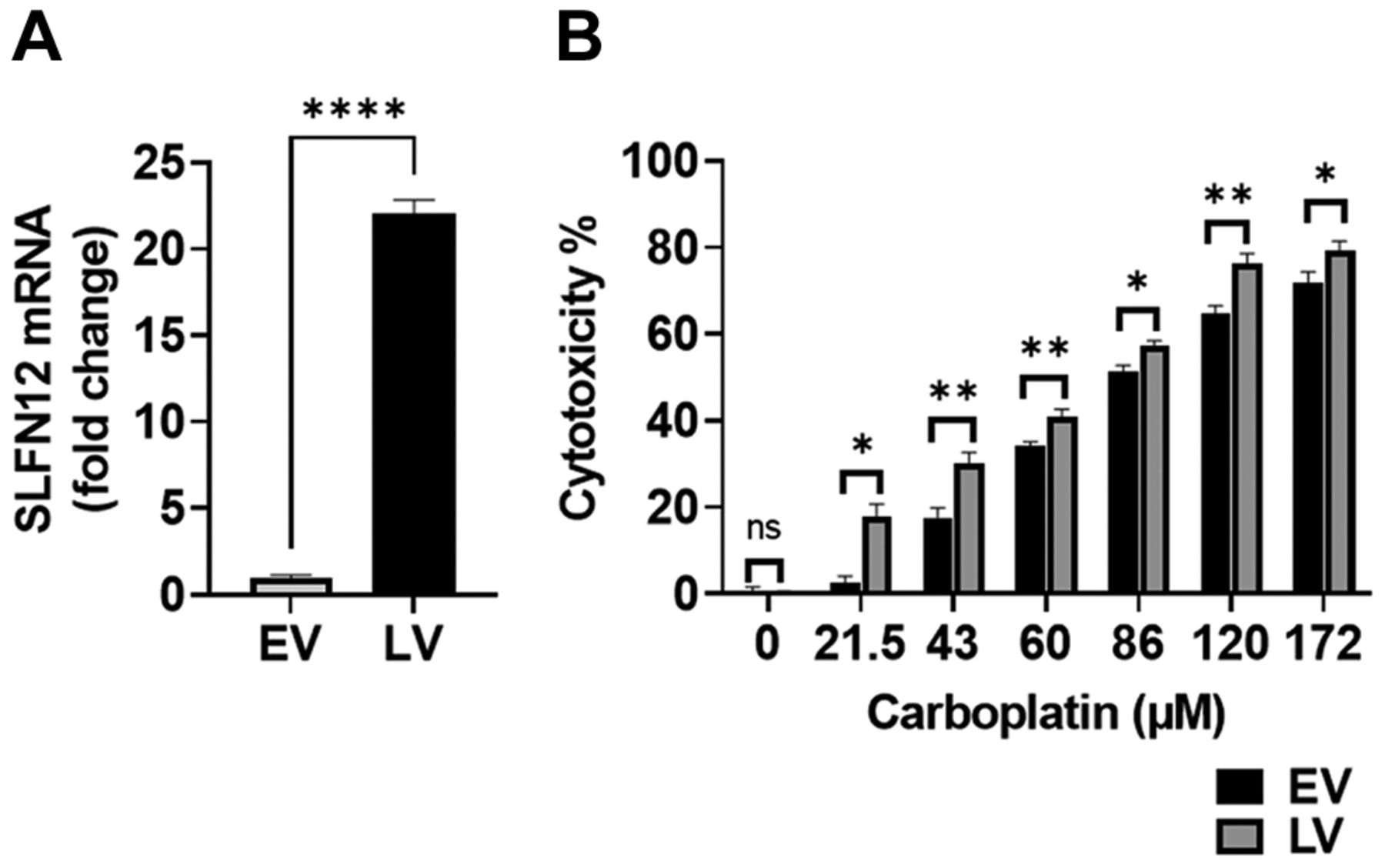
LV are more sensitive to carboplatin than EV. SLFN12-over-expressing cells (LV) (Figure 1A) and control cells (EV) were treated with 0-172 μM carboplatin for 72 h. Carboplatin cytotoxicity in LV was significantly higher than that observed in EV at all doses studied (Figure 1B). For instance, at 21.5 μM carboplatin, we observed 2.5±1.7% cytotoxicity for EV and 17.7±3.1% for LV (n=3, p<0.05), whereas at 172 μM, EV cytotoxicity was 71.7±2.7% compared to LV cytotoxicity of 79.4±2.1%. (n=6, p<0.05).

SLFN12 over-expressing cells are more sensitive to carboplatin. (A) RT-qPCR analysis demonstrates over-expression of SLFN12 in cells stably transfected with SLFN12 using a lentivirus that encodes for SLFN12 (LV) as opposed to cells infected with an empty vector lentivirus (EV). (B) Cytotoxic effect of treatment with 0-172 μM carboplatin for 72 h on control cells infected with an empty vector lentivirus (EV, black bars) and cells over-expressing SLFN12 after infection with a lentivirus encoding SLFN12 (LV, grey bars). LV cytotoxicity was higher than EV cytotoxicity for all carboplatin doses studied (n=3-36, ns: not significant, *p<0.05, **p<0.01).
LV are more sensitive to paclitaxel than EV. LV and EV were treated with 0-80 nM paclitaxel for 24 h. There were less viable LV compared to EV after treatment at all doses except 80 nM, which was the highest dose used (Figure 2). For instance, at 5 nM paclitaxel, we observed 2.9±1.6% cytotoxicity for EV and 11.3±1.0% for LV (n=6, p<0.01). At 60 nM, cytotoxicity was 50.3±0.8% for EV but 55.8±0.4% for LV (n=6, p<0.001). At 80 nM, cytotoxicity was similarly high in both LV and EV.

SLFN12 over-expressing cells are more sensitive to paclitaxel. Cytotoxic effect of treatment with 0-80 nM paclitaxel for 24 h on control cells infected with an empty vector lentivirus (EV, black bars) and cells over-expressing SLFN12 after infection with a lentivirus encoding SLFN12 (LV, grey bars). LV cytotoxicity was higher than EV cytotoxicity for all paclitaxel doses studied except 80 nM (n=6-51, ns: not significant, *p<0.05, **p<0.01, ***p<0.001).
LV are more sensitive to zoledronic acid than EV. LV and EV were treated with 0-30 μM zoledronic acid for 24 h. The LV were less viable than EV after treatment at all doses except for 7.5 μM, which was the lowest dose used (Figure 3). For instance, at 15 μM zoledronic acid, we observed 5.4±0.8% cytotoxicity for EV and 10.0±1.1% for LV (n=6, p<0.01). At 30 μM, cytotoxicity was 26.3±2.8% for EV but 36.5.8±1.2% for LV (n=6, p<0.01). At 7.5 μM, cytotoxicity was similarly low in both LV and EV.

SLFN12 over-expressing cells are more sensitive to zoledronic acid. Cytotoxic effect of treatment with 0-30 μM zoledronic acid for 24 h on control cells infected with an empty vector lentivirus (EV, black bars) and cells over-expressing SLFN12 after infection with a lentivirus encoding SLFN12 (LV, grey bars). LV cytotoxicity was higher than EV cytotoxicity for all zoledronic acid doses studied except for the 7.5 μM dose (n=3-21, ns: not significant, **p<0.01).
LV are more sensitive to camptothecin (CPT) than EV. LV and EV were treated with 0-80 μM CPT for 24 h. CPT cytotoxicity for LV was significantly higher than that for EV at all doses studied (Figure 4). For instance, at 40 μM CPT, we observed 21.0±1.6% cytotoxicity for EV and 28.6±1.3% for LV (n=12, p<0.01), whereas at 80 μM, EV cytotoxicity was 30.9±1.5% compared to 37.0±1.0% in LV (n=6, p<0.01).

SLFN12 over-expressing cells are more sensitive to camptothecin (CPT). Cytotoxic effct of treatment with 0-80 μM CPT for 24 h on control cells infected with an empty vector lentivirus (EV, black bars) and cells over-expressing SLFN12 after infection with a lentivirus encoding SLFN12 (LV, grey bars). LV cytotoxicity was higher than EV cytotoxicity for all CPT doses studied (n=6-24, ns: not significant, **p<0.01).
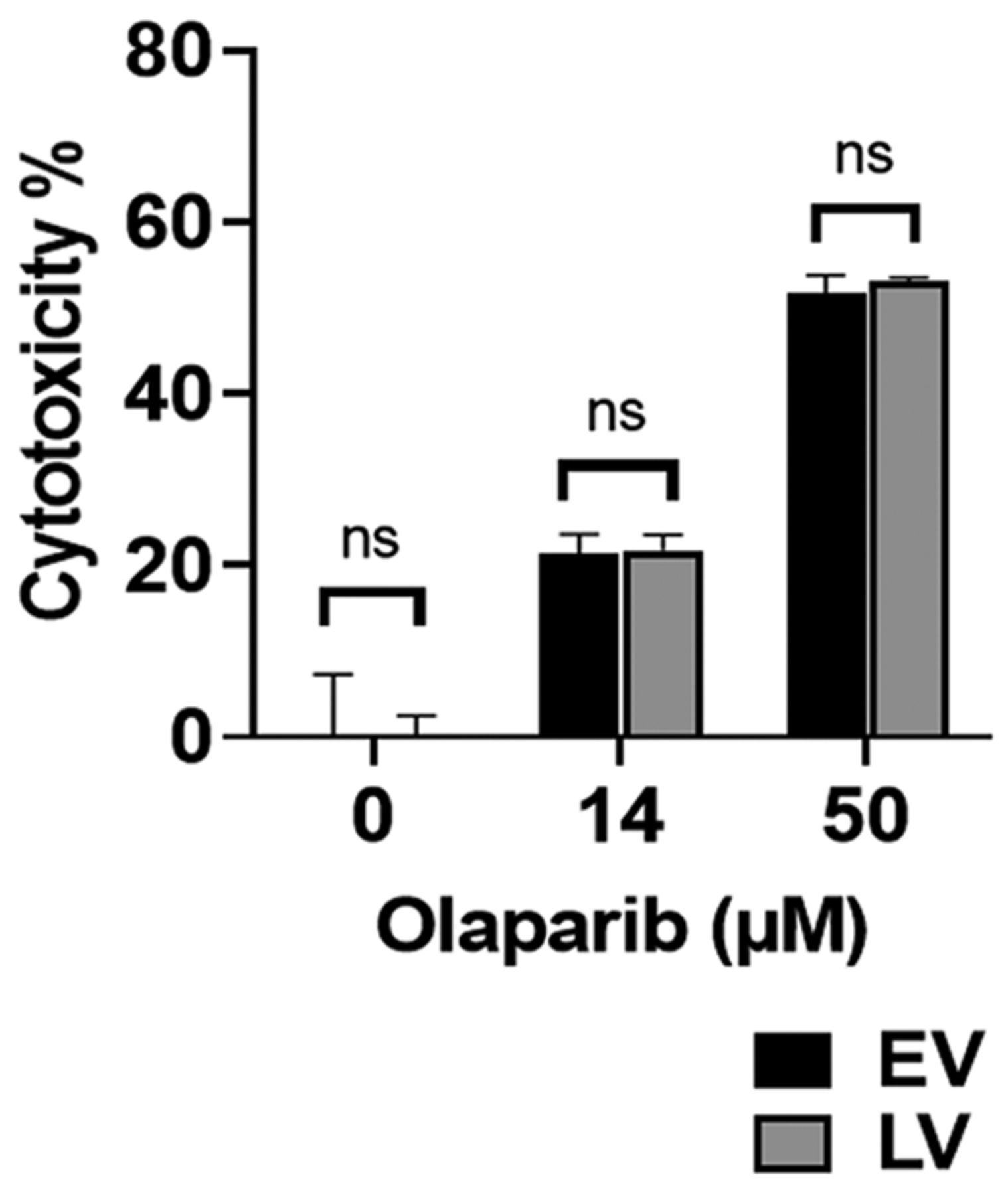
LV and EV are similarly sensitive to olaparib. LV and EV were treated with 0-50 μM olaparib for 72 h. Olaparib cytotoxicity for LV was similar to that for EV with no significant difference at any dose studied (Figure 5). For instance, at 15 μM olaparib, we observed 21.3±2.3% cytotoxicity for EV and 21.8±1.8% for LV (n=3), while at 50 μM EV cytotoxicity was 51.8.±2.0% compared to LV cytotoxicity of 53.2±0.4% (n=3).

SLFN12 over-expressing cells are as sensitive to olaparib as baseline expressing cells. Cytotoxic effect of treatment with 0-50 μM olaparib for 72 h on control cells infected with an empty vector lentivirus (EV, black bars) and cells over-expressing SLFN12 after infection with a lentivirus encoding SLFN12 (LV, grey bars). LV cytotoxicity was similar to EV cytotoxicity for all olaparib doses studied with no significant difference (n=3-6, ns: not significant).
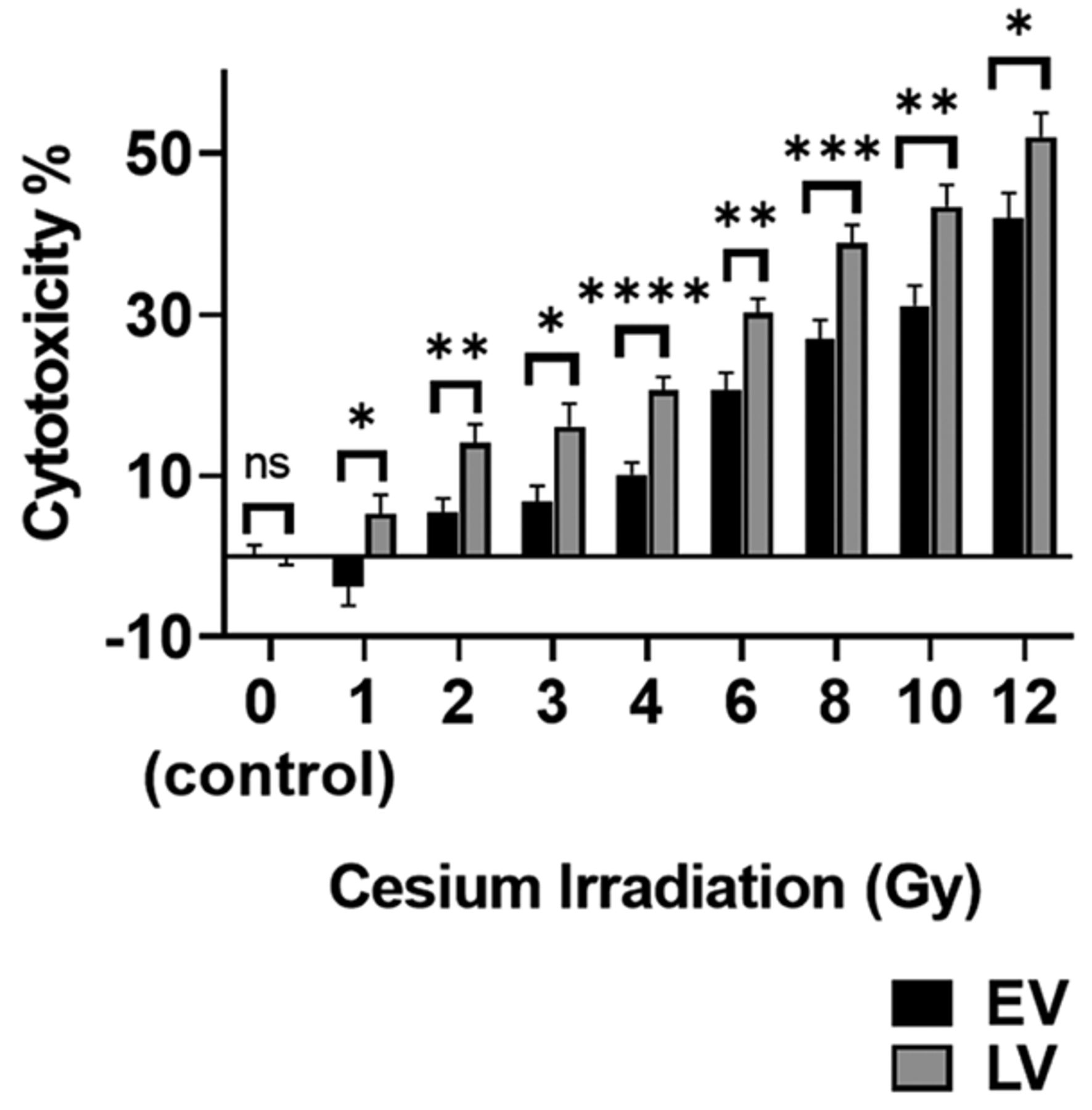
LV are more sensitive to Cesium irradiation than EV. LV and EV were exposed to 0-12 Gy cesium irradiation. Cesium irradiation caused significantly more cytotoxicity in LV than in EV at all doses studied (Figure 6). For instance, at 2 Gy, we observed 5.5±1.7% cytotoxicity for EV and 14.1±2.2% for LV (n=24, p<0.01), while at 12 Gy EV cytotoxicity was 41.9±3.0% compared to LV cytotoxicity of 52.0±2.9%. (n=12, p<0.05).

SLFN12 over-expressing cells are more sensitive to cesium irradiation. Cytotoxic effect of treatment with 0-12 Gy cesium irradiation after 24 h on control cells infected with an empty vector lentivirus (EV, black bars) and cells over-expressing SLFN12 after infection with a lentivirus encoding SLFN12 (LV, grey bars). LV cytotoxicity was higher than EV cytotoxicity for all cesium irradiation doses studied (n=12-36, ns: not significant, *p<0.05, **p<0.01, ***p≤0.001, ****p≤0.0001).
Over-expression of SLFN12 decreases the phosphorylation of CHK1 and CHK2 in comparison to low level expressing cells. The heightened sensitivity of SLFN12-over-expressing cells to DNA-damaging agents suggested that over-expression of SLFN12 might influence the DNA damage response. Therefore, we analyzed the kinetics of CHK1 and CHK2 protein phosphorylation in LV and EV after DNA damage was induced by 1 h of CPT treatment. Cells were treated with CPT for 1 h. The phosphorylation of CHK1 and CHK2 at 0 h (prior to treatment) and at 1 h after CPT treatment was compared by western blot. All data were normalized to EV at 1 h. CHK1 phosphorylation in LV at 0 h prior to treatment was significantly higher than that observed in EV (0.4±0.05 with LV vs. 0.23±0.03 with EV, n=5, p<0.05) suggesting a reduced basal proliferation of LV compared to that of EV (Figure 7A). In contrast, basal CHK2 phosphorylation did not differ significantly between EV and LV (Figure 7B). The phosphorylation of both CHK1 and CHK2 in LV 1 h after treatment was significantly lower than that in EV, indicating a decrease in the phosphorylation of CHK1 and CHK2 in response to DNA damage. For instance, LV CHK1 phosphorylation at 1 h was 83±3% of EV CHK1 phosphorylation (n=23, p<0.0001) (Figure 7A), whereas LV CHK2 phosphorylation 1 h after treatment was 74±4% of EV CHK2 phosphorylation (n=12, p<0.0001) (Figure 7B).
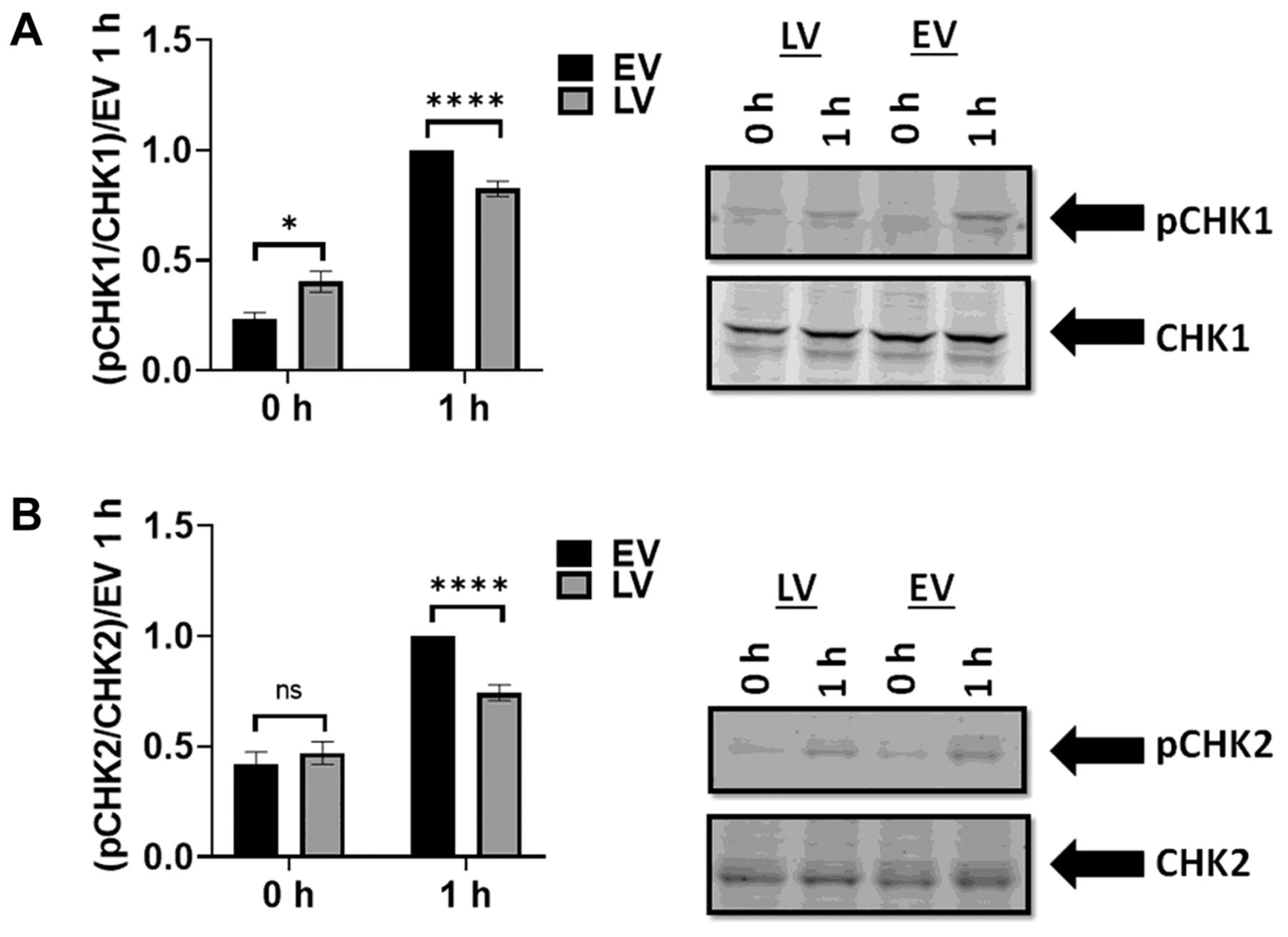
SLFN12 over-expression reduces phosphorylation of CHK1 and CHK2 in response to a DNA damaging agent. (A) Western blot analysis of phosphorylated CHK1 and total CHK1. Representative blot images of EV and LV without CPT treatment (denoted 0-h) and 1 h after CPT treatment are shown. (Data are normalized to EV at 1 h). Basal phosphorylation of CHK1 (0-h) is less in EV than in LV (n=5, *p<0.05), while CHK1 phosphorylation increases more in EV than in LV at 1 h after CPT treatment (n=23, ****p<0.0001). (B) Analysis of phosphorylated and total CHK2 by western blot. Representative blot images of EV and LV with no CPT treatment (denoted 0-h) and 1 h after CPT treatment are shown. (Data are normalized to EV at 1 h). There was no significant difference between basal EV and LV CHK2 phosphorylation (0-h) (n=5, p=0.5, ns: not significant). In contrast, phosphorylation of CHK2 in EV is significantly greater than in LV 1 h after CPT treatment (n=12, ****p<0.0001).
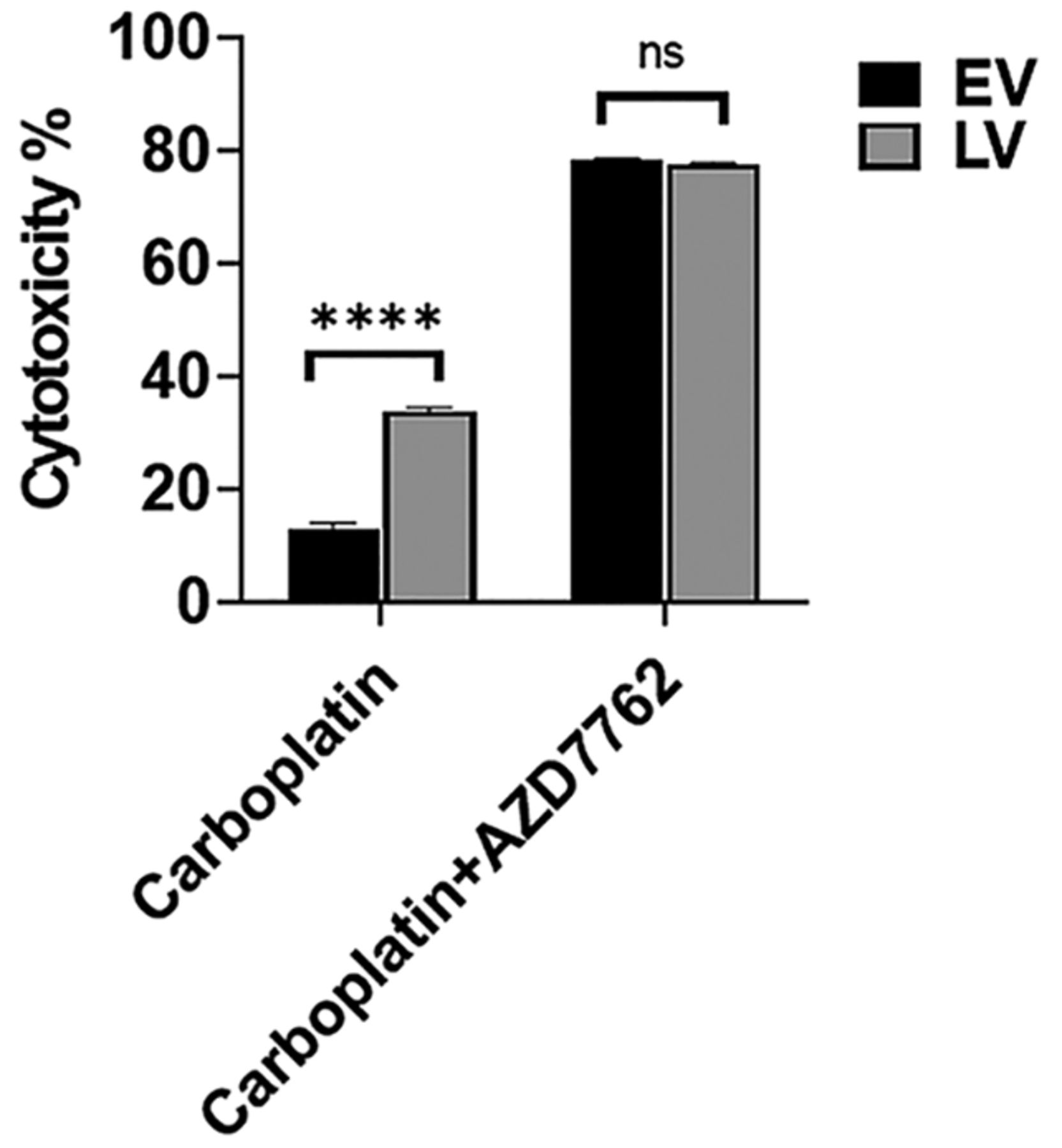
CHK1/CHK2 inhibition abolished differences in sensitivity to carboplatin between LV and EV. To confirm that the effect of SLFN12 on phosphorylation of CHK1 and CHK2 might contribute to the sensitization of cells to DNA damaging agents, LV and EV were treated with the DNA damaging agent carboplatin (43 μM) in the presence or absence of the CHK1/CHK2 inhibitor AZD7762 (1.25 μM) for 72 h. Carboplatin again caused significantly more cytotoxicity in LV than EV (n=20, p<0.0001), but this difference was abolished by treatment with AZD7762 (Figure 8).

The increased sensitivity to carboplatin induced by SLFN12 over-expression is abolished by CHK1/CHK2 inhibition. Cytotoxic effect of treatment with 43 μM carboplatin for 72 h in the absence or presence of 1.25 μM AZD7762 on control cells infected with an empty vector lentivirus (EV, black bars) and cells over-expressing SLFN12 after infection with a lentivirus encoding SLFN12 (LV, grey bars). LV cytotoxicity was higher than EV cytotoxicity for carboplatin 43 μM in the absence of AZD7762 (n=20, ****p≤0.0001). In contrast, LV cytotoxicity after 43 μM carboplatin was similar to that observed in EV when both had been treated with 1.25 μM AZD7762 (n=20).
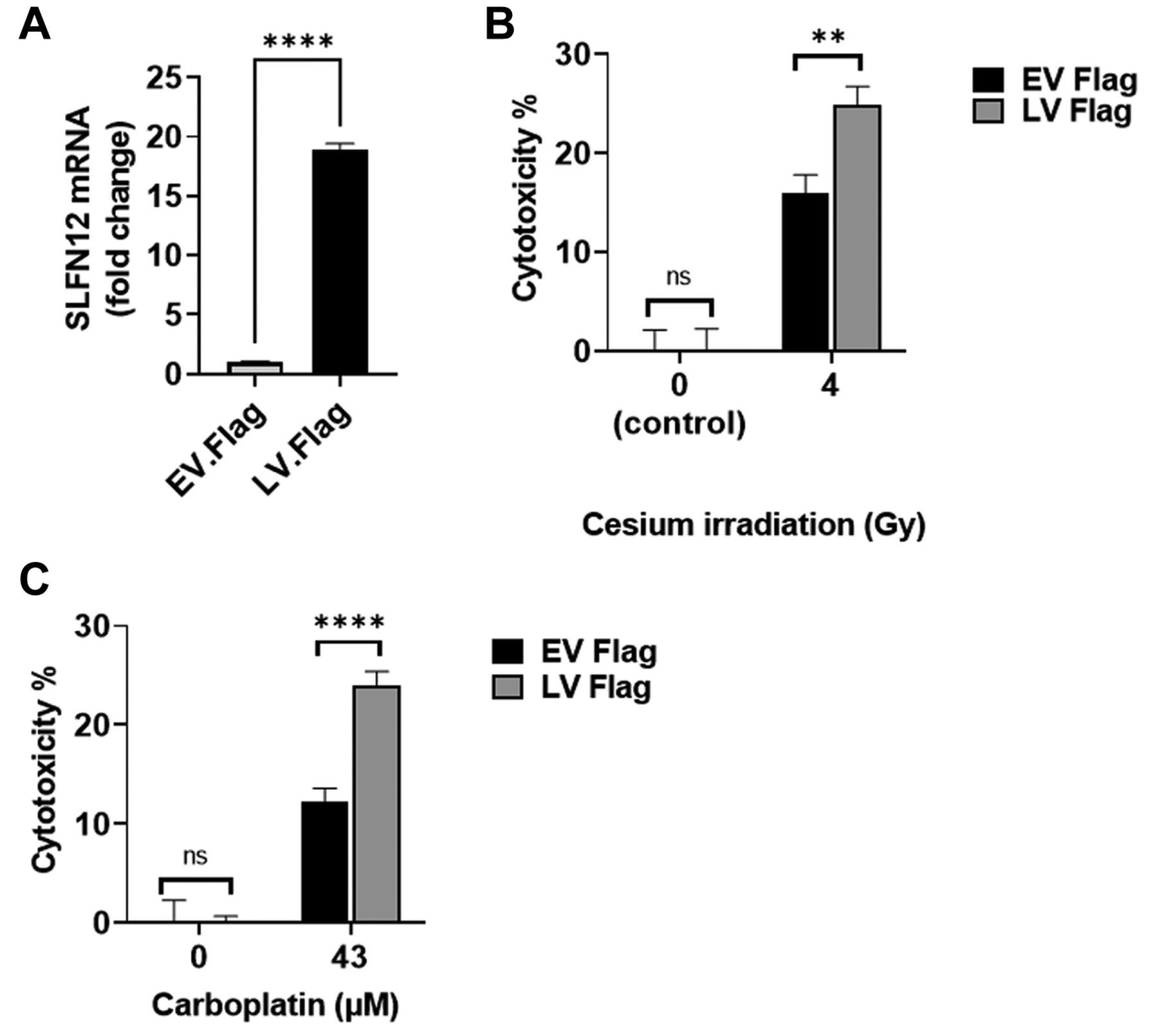
LV-Flag are more sensitive to carboplatin and cesium irradiation than EV-Flag. To confirm our previous results in a different SLFN12 over-expressing cell clone and with a different SLFN12 construct, we treated MDA-MB-231 cells over-expressing SLFN12 (LV-Flag) and baseline expressing cells (EV-Flag) (Figure 9A) with 43 μM carboplatin for 72 h or with 4 Gy cesium irradiation. We observed increased cytotoxicity in LV-Flag cells compared to that in EV-Flag cells in response to either cesium irradiation or carboplatin. For instance, after 4 Gy cesium irradiation, we observed 16.0±1.8% cytotoxicity in EV-Flag and 24.9±1.8% in LV-Flag (Figure 9B, n=24, p<0.01). After treatment with 43 μM carboplatin, EV-Flag cytotoxicity was 12.2±1.3% compared to 24.0.±1.3% in LV-Flag (Figure 9C, n=15, p<0.0001).
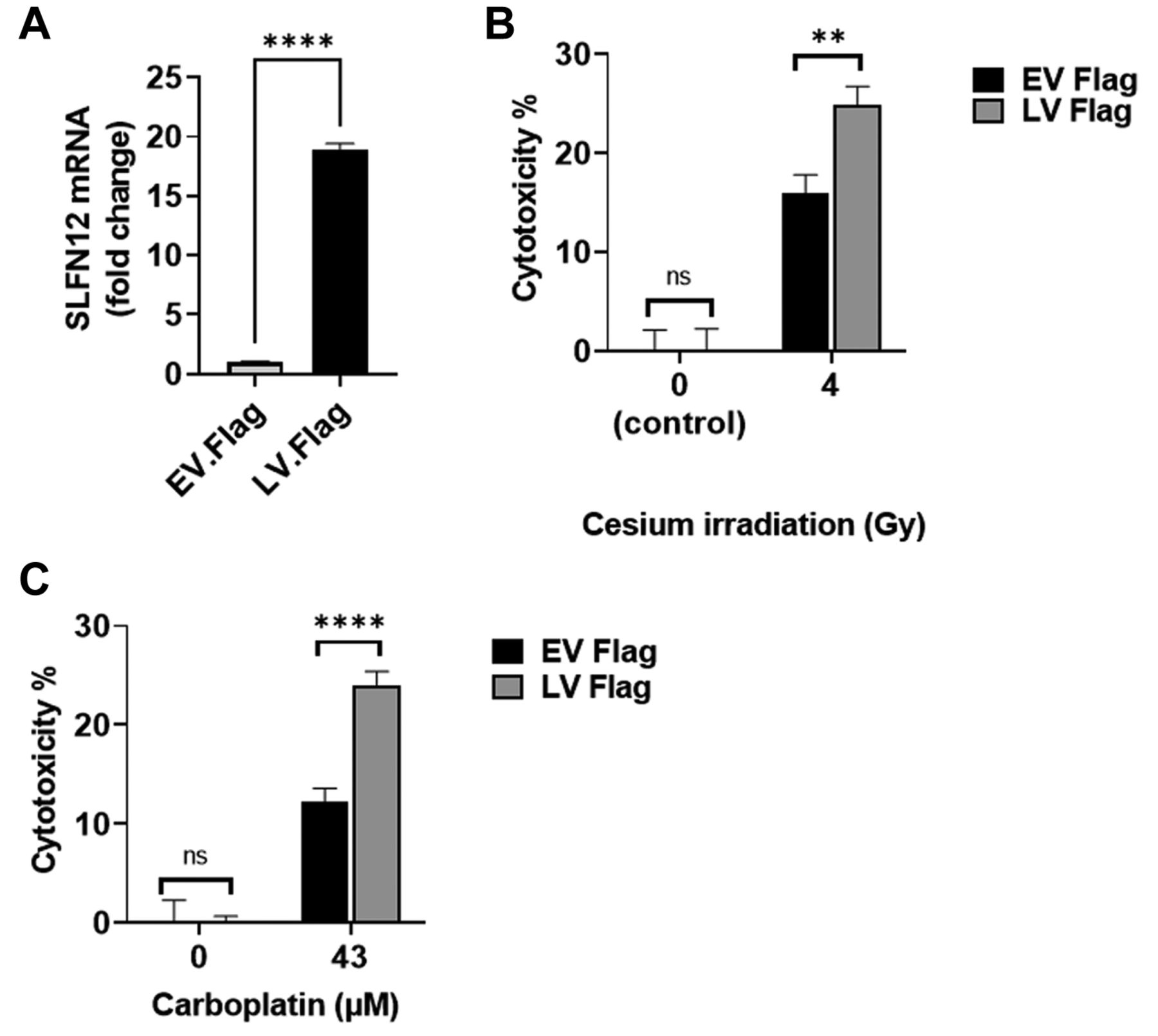
FLAG tagged SLFN12 also increases sensitivity of MDA-MB-231 cells to cesium irradiation or carboplatin. (A) RT-qPCR analysis demonstrates over-expression of SLFN12 in MDA-MB-231 cells stably transfected with SLFN12 using a lentivirus that encodes for SLFN12 (LV-Flag) as opposed to cells infected with an empty vector lentivirus (EV-Flag). (B) Cytotoxic effect of 4 Gy cesium irradiation for 24 h on control cells infected with an empty vector lentivirus (EV-Flag, black bars) and cells over-expressing SLFN12 after infection with a lentivirus encoding SLFN12 (LV-Flag, grey bars). LV-Flag cytotoxicity was higher than EV-Flag cytotoxicity (n=24, **p<0.01). (C) Cytotoxic effect of treatment with 43 μM carboplatin for 72 h on control cells infected with an empty vector lentivirus (EV-Flag, black bars) and cells over-expressing SLFN12 after infection with a lentivirus encoding SLFN12 (LV-Flag, grey bars). LV-Flag cytotoxicity was higher than EV-Flag cytotoxicity (n=15, ****p<0.0001).
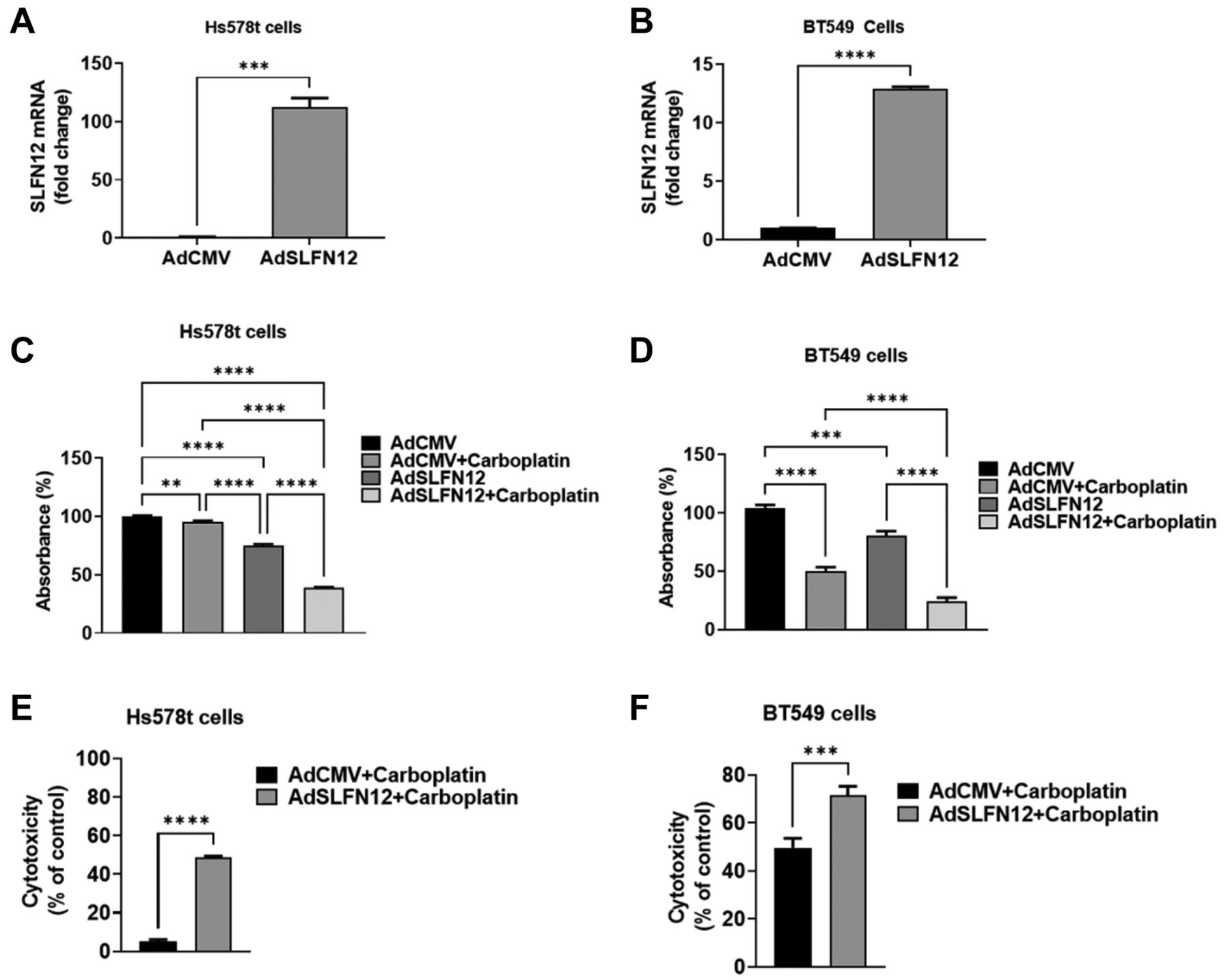
SLFN12 transient over-expression sensitizes Hs578t and BT549 TNBC cells to carboplatin. To confirm that the effect of SLFN12 stable over-expression in sensitizing MDA-MB-231 cells was not idiosyncratic to a single cell line, we used an adenoviral vector to transiently over-express SLFN12 in Hs578T and BT-549 TNBC cells (Figure 10A and B). Infection with the SLFN12 adenoviral vector reduced Hs578t (Figure 10C) and BT549 cell numbers (Figure 10D) compared with infection with the empty vector control, consistent with an overall slowing of proliferation. However, SLFN12 also increased the sensitivity of both BT549 and Hs578T cells to the cytotoxic effect of carboplatin. When we calculated the ratios of cell numbers after carboplatin treatment for each cell line to cell numbers prior to carboplatin treatment, we found that SLFN12 over-expression significantly increased the cytotoxic effect of carboplatin in Hs578t (Figure 10E) and BT549 cells (Figure 10F).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
SLFN12 increases the cytotoxic effect of carboplatin in Hs578t and BT549 cells. (A, B) Primer-probe RT-qPCR analysis of SLFN12 mRNA levels, 48-h after transduction with adenoviral vector expressing SLFN12 (AdSLFN12) or control empty vector (AdCMV) in Hs578t (A) and BT549 (B) cells. HPRT was used as a reference gene. (n=3, *** p≤0.001, **** p≤0.0001). (C, D) Number of Hs588T (C) and BT549 (D) cells, measured by crystal violet uptake, after 72 h of transduction with viral vector expressing SLFN12 (AdSLFN12) or the control vector (AdCMV) in combination with 43 μM of carboplatin (added simultaneously with the viral vector transduction). AdSLFN12 transduction reduces cell numbers, while the simultaneous addition of carboplatin further reduces cell number (data normalized to AdCMV group, n=15, **p<0.01, ***p≤0.001, ****p≤0.0001, one-way ANOVA). (E, F) Cell cytotoxicity calculated from the cell number data shown in Figures C and D by measuring the ratio of each carboplatin-treated group to its own control without carboplatin shows that the combination of carboplatin (43 μM) and SLFN12 over-expression (AdSLFN12) increases the cytotoxic effect of carboplatin in (E) Hs578t, and (F) BT549 cells. (***p≤0.001, ****p≤0.0001, Student’s t-test).
Discussion
TNBC is a uniquely aggressive type of breast cancer with poor prognosis and limited treatment options. These tumors are not only unresponsive to endocrine or other targeted therapy but also relatively chemoresistant and radioresistant (3-5). SLFN12 is a novel protein that correlates with prognosis in TNBC patients, an effect previously attributed to the intrinsic effects of SLFN12 on tumor cell proliferation and differentiation (4). Our study suggests that in addition to altering intrinsic tumor biology, SLFN12 increases the sensitivity of TNBC cell to radiation and cytotoxic drugs by reducing the phosphorylation of both CHK1 and CHK2, perhaps further contributing to differences in survival between patients with high SLFN12 and low SLFN12 tumors.
Gene-chip microarray analysis of human TNBC tumors demonstrates that SLFN12 expression can vary more than 200-fold, with probe expression in one study varying from 3-730 (18). Such differences in SLFN12 expression are known to be prognostically important (4), although the impact of chemotherapy choice on survival has not yet been elucidated. Nevertheless, such variability in human tumors is somewhat reassuring that the 30-fold difference in SLFN12 over-expression levels in the cells that we studied here may well be pathophysiologically relevant to human TNBC.
Although no other Schlafen has been reported to sensitize cancer cells to zoledronate, our data shows that SLFN12 sensitizes the TNBC cells to zoledronic acid, a bisphosphonate reported to have anticancer effects in breast cancer (19, 20). This raises the possibility that zoledronate might be more effective in TNBC tumors that express high levels of SLFN12.
The effect of SLFN12 on chemosensitivity and radiosensitivity has not previously been investigated, but there are reports describing SLFN11 effects. Similar to our SLFN12 observations, SLN11 sensitizes cancer cells to radiation (21), carboplatin, and camptothecin (22, 23). However, SLFN11 also sensitizes cancer cells to the poly (ADPribose) polymerase (PARP) inhibitor olaparib (22), but not to the microtubule inhibitor paclitaxel (24). In contrast, our data shows that SLFN12 sensitizes TNBC cancer cells to paclitaxel, but not to olaparib. This likely reflects the differences in the mechanisms by which SLFN12 and SLFN11 act. SLFN11 is a long Schlafen, which possesses an extra c-terminal domain that harbors a nuclear import signal, with characteristic ability of DNA binding in the nucleus (4, 11). In contrast, SLFN12 lacks such a domain and localizes in the cytoplasm (4, 11). Mechanistically, SLFN11 works by accessibility induction of the genome-wide chromatin during replication stress in response to DNA damaging agents at promoter regions (9, 25). In addition, SLFN11 is recruited to the stressed replication forks where nascent DNA binding and replication helicase subunit minichromosome maintenance complex component 3 (MCM3) initiation takes place (9, 26). In contrast, SLFN12 is a multifunctional protein that has been reported to induce differentiation by reducing ZEB1 translation in TNBC (4, 9) as well as to reduce c-myc translation in lung adenocarcinoma (9, 10) and to induce differentiation by binding serpinb12 to modulate deubiquitylases (USP14 and UCHL5) in a colon cancer cell line (9).
Our current work suggests that CHK1 is more phosphorylated in cancer cells that over-express SLFN12 even at baseline, before treatment with any DNA-damaging agent. Unlike CHK1, phosphorylation of CHK2 was not altered by SLFN12 over-expression prior to DNA damage, indicating a specific effect of SLFN12 on CHK1. In normal cells, CHK1 controls DNA replication during S-phase, spindle checkpoint during M-phase and mitotic entry (27, 28). Inhibition of CHK1 increases DNA synthesis and replication (27, 28), while CHK1 activation in normal cells slows their proliferation because of decreased initiation of DNA replication. Thus, this increased CHK1 phosphorylation may contribute to the slower proliferation observed in TNBC cells with high SLFN12 levels (4), potentially contributing to intrinsically less aggressive behavior even independently of the response to therapy in high SLFN12 TNBC.
However, in the setting of DNA damage, the phosphorylation of both the checkpoint maintenance proteins CHK1 and CHK2 is critical for initiating the DNA repair response to counteract the effect of the DNA damage (29-32) in both normal and cancer cells. Inhibiting CHK1/CHK2 phosphorylation sensitizes cancer cells to DNA damage induced by agents like cytotoxic drugs and radiation (29, 33). Our data showed that after treatment with DNA damaging agents, CHK1 and CHK2 phosphorylation is lower in SLFN12 over-expressing TNBC cells. As this would indicate a reduced DNA repair response in cancer cells that express relatively higher levels of SLFN12, it may explain the increased sensitivity of SLFN12 over-expressing cancer cells to DNA damaging drugs (carboplatin and camptothecin) and irradiation. Although paclitaxel is a microtubule inhibitor, it has been reported that CHK1 plays a role in M-phase checkpoint protection; furthermore, CHK1 inhibition attenuates the activation of p42/p44 (anti-apoptotic), which contributes to the sensitization of cancer cells to paclitaxel-induced cell death (34). Such a mechanism aligns with our finding of paclitaxel-sensitization in SLFN12 over-expressing TNBC cells.
Conclusion
Taken together with previous analyses of SLFN12 effects on TNBC survival (4), these results would be consistent with a model in which SLFN12 levels affect not only intrinsic tumor biology but also the tumor’s response to treatment. While the correlation between SLFN12 levels and individual chemotherapeutic responses awaits further study, these results raise the possibility that in the future, SLFN12 levels might be used to choose the most appropriate chemotherapeutic strategy for patients with TNBC. Moreover, as we continue to explore the mechanisms by which SLFN12 exerts its effects, these results raise the possibility that targeting and activating the SLFN12 pathway might act synergistically with conventional cytotoxic chemotherapy or radiation in TNBC that expresses only low SLFN12 levels.
Acknowledgements
The Authors would like to thank Dr. David Bradley and Steven Adkins for their assistance and use of the Gammacell 3000 Elan cesium irradiator.
Footnotes
Authors’ Contributions
Conceptualization, A.A.R.E, S.A.-M., E.E.V.-D.,M.D.B; methodology, A.A.R.E, S.A.-M.,E.E.V.-D.,M.D.B; formal analysis, A.A.R.E, S.A.-M., E.E.V.-D., M.D.B; investigation, A.A.R.E, S.A.-M.; data curation, A.A.R.E, S.A.-M.,M.D.B; writing—original draft preparation, A.A.R.E, S.A.-M.,M.D.B; writing—review and editing, A.A.R.E, S.A.-M., E.E.V.-D. M.D.B; visualization, A.A.R.E, S.A.-M., E.E.V.-D. M.D.B. All Authors have read and agreed to the published version of the manuscript.
Conflicts of Interest
The Authors declare no conflicts of interest in relation to this study.
- Received January 19, 2022.
- Revision received January 31, 2022.
- Accepted February 2, 2022.
- Copyright© 2022, International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved
This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY-NC-ND) 4.0 international license (https://creativecommons.org/licenses/by-nc-nd/4.0).