


Abstract
The nuclear factor of activated T cells (NFAT) mediates a cyclosporin A (CsA)- and FK506-suppressible transcriptional program in lymphocytes after antigen-stimulated phospholipase C activation. Nonlymphoid cells also express NFAT isoforms, raising the possibility that these isoforms can be regulated by other extracellular stimuli. This study sought to determine whether histamine can trigger NFAT-mediated transcription in human umbilical vein endothelial cells (HUVEC), using a retrovirus-based luciferase reporter driven by a well characterized, NFAT-specific enhancer. Luciferase levels are induced up to 60-fold over basal levels after costimulation of HUVEC with Ca2+-mobilizing drugs and a phorbol ester, a response that is 20-fold greater than that observed when HUVEC are stimulated with either drug alone. These synergistic responses are inhibited in cells treated with CsA. CsA and FK506 also inhibit the luciferase response to histamine, indicating that histamine can induce NFAT-mediated transcription in HUVEC. To identify candidate genes in HUVEC that might be regulated by NFAT, the expression of several chemokine mRNAs was measured after histamine treatment. Of the mRNAs tested, only those encoding monocyte chemotactic protein-1 (∼2-fold over basal) and interleukin-8 (∼6-fold over basal) are induced by histamine; both of these responses are suppressed by CsA and FK506. The H1histamine receptor antagonist chlorpheniramine, but not the H2 receptor antagonist ranitidine, blocks the effects of histamine in this preparation. These data provide the first evidence for a physiological inducer of NFAT-mediated transcription in endothelial cells and support the hypothesis that NFAT participates in H1 histamine receptor-induced interleukin-8 gene expression.
Vascular tone can be acutely modulated by release of endothelium-derived vasotropic factors in response to cell surface receptor activation. Stimulation of these same receptors can have a more prolonged effect on vascular function by modulating endothelial cell gene expression. For example, activated endothelial cells transiently express factors involved in the trafficking of circulating immune cells across the endothelial cell barrier into underlying tissues (Springer, 1995; Ley, 1996; Luscinskas and Gimbrone, 1996). One general mechanism to control gene expression that is used in endothelium, as in all cells, is the convergence of intracellular signaling cascades to stimulate a wide variety of ubiquitous transcriptional coactivators. Transcription factors such as the NFκB and signal transducer andtrans-activator proteins (Collins et al., 1995), as well as the basic leucine zipper proteins, represented by the c-Fos, c-Myc, activating transcription factor, and cAMP response element-binding protein families of trans-activators, are involved in the induction of numerous genes in endothelial cells.
A family of transcription factors that have received little attention regarding the control of vascular gene expression are the NFATs. NFAT was first described as a T cell protein, induced by antigen receptor stimulation, that binds to a major upstream enhancer in the IL-2 cytokine gene (Durand et al., 1988). NFAT is now known to participate in the coordinate expression of several genes induced in lymphocytes during immune responses (Rao et al., 1997). Immunosuppressive agents such as CsA and FK506 inhibit NFAT-mediated transcription in lymphocytes (Clipstone and Crabtree, 1992; Bramet al., 1993), which is the principle mechanism thought to account for the clinical efficacy of these drugs (Schreiber and Crabtree, 1992). The control of NFAT-mediated transcription in lymphocytes is well understood and is strictly dependent on simultaneous signaling through Ca2+ and the MAP kinase pathways (Wu et al., 1995). In resting cells, inactive NFAT is heavily phosphorylated and sequestered in the cytoplasm. Signaling through the Ca2+ pathways activates the phosphatase calcineurin, resulting in dephosphorylation and nuclear translocation of NFAT. Calcineurin is inhibited by a complex formed between intracellular immunophilin proteins and the macrolide immunosuppressants, accounting for the mechanism whereby these drugs inhibit NFAT-mediated transcription (Flanagan et al., 1991; Clipstone and Crabtree, 1992; Jain et al., 1993; Ruff and Leach, 1995). Whether nuclear NFAT is capable of functioning as a transcriptional activator alone is presently uncertain. However, it is clear that NFAT assembles in a multimeric complex with AP1 transcriptional partners on composite, purine-rich/AP1-like, enhancer elements in target genes. In most instances, this is reflected as synergistic transcriptional induction that is markedly greater than that achieved by signaling through Ca2+ or MAP kinase cascades alone (Jain et al., 1992, 1993; Northrop et al., 1993).
Early notions that NFAT expression is restricted to lymphocytes are no longer tenable, but very little is known about the activation, targets, or roles of NFAT in nonlymphoid cells. Humans and mice possess at least four genes encoding distinct NFAT isoforms (for review, see Raoet al., 1997). As a group, the mRNAs transcribed from these genes are expressed widely, but differentially, in several tissues (for example, see Hoey et al., 1995). Nevertheless, the specific nonlymphoid cell types in which NFAT proteins are expressed are for the most part unknown. Both in human subjects and in animal models (Younget al., 1995), CsA and FK506 invariably induce cardiovascular toxicity, notably including the development of reversible hypertension of unknown mechanism (First et al., 1994). Taken together, an intriguing possibility raised by these observations is that disruption of NFAT-mediated processes in vascular cells might be responsible, in part, for the side effects associated with immunosuppressive therapy.
A recent study showed that CsA-sensitive induction of NFAT-mediated transcription can be elicited in HUVEC and that NFAT participates in the control of the GM-CSF/IL-6 locus and in E-selectin gene transcription in HUVEC (Cockerill et al., 1995). However, that study used the combined application of a strong Ca2+ ionophore and a phorbol ester, as stimulants of the calcineurin and MAP kinase pathways, respectively, necessary for activation of NFAT-mediated transcription. A crucial question that is still unanswered is whether physiological agonists, acting through natively expressed endothelial cell surface receptors, are capable of triggering NFAT-mediated transcription in these cells. The experiments shown here demonstrate that this is the case. Using a novel reporter gene strategy, we provide evidence entirely consistent with histamine-induced NFAT-mediated transcription in HUVEC and also consistent with the hypothesis that NFAT may participate in control of IL-8 gene expression in response to histamine stimulation.
Experimental Procedures
Materials.
The retroviral plasmid pLNCX was a gift from A. D. Miller (Fred Hutchinson Cancer Center, Seattle, WA). A plasmid encoding NFATc1 (pSH107c) was a gift from G. Crabtree and S. Ho (Standford University, Palo Alto, CA). Dr. R. J. Bram (St. Jude Children’s Hospital, Memphis, TN) kindly provided pNFAT-Luc. HUVEC were obtained from the Tissue Core Facility of the Emory Skin Diseases Research Center, at passage 2. Retroviral producer cells (Bing-CAK8; American Type Culture Collection CRL-11554) were obtained from the American Type Culture Collection (Rockville, MD). A mouse monoclonal antibody against NFATc1 (clone 7A6) (Ho et al., 1995) was purchased from Affinity Bioreagents (Golden, CO). Secondary antibodies were purchased from Jackson Immunoresearch Laboratories, (West Grove, PA). EGF, PDGF-BB, glutamine, and antibiotics for tissue culture were purchased from GIBCO-Life Technologies (Gaithersburg, MD). CsA was a gift from Sandoz Pharmaceuticals (East Hanover, NJ), and FK506 was generously provided by S. Thomas, Fujisawa USA (Melrose Park, IL). Purified human thrombin was a gift from S. Krishnaswamy (Department of Medicine, Emory University, Atlanta, GA). Sera were purchased from Irvine Scientific (Irvine, CA). Ranitidine was purchased from Research Biochemicals (Natick, MA). (+)-Chlorpheniramine maleate and other reagents were purchased from Sigma Chemical Co. (St. Louis, MO), unless specified otherwise.
Retroviral plasmid production and HUVEC infection.
Retroviral plasmids are maintained in Escherichia coli Top 10F′ cells (Invitrogen, Carlsbad, CA), using 100 μg/ml ampicillin and 12.5 μg/ml tetracycline in 2× YT medium, and grow poorly in other standard strains of E. coli. Plasmid pKA7 is a retroviral NFAT- luciferase reporter plasmid constructed in a modification of the vector pLNCX. It contains three copies of the distal NFAT enhancer element from the human IL-2 gene (Durand et al., 1988), which are placed upstream from a minimal human IL-2 promoter [base pairs −72 to +48 around the transcription start site (+1)]. These elements were derived as aBamHI-HindIII fragment from the plasmid vector pNFAT-Luc (Northrop et al., 1993). The luciferase coding sequence, including downstream polyadenylation signals, were derived as a HindIII-SacI fragment from the plasmid poLUC (Braiser et al., 1989). This NFAT-responsive luciferase cassette is expressed as an internal gene within the retroviral LTR in an opposite strand orientation, with its transcription directed toward the viral 5′ LTR promoter. Except for the retroviral platform, this reporter transcriptional cassette is identical to that previously described (Boss et al., 1996). The plasmid and sequence are available upon request. To produce infectious retrovirus, pKA7 was transfected (using CaPO4) into Bing Cak-8, helper virus-free, amphotropic, producer cells (Pear et al., 1993) grown in 10% fetal bovine serum in DMEM/bicarbonate with penicillin (100 units/ml) and streptomycin (100 μg/ml), in 100-mm-diameter culture dishes. Beginning 24 hr after transfection, the producer cells were placed in a 32° incubator at 5% CO2, in 6 ml of fresh culture medium. The supernatant containing viral particles was collected three times at 12-hr intervals thereafter, filtered through sterile 0.4-μm cellulose acetate disks, frozen in an ethanol/dry ice bath, and stored at −80° until used.
Normal HUVEC growth medium consisted of Cellgro medium M199 (purchased from Fisher Scientific). This was supplemented with 20% heat-inactivated fetal bovine serum, 8000 units/ml heparin (Elkins-Sinn, Cherry Hill, NJ), 25 mg of endothelial mitogen (Biomedical Technologies, Stoughton, MA), 2 mm glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin, and 250 ng/ml fungizone. To create NFAT-luciferase reporter cells, HUVEC were seeded onto gelatin-coated, 6- × 35-mm, multiwell, tissue culture plates and were infected with retroviral supernatants at 40–50% confluency the following day. Frozen retroviral supernatant was thawed and warmed to 37°, after which polybrene was added to achieve a final concentration of 8 μg/ml. This mixture was applied at 2 ml/well to the endothelial cells for 20–30 min, in a 37° incubator with 5% CO2. The plates were then centrifuged in a Beckman model GS-6R refrigerated tabletop centrifuge at 30–32° at 2500 rpm for 30 min, after which the retroviral medium was replaced with 2 ml/well of normal growth medium. This infection procedure was repeated three more times at 6–10 hr, 24 hr, and 30–32 hr after the initial infection. The pKA7-infected cells, referred to as pKA7/HUVEC, were grown to confluency, pooled, and assayed for luciferase activity at passages 4–8.
To estimate the efficiency of retroviral infection, one separate well on the culture plate was infected with retrovirus prepared from the plasmid MFG-lacZ, which carries a β-galactosidase reporter gene (Dwawan et al., 1991), each time cells were infected with pKA7. Infected cells expressing β-galactosidase were detected using a standard histochemical colorimetric assay (Dwawan et al., 1991). Based on these observations, the infection efficiency for HUVEC was estimated to vary between 40 and 90%.
Luciferase assays.
pKA7/HUVEC were grown on gelatin-coated, 12-well plates for 3–4 days, until confluent. CsA was delivered as a 10-fold concentrated solution in vehicle consisting of 0.025% ethanol and 0.001% Tween 80 (v/v) in normal growth medium. Other drugs were applied as 20-fold concentrated stock solutions in phosphate-buffered saline. After an incubation of 6–8 hr at 37° in 5% CO2, the medium was aspirated and the lysed cell supernatants were assayed for luciferase activity as described previously (Takeuchi et al., 1993).
Western blots.
Control cell extracts were prepared from COS-7 cells transfected with plasmid pSH107c (Ho et al., 1995), which expresses the human NFATc1 isoform, or with a sham plasmid. These and the endothelial cell extracts were prepared for SDS-polyacrylamide gel electrophoresis by lysis in RIPA-2 buffer (150 mm NaCl, 1% Nonidet P-40, 0.5% deoxycholate, 0.1% SDS, 50 mm Tris·HCl, pH 8) containing a cocktail of protease inhibitors. Protein concentrations were measured with a kit from Bio-Rad (Richmond, CA), using bovine γ-globulin as the protein standard. The proteins were transferred from the SDS-polyacrylamide gels to Immobilon P membranes (Millipore, Bedford, MA) in transfer buffer containing 0.05% SDS and 10% methanol in 50 mmTris-glycine. Membranes were blocked with 5% nonfat dried milk and 0.1% Tween 20 in 0.02 m Tris-buffered 0.5 msaline, pH 7.5, for 1 hr and were then incubated overnight at 4° with a 1/2000 dilution (in blocking solution) of anti-NFATc1 monoclonal antibody. Blots were developed using a chemiluminescence kit (Phototope-HRP; New England Biolabs, Natick, MA) with a horseradish peroxidase-conjugated anti-mouse IgG secondary antibody diluted (1/20,000) in Tris-buffered saline with 5% horse serum.
Intracellular Ca2+ measurements.
Confluent pKA7/HUVEC (passages 4–6) were washed with Ca2+/Mg2+-free HBSS, treated with 5–10 mg/ml type 1 collagenase (Worthington Biochemical, Freehold, NJ), gently scraped into centrifuge tubes, and washed with Ca2+/Mg2+-containing HBSS. After the cells were pelleted by centrifugation at 500 rpm in a tabletop centrifuge, they were resuspended in HEPES-buffered DMEM containing 0.5 mg/ml bovine serum albumin, pH 7.4. Fura-2/acetoxymethyl ester was added to a final concentration of 1 μm, and the cells were incubated in the dark at 37° for 15 min. They were then brought to a volume of 50 ml in DMEM with bovine serum albumin and were centrifuged at 500 rpm in a tabletop centrifuge at 4° for 10 min; the pellet was resuspended in Ca2+/Mg2+-containing HBSS. The cells were diluted with Ca2+/Mg2+-containing HBSS to 2–5 × 105 cells/ml and were distributed into 3-ml aliquots, which were kept on ice. Immediately before the assay, each aliquot was warmed for 1 min at 37° and collected by centrifugation. The pellet was resuspended in 1 ml of warmed (37°) Ca2+/Mg2+-containing HBSS, before transfer to a cuvette containing 2 ml of warmed Ca2+/Mg2+-containing HBSS. Drugs were added, with stirring, as 100-fold concentrated stock solutions, and the emission intensity at 510 nm was measured (at an excitation ratio of 340/380 nm) with a Perkin Elmer LS50 spectrometer.
Inositol phosphate assays.
Confluent HUVEC (passages 5–6) grown on 35-mm-diameter culture dishes were pretreated for 16–24 hr with 1 ml of growth medium containing 2.5 μCi ofmyo-[3H]inositol. After aspiration, the cells were incubated for 10 min at 37°, in the absence or presence of antagonists, in medium containing 5 mm LiCl. Histamine was then added to a final concentration of 100 μm, and the cells were stimulated for 45 min at 37° before medium aspiration and the addition of 1 ml of 20 mmformic acid to each well. Total inositol phosphates were subsequently isolated by chromatography on Dowex AG 1X8 resin (Bio-Rad), as described previously (Murphy et al., 1993).
Ribonuclease protection assays.
Confluent HUVEC (passages 4–7) grown on 35-mm-diameter culture dishes were pretreated for 30 min with antagonists (or their vehicle) and CsA or FK506 (or their vehicle) before the addition of histamine. As indicated in the figure legends, at times after this the medium was aspirated and 1 ml of Trizol (GIBCO-Life Technologies) was added to each well. RNA was extracted from the samples, resuspended in 50 μl of RNase-free water, and quantified by measurement of absorbance at 260 nm. Aliquots of total RNA (8–10 μg) were lyophilized under vacuum, in 200-μl microcentrifuge tubes, before resuspension in 8 μl of hybridization solution supplied with the Riboquant Multiprobe RPA kit (Pharmingen, San Diego, CA). Two microliters of [α-32P]UTP-labeled riboprobes in hybridization solution were then added, and the samples were overlaid with mineral oil before brief heating to 90° and then heating to 56° for 12–16 hr. The riboprobes were synthesized using T7 RNA polymerase, in a single reaction, from a mixture of templates supplied with the Pharmingen kit; they included probes for the chemokines Ltn (433 bases), RANTES (390 bases), IP-10 (349 bases), MIP-1β (314 bases), MIP-1α (256 bases), MCP-1 (231 bases), IL-8 (204 bases), I-309 (191 bases), and the ubiquitously expressed genes L32 (141 bases) and GAPDH (125 bases). After hybridization, the samples were digested for 45 min at 30° with a mixture of RNase A and RNase T1 (as recommended in the kit), treated with proteinase K, extracted with phenol/chloroform/isoamyl alcohol (50:50:0.5), precipitated with ethanol, and resolved on vertical 5% polyacrylamide/urea sequencing gels. The gel was developed on a Molecular Dynamics PhosphorImager, and the mRNA hybridization signals were measured by a volume integration protocol using ImageQuant software. To control for sample processing artifacts, the chemokine hybridization signals were divided by the signal for GAPDH in each sample, to obtain a ratio value. Neither CsA nor histamine affected GAPDH hybridization signals, as determined by independent analysis of the GAPDH data. The data are expressed as fold increases over basal levels of expression; the chemokine/GAPDH signal ratios were normalized to the ratio for cells not exposed to histamine.
Results
A selective monoclonal antibody against NFATc1 (Ho et al., 1995) recognizes a protein extracted from HUVEC cultures, as detected by Western blotting (Fig. 1). This antibody also recognizes a protein in control extracts from COS-7 cells transfected with a cloned NFATc1 plasmid but recognizes nothing in extracts from sham-transfected COS-7 cell controls. No immunoreactivity is observed in control blots upon omission of the primary antibody. The immunoreactive protein from HUVEC is heterogeneous in size, in the molecular weight range of 95,000–115,000. This heterogeneity is common in cells expressing NFAT isoforms and may reflect alternative splicing, multiple phosphorylation states of the protein (Ruff and Leach, 1995), or other post-translational modifications (Rao et al., 1997). These data extend previous studies by showing that HUVEC are capable of expressing the NFATc1 isoform (Cockerill et al., 1995). We were unable to determine by Western blot analysis, using commercially available antibodies, whether other NFAT isoforms are also present in HUVEC. Expression of additional isoforms in these cells remains a possibility.

NFATc1 immunoreactivity in HUVEC extracts. Western blots, using a selective monoclonal antibody against NFATc1, detected a protein in HUVEC (100 μg of protein) (lane 1) and COS-7 cells transfected with a NFATc1 expression plasmid (1 μg of protein) (lane 3) but detected nothing in extracts of COS-7 cells transfected with a sham plasmid (1 μg of protein) (lane 2). The relative molecular masses (Mr) of standard markers are shown. This result is representative of four experiments.
To measure NFAT-mediated transcription in HUVEC, the vector pKA7 was created; this vector has a NFAT-responsive enhancer coupled to the minimal IL-2 gene promoter driving a luciferase reporter. This was placed as an internal gene in a retroviral LTR. Construction of this reporter into a retroviral vector allows for much more efficient analysis of reporter activity in HUVEC cultures than is possible with standard plasmid DNA transfection approaches, which are limited by cellular toxicity of the transfection procedure, poor reproducibility, and low transfection efficiency. The NFAT-responsive promoter used here was subcloned from a well characterized plasmid reporter that responds specifically only to NFAT (Northrop et al., 1993; Bram and Crabtree, 1994; Boss et al., 1996), and this fidelity is conserved in the retroviral vector (see below). Inducible luciferase activity, consistent with a NFAT-mediated response, is expressed from this vector but not from control retroviral luciferase reporters that lack the NFAT enhancer triplet or the minimal IL-2 promoter (data not shown).
The prototypical lymphocyte NFAT-mediated transcriptional response is elicited in pKA7/HUVEC cultures by concomitant increases in intracellular Ca2+ concentrations and MAP kinase activity and is inhibited by CsA (Fig.2). Thapsigargin, which inhibits the reuptake of Ca2+ into intracellular stores, and the Ca2+-releasing ionophore ionomycin elicit 4–5-fold greater luciferase activities, compared with basal levels, when applied alone to pKA7/HUVEC, even at maximal doses. A similar small luciferase response is incurred using the protein kinase C activator PMA alone, to stimulate the MAP kinase pathways. However, costimulation of pKA7/HUVEC with PMA (100 nm) markedly potentiates dose-dependent luciferase responses induced by either thapsigargin (maximal response at 30 nm, 29-fold) or ionomycin (maximal response at 3 μm, 60-fold) (Fig. 2A). The responses to Ca2+ mobilization together with stimulation with PMA are inhibited by CsA treatment, in a dose-dependent manner (Fig. 2B). The synergistic responses and CsA sensitivity are entirely consistent with NFAT-mediated transcription.

Induction of a NFAT-responsive luciferase reporter gene in pKA7/HUVEC. A, Synergistic effects of Ca2+-mobilizing drugs and phorbol ester. HUVEC cultures containing a NFAT-responsive luciferase reporter were stimulated for 6 hr with various doses of ionomycin (▿), thapsigargin (○), or PMA (▵), with each inducing slight dose-dependent luciferase responses. Costimulation with PMA (100 nm) potentiates the responses elicited by ionomycin (•) or thapsigargin (♦). B, Suppression of NFAT-mediated transcription by CsA. Luciferase responses induced by costimulation with 3.2 μmionomycin and 100 nm PMA (•), or with 10 nmthapsigargin and 100 nm PMA (○), are blocked by CsA, when CsA is applied 30 min before and during exposure to the drugs.Points, mean ± standard error of three experiments, each performed in duplicate.
NFAT-mediated transcription is known to respond differentially to distinct patterns of Ca2+ signaling in lymphocytes (Timmerman et al., 1996). Because thapsigargin and ionomycin do not equivalently induce NFAT-mediated transcription in pKA7/HUVEC, we analyzed the changes in intracellular Ca2+ levels evoked by each compound. Thapsigargin elicits a delayed rise in intracellular Ca2+levels that does not return to base-line, whereas ionomycin elicits a more rapid and much larger Ca2+ transient that decays to a sustained level higher than that elicited by thapsigargin (Fig. 3A). Therefore, the differences in NFAT responsiveness in pKA7/HUVEC induced by maximal concentrations of thapsigargin and ionomycin may reflect the distinct methods of these compounds for elevating intracellular Ca2+ levels.

Mobilization of intracellular Ca2+ in HUVEC. Representative tracings of Ca2+ release, assessed by fura-2 spectrofluorimetry, in collagenase-dispersed HUVEC are shown. A, Ionomycin (3 μm) and thapsigargin (10 nm) induce sustained elevations of intracellular Ca2+ levels. B, Histamine (100 μm), like ionomycin, elicits a sharp early rise in intracellular Ca2+ levels, in addition to the sustained response. C–F, Neither carbachol (1 mm) (C), thrombin (1 μg/ml) (D), EGF (50 ng/ml) (E), nor PDGF-BB (50 ng/ml) (F) induces Ca2+ mobilization (first arrow in each panel), although histamine evokes robust Ca2+responses in the same cells (second arrow in each panel). The data shown are representative of three or four independent responses for each condition.
Little information exists in the literature regarding the control of NFAT-mediated transcription by endogenous physiological agonists. Therefore, histamine and other agonists known to stimulate endothelial cell phospholipase C activity were screened for their abilities to activate NFAT-mediated luciferase activity in cultured HUVEC. Histamine induces a robust, dose-dependent, luciferase response in pKA7/HUVEC, with an EC50 of 1.4 ± 1.1 μm(three experiments). The maximal response is induced by 30 μm histamine (Fig. 4A) and varies, with different HUVEC/retroviral reporter preparations, between 5- and 15-fold over basal levels (data not shown). This effect of histamine is completely blocked by either 1 μm CsA or 100 nm FK506 (Fig. 4B). As shown in Fig.5, the H1 histamine receptor-selective antagonist (+)-chlorpheniramine (Hill, 1990) completely inhibits both histamine-induced luciferase induction (IC50 = 543 ± 151 nm; mean ± standard error) and histamine-induced inositol phosphate production (IC50 = 394 ± 119 nm; mean ± standard error), with the same dose-response relationships. In contrast, the H2 histamine receptor-selective antagonist ranitidine (Hill, 1990) fails to inhibit either effect of histamine (Fig. 5). Taken together, these experiments indicate that H1 histamine receptors regulate NFAT-mediated transcription in endothelial cells.

Histamine induction of CsA-sensitive NFAT-mediated transcription in pKA7/HUVEC. A, Dose-response relationship for histamine. pKA7/HUVEC were stimulated with the indicated doses of histamine for 6 hr before luciferase was measured by luminometry. B, Immunosuppressant block of luciferase activity induced by histamine. CsA (1 μm) or FK506 (100 nm) was applied 30 min before and during exposure to 100 μm histamine for 6 hr. Points or bars, mean ± standard error of three or four experiments, each performed in duplicate.
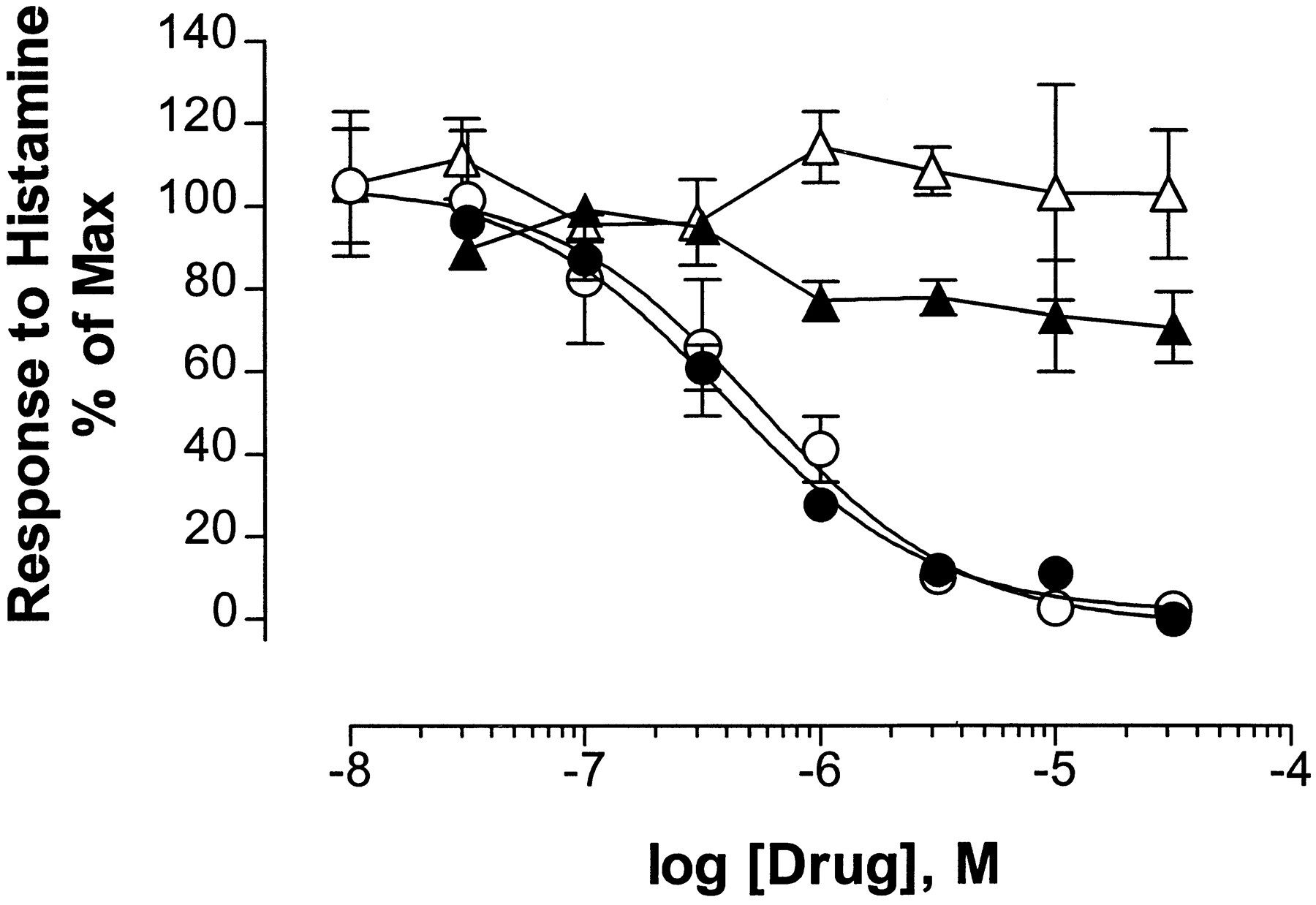
Mediation of histamine responses by H1receptors. Inositol phosphate production (closed symbols) or NFAT-mediated luciferase induction (open symbols) was measured in response to 100 μmhistamine with the indicated concentrations of the H1receptor-selective antagonist chlorpheniramine (circles) or the H2 receptor-selective antagonist ranitidine (triangles). Only chlorpheniramine inhibits the measured effects of histamine. Points, mean ± standard error of three or four experiments, each performed in duplicate.
The following compounds do not evoke luciferase responses in these cells: thrombin (1 μg/ml), EGF (50 ng/ml), PDGF-BB (50 ng/ml), and carbachol (1 mm). However, measurements of Ca2+ signaling indicate why these different vasoactive mediators are ineffective in controlling NFAT-mediated transcription in these cells. Histamine (100 μm) consistently produces a rapid rise in internal Ca2+ levels, followed by a sustained elevation of Ca2+ levels (Fig. 3B), in agreement with previous reports (for review, see Hill, 1990). In contrast, thrombin (1 μg/ml), EGF (50 ng/ml), PDGF-BB (50 ng/ml), and carbachol (1 mm) do not detectably elevate intracellular Ca2+ levels in our HUVEC preparation (Fig. 3, C–F). Therefore, either these HUVEC do not express receptors for these compounds under the conditions used in this study or the receptors are not as efficiently coupled to phospholipase C as are histamine receptors in these cells.
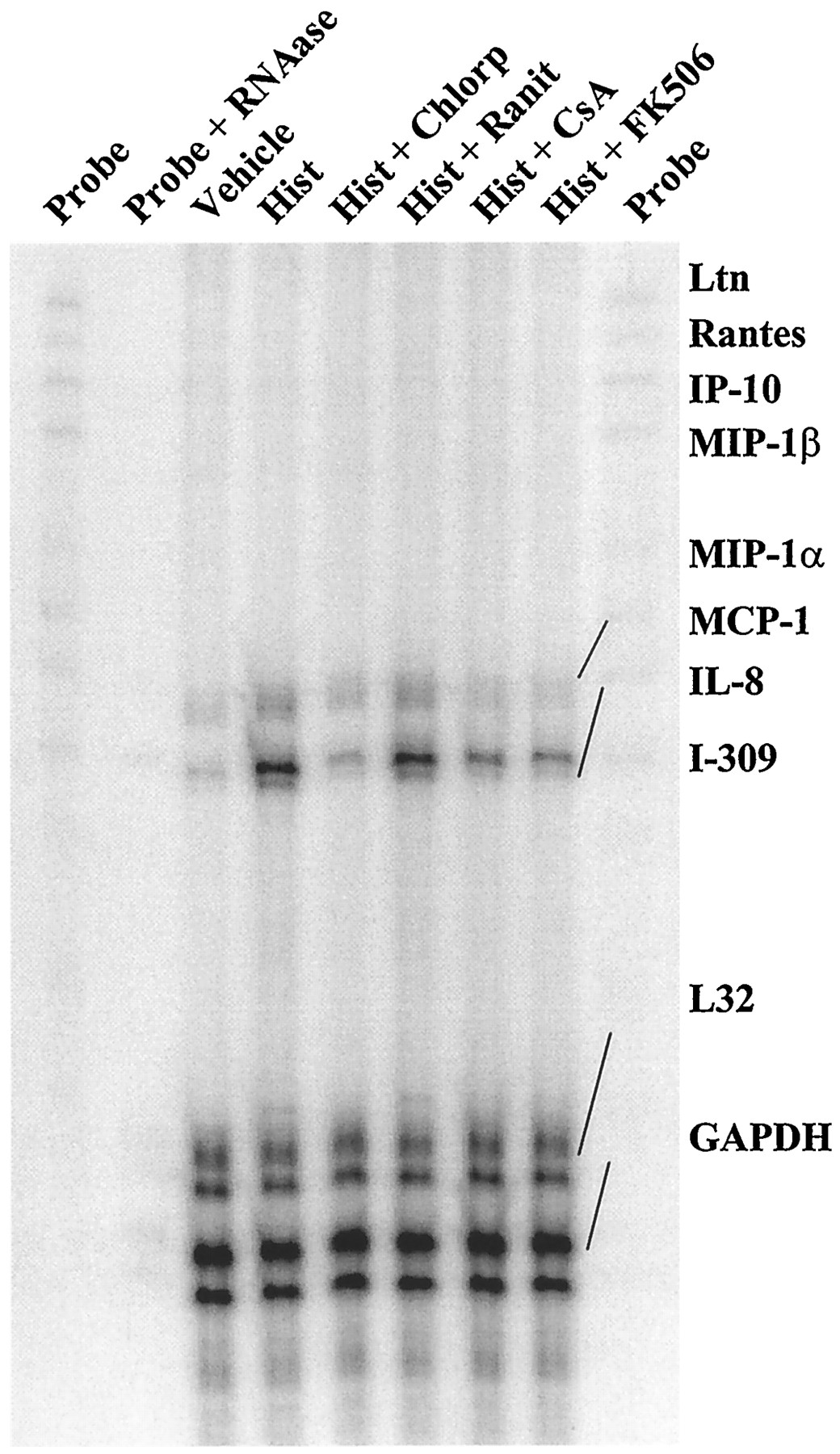
A multiplex RNase protection assay was used to identify candidate genes regulated by NFAT in HUVEC. The expression of several chemokine mRNAs was surveyed because these chemokines are among the first signals expressed in endothelium activated by proinflammatory stimuli such as histamine and are crucial to the inflammatory response (Springer, 1995). Of the eight chemokine mRNAs screened, only two (i.e., MCP-1 and IL-8), yield a detectable hybridization signal at various times after the addition of histamine (Fig. 6). Both of these mRNAs are detectable in cells treated in the absence or presence of CsA, but they are differentially induced by histamine. The results of three independent experiments are shown in Fig.7 and demonstrate that, after 1 hr of treatment, histamine induces IL-8 mRNA expression to 6.4 ± 1.3-fold over basal levels but MCP-1 mRNA induction by histamine is significantly less, at 2.0 ± 0.4-fold over basal levels (mean ± standard error, three experiments). CsA treatment abolishes the induction of both of these mRNAs by histamine throughout the time course of the experiment.

Histamine-induced expression of chemokine mRNAs in HUVEC. The PhosphorImage of a multiplex RNase protection assay shows the time course for induction of HUVEC MCP-1 and IL-8 mRNA expression after the addition of 100 μm histamine (Hist), in the absence (−) or presence (+) of 1 μm CsA. Rightmost lane, approximately 1% of the total amount of undigested probe hybridized with the HUVEC mRNA samples. The probes were completely digested when hybridized to yeast tRNA control. The image is representative of three experiments.

Quantitative analysis of histamine-induced expression of IL-8 and MCP-1 mRNAs in HUVEC. HUVEC were stimulated for the indicated times in the absence (open symbols) or presence (closed symbols) of 1 μm CsA. Hybridization signals in PhosphorImages were quantified by volume integration. The ratios of IL-8 (circles) or MCP-1 (diamonds) mRNA signals to GAPDH mRNA signals in the samples were normalized to the ratios of signals in samples from cells not treated with histamine in the absence or presence of CsA.Points, represents the mean ± standard error of three experiments.
The pharmacological characteristics of this response were examined further by measuring the effects of various inhibitors on histamine-induced IL-8 and MCP-1 mRNAs. As shown in Fig.8, the induction elicited by a 60-min stimulation with 100 μm histamine is suppressed by treatment with (+)-chlorpheniramine, CsA, or FK506 but not ranitidine. The sensitivity of these responses to CsA and FK506 is consistent with a role for NFAT in the induction of these responses by histamine. Furthermore, the inhibition of expression of IL-8 mRNA, and to a lesser extent MCP-1 mRNA, by an H1-selective antagonist indicates that, like luciferase activation, this response is mediated through H1 histamine receptors.

Block of H1 histamine receptor-induced chemokine mRNAs by immunosuppressants. HUVEC were left unstimulated (Vehicle) or were stimulated for 60 min with 100 μm histamine alone (Hist) or in the presence of 32 μm chlorpheniramine (Chlorp), 32 μm ranitidine (Ranit), 1 μm CsA, or 100 nmFK506. This PhosphorImage represents an RNase protection experiment that was repeated once, with similar results.
Discussion
Much of the focus on NFAT has been on elucidating its role in regulating genes involved in immune cell function. The role of NFAT in other cell types has not been examined as extensively and is far from clear. NFAT has long been acknowledged as a crucial regulator of cytokine expression in lymphocytes, where it mediates responses that are initiated via T cell or B cell antigen receptors by signaling through phospholipase Cγ (Desai et al., 1990; Wu et al., 1995). NFAT has been implicated quite strongly in the control of several genes, including those encoding IL-2, IL-3, IL-4, IL-5, IL-13, GM-CSF, tumor necrosis factor-α, and interferon-γ (for review, see Rao et al., 1997). Mice and humans have at least four NFAT genes (for review, see Rao et al., 1997), which mRNA distribution studies indicate are widely expressed in many tissues. To date, however, NFAT expression has been found in only a few types of nonlymphoid cells including mast cells (Weiss et al., 1996), in central nervous system neurons (Ho et al., 1994), and in the PC-12 cell line (Ho et al., 1994; Boss et al., 1996). One report has implicated NFAT in the control of the GM-CSF, IL-6, and E-selectin gene expression that occurs after costimulation of endothelial cells with ionomycin and PMA (Cockerill et al., 1995). In addition to this evidence, we have identified NFAT protein and transcriptional function in vascular smooth muscle (Boss et al., 1998) and skeletal muscle (Abbot KL, Friday B, Thaloor D, Murphy TJ, and Pavlath G, Activation and cellular localization of the cyclosporin A-sensitive transcription factor NFAT in skeletal muscle cells, submitted for publication). The likelihood is high that NFAT will be identified in even more cell types. An understanding of the extracellular factors and cell surface receptors that can regulate NFAT activity and of the identity of the genes that NFAT regulates in nonlymphoid cells is crucial for understanding the physiological role of NFAT outside the immune system.
CsA and related immunosuppressants such as FK506, when used at therapeutic levels, are toxic to several organ systems (Faulds et al., 1993). The mechanisms associated with this toxicity are largely unknown. Because NFAT-mediated transcription is a process regulated by calcineurin, we speculate that inhibition of transcription in nonlymphoid cells by CsA might partly explain the toxicity associated with immunosuppressant therapy. An implication of this conjecture is that NFAT-mediated transcription might be regulated by the tonic effects of hormones and autocoids on nonimmune cells in the course of normal physiological functioning. Alternatively, NFAT may participate in the ability of the body to adapt to physiological stresses. The present study supports these as possibilities, but additional studies will be necessary to test these notions in vivo. If such roles are indicated, however, it is likely that genes regulated by NFAT in nonimmune tissues would have functions distinct from those of genes regulated by NFAT in immune cells. Clarification of these issues will be important in developing strategies to identify new generations of immunosuppressive drugs that have less toxicity than CsA.
The present study provides the first evidence that an endogenous autacoid is capable of inducing NFAT-mediated transcription in endothelial cells, presumably through activation of the heterotrimeric G protein-coupled H1 histamine receptor. These data are consistent with those of a previous study, which used a heterologous expression system to demonstrate that stimulation of phospholipase C through activation of receptors coupled to Gαq is sufficient for activation of NFAT in PC-12 cells (Boss et al., 1996). Although the response to histamine in HUVEC is robust, it is <30% of that which can be achieved maximally with ionomycin and PMA. However, perhaps this response to the autacoid, rather than that to strong drugs that have no physiological counterparts, should be taken as the standard of measure. Furthermore, this artificial culture environment certainly fails to approach the complexity of in vivo conditions. Endothelial cells at a site of inflammation would be exposed to a much more complex milieu of extracellular signals acting on receptors. We speculate that the sum of their activities on phospholipase C stimulation could result in synergistic effects on NFAT-mediated transcription, as we have seen recently in vascular smooth muscle cells (Boss et al., 1998). Unfortunately, we did not find any endothelial stimulants, other than histamine, to test this notion in our preparation. The observation that CsA effectively suppresses the induction by histamine of an intrinsic mRNA in HUVEC (as effectively as it suppresses an artificial reporter gene) also suggests a meaningful physiological role for autocoids in regulating NFAT-mediated transcription in the endothelium.
Recent studies performed in T cells indicate that profound and sustained Ca2+ responses, such as those triggered by antigen presentation, are required for NFAT dephosphorylation and the induction of NFAT-dependent transcription (Timmerman et al., 1996). This has been interpreted to suggest that NFAT might act as a filter that passes on information from strong signals such as those initiated by antigen presentation but eliminates information from the presumably weaker but more constant signals initiated by autocoids, hormones, and neurotransmitters. Whether this notion holds true in lymphoid cells remains to be seen; however, this work clearly shows that engagement of histamine receptors both elicits a strong Ca2+ response and induces NFAT-mediated transcription in endothelial cells. Although several other receptor agonists, including thrombin and carbachol, are known to substantially elevate Ca2+ levels via phospholipase C-coupled receptors in endothelial cells and were therefore expected to stimulate NFAT-mediated transcription, neither effect was observed in our HUVEC preparation. This discrepancy can probably be attributed to differences in HUVEC culture conditions, and we have no reason to suppose that other agonists of phospholipase C-coupled receptors, including growth factors, would fail to activate NFAT in these cells, provided that they produce a sufficient Ca2+ response and activate MAP kinase.
One of the most well characterized actions of histamine in the endothelium is its ability to enhance extravasation of circulating immune cells. IL-8 and MCP-1 are two chemoattractants involved in neutrophil recruitment to activated endothelium (Kilgore et al., 1996). For this reason, we sought to determine whether expression of their mRNAs or other chemokine mRNAs is induced by histamine in our preparation and whether CsA can block this induction. The 2-fold response of the MCP-1 mRNA to histamine is rather weak and approaches our lower limit of confidence with this particular mRNA expression assay. Because of this, it would be premature to conclude that NFAT participates in the control of MCP-1 expression, but it is a matter than will require additional investigation.
In contrast, there is a more robust effect of both histamine and CsA treatment on HUVEC IL-8 mRNA levels. These data extend certain key findings in the literature, which together allow us to draw some inferences. Histamine was shown previously to transiently increase the expression of IL-8 in endothelial cells (Jeannin et al., 1994), but our data provide the first evidence that this induction in endothelium can be inhibited by CsA. Induction of the IL-8 gene in Jurkat T lymphocytes (a cell line in which NFAT regulation is well understood) by ionomycin and PMA is inhibited by FK506, which suggests a role for NFAT (Okamoto et al., 1994). The enhancer element responsible for this effect in T cells appears to be in the proximal promoter region of the human IL-8 gene, as a closely spaced composite of NFκB-like and AP1-like binding sites (Okamoto et al., 1994). However, Jurkat NFκB-related proteins do not seem to bind to this IL-8 gene enhancer under these inductive conditions, as assessed by immunoshift gel mobility DNA binding assays. Furthermore, macrolide immunosuppressants are known not to block NFκB-mediated transcriptional induction in Jurkat cells (Mattila et al., 1990). Therefore, although there are no published data showing that NFAT interacts with the IL-8 promoter NFκB-like site, this is most likely the case. Our data and the considerations cited above support this conjecture and indicate that this site bears reasonable similarity to known NFAT-responsive elements in other genes (Rao et al., 1997). Taken together, this proximal element of the IL-8 gene represents a reasonable candidate site for coordinating the CsA-sensitive, histamine-induced, IL-8 mRNA expression shown in these HUVEC. Further experiments will help to clarify this issue.
Several interesting implications are raised by the present results. First, we show that NFAT can serve as a downstream effector of receptors coupled to Gαq. Therefore, it is worthwhile to consider the possibility that NFAT participates in specific gene expression responses known to be regulated by this large class of receptors in endothelial cells, and perhaps in other cells as well. The use of macrolide immunosuppressants as potential inhibitors of such responses provides a simple pharmacological approach to test this. Second, the blockade of an important endothelium-derived neutrophil chemotactic factor by CsA raises the possibility that inhibition of NFAT-mediated endothelial cell processes may contribute to the total therapeutic efficacy of macrolide immunosuppressive drugs. If this is so, then new generations of immunosuppressant agents that fail to address the role of the endothelium in the immune response might prove to be less effective than the drugs currently used for this purpose. Third, the extent to which the vascular pathological changes associated with immunosuppressant therapy might result from interference with NFAT-mediated transcription suggests that some of the genes targeted by NFAT in blood vessels may play salubrious roles in maintaining normal vascular homeostasis. Discovery of these putative genes may provide new insights into how vascular disorders develop.
Acknowledgments
We thank A. D. Miller (Fred Hutchinson Cancer Research Center, Seattle, WA), G. Crabtree, S. Ho (Stanford University, Stanford, CA), and R. J. Bram (St. Jude’s Children’s Research Hospital, Memphis, TN) for supplying reagents, Neera Bahl (Emory Skin Diseases Research Center, Emory University, Atlanta, GA) for culturing HUVEC, and Karen Abbott (Emory University) for constructing pKA7.
Footnotes
- Received January 12, 1998.
- Accepted May 7, 1998.
-
Send reprint requests to: Dr. T. J. Murphy, Department of Pharmacology, 5031 O. W. Rollins Research Building, Emory University School of Medicine, Atlanta, GA 30322. E-mail:tmurphy{at}pharm.emory.edu
-
This work was supported by National Institutes of Health Grants HL52810, NS32706, and AR42687. T.J.M. is an Established Investigator of the American Heart Association. An abstract of this work was presented at the Experimental Biology Meeting 1998 (San Francisco, CA).
Abbreviations
- NFκB
- nuclear factor κB
- CsA
- cyclosporin A
- NFAT
- nuclear factor of activated T cells
- HUVEC
- human umbilical vein endothelial cell(s)
- MCP-1
- monocyte chemotactic protein-1
- IL
- interleukin
- EGF
- epidermal growth factor
- PDGF-BB
- platelet-derived growth factor BB
- MAP
- mitogen-activated protein
- GM-CSF
- granulocyte/macrophage-colony-stimulating factor
- LTR
- long terminal repeat
- DMEM
- Dulbecco’s modified Eagle medium
- SDS
- sodium dodecyl sulfate
- HBSS
- Hanks’ balanced salt solution
- GAPDH
- glyceraldehyde phosphate dehydrogenase
- PMA
- phorbol myristate acetate
- HEPES
- 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- AP1
- activator protein 1
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}