Article Text

Abstract
Germline mutations of the BRCA1 gene predispose individuals mainly to the development of breast and/or ovarian cancer. However, the exact function of the gene is still unclear, although the encoded proteins are involved in various cellular processes, including transcriptional regulation and DNA repair pathways. Several BRCA1 splice variants are found in different tissues, but in spite of intense investigations, their regulation and possible functions are poorly understood at the moment. This review summarises current knowledge on the roles of these splice variants and the mechanisms responsible for their formation. Because alternative splicing is now widely accepted as an important source of genetic diversity, elucidating the functions of the BRCA1 splice variants would help in the understanding of the exact role(s) of this tumour suppressor. This should help to resolve the current paradox that, despite its seemingly vital cellular functions, mutations of this gene are associated with tissue specific tumour formation predominantly in the breast and the ovary.
- BRCA1
- alternative splicing
- breast cancer
- ovarian cancer
Statistics from Altmetric.com
The BRCA1 gene was cloned in 1994,1 and from that date, numerous studies have been undertaken with the aim of understanding its function(s). Germline mutations of the gene predispose individuals mainly to breast and ovarian cancer,2–5 but they also increase the risk of Fallopian tube cancer6,7 and, to a lesser extent, of pancreatic and prostate cancer.8 Based on these observations, the gene product was first expected to be involved in tissue specific processes in these organs, especially in the breast and the ovary. However, on the contrary, many functions of the protein have been found that are not tissue specific. First, the expression of the gene was proved to be strongly cell cycle dependent,9–12 and the gene product was shown to be involved in the control of the G1–S and the G2–M transition checkpoints.11,13–19 Other studies revealed that the brca1 protein is involved in transcriptional regulation20,21 and in chromatin remodelling,22,23 and the observation that it might be a part of the RNA polymerase II holoenzyme complex supported these findings.24 In the meantime, the protein was found to be involved in various DNA repair processes, such as the control of homologous recombination, non-homologous end joining, and transcription coupled repair.2–5 In addition, many DNA repair proteins and protein complexes were shown to interact with brca1, including rad51, the rad50–mre11–nbs1 complex, and the BASC supercomplex containing, among others, mismatch repair proteins and the atm protein,25–27 which strengthened the idea of BRCA1 being a DNA repair gene. The protein can also interact with the product of the other breast cancer susceptibility gene, BRCA2, which was recently shown to be a strong candidate for the gene encoding Fanconi protein D1.28,29 The connection between BRCA1 and the Fanconi anaemia pathway further supports its role in cell cycle checkpoints and in DNA repair mechanisms.30
“The brca1 protein is involved in transcriptional regulation and in chromatin remodelling, and the observation that it might be a part of the RNA polymerase II holoenzyme complex further supported these last findings”
Initially undertaken as a separate line of investigation, among many other protein species, the brca1 protein was shown to dimerise with the bard1 protein,31,32 and the complex was found to have ubiquitin ligase activity.33 Interestingly, this last observation gives rise to a hypothesis that could integrate some of the so far seemingly different cellular pathways of brca1. An idea was proposed that, as part of the RNA polymerase II holoenzyme, the brca1–bard1 dimer could somehow “sense” the presence of a mutation in front of the transcription complex, and could stop transcription by tagging the proteins for degradation through the ubiquitin–proteasome pathway. The brca1 protein could then recruit different protein complexes to repair the DNA damage before transcription is allowed to proceed.34 Many details of this hypothesis still need further experimental support, although this intriguing idea could clearly integrate many, up to now seemingly independent, observations.
Nonetheless, the above stated observations have not resolved the paradox of why germline mutations of the gene can lead to tissue specific tumorigenesis. Because somatic mutations of the gene occur at much lower frequencies in breast and ovarian tumours than had been expected,35–39 many researchers hypothesised that perhaps certain processes disturbing the expression pattern of the gene could lie behind the malignant transformation in many, mainly sporadic, tumours.40–45 By examining the expression pattern of the gene, more and more evidence was gathered indicating that there are a large number of splice variants present in different tissues, with remarkably different expression patterns.1,46–49 In addition, because alternative splicing is now widely recognised as an important source of genetic diversity,50–56 investigators started to question whether all the described cellular functions and protein partners still hold true for the different species encoded by different BRCA1 splice variants, and to what extent the regulation and function of these variants differ from those of the full length species. This review is intended to provide a current picture concerning BRCA1 alternative splicing because this area of genetics is presently under intense investigation.
THE COMPLEX UNIVERSE OF BRCA1 SPLICE VARIANTS
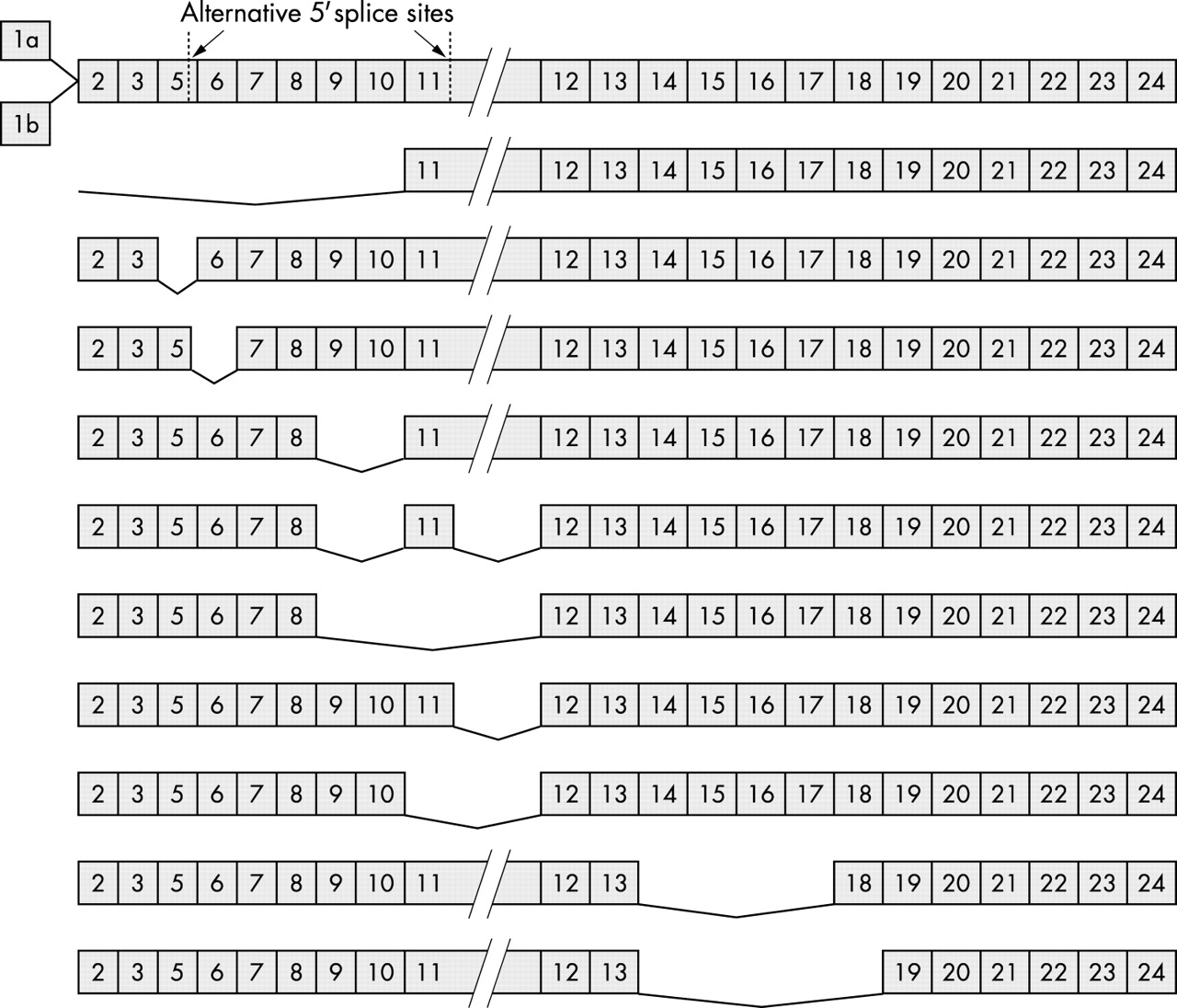
As shown in table 1, the number of known BRCA1 mRNA variants is relatively high. Nevertheless, these mRNA species might not all be functional because many of them were detected in tumour samples, and might represent aberrant splicing products as a result of genetic instability in the tumour.63–65 Keeping the original reading frame of the protein might be a good indication of those species that are truly functional, and selecting variants using this criterion would lower the number to be investigated (fig 1). Those variants with premature termination codons could be the result of subtle cellular regulation, because it was shown that the concentrations of certain proteins and/or mRNA molecules could be downregulated by changing the alternative splicing pattern of the gene, leading to the formation of mRNA variants that can be degraded by the pathway of nonsense mediated mRNA decay.66 These possible regulatory mechanisms all need to be considered when characterising a given splice variant to understand its possible function(s) in different physiological conditions. At present, as far as we are aware, no study has specifically been directed towards a complete enumeration of all functional BRCA1 variants. However, several studies have claimed that four mRNA variants—the full length, the Δ(9,10), the Δ(11q), and the Δ(9,10,11q) variants (table 1; fig 1)—are expressed in a variety of tissues, under different conditions, and hence are called predominant splice variants.12,46,49
The known BRCA1 splice variants in humans, indicating the exon(part)s missing, the tissues where a variant is most abundant, and also whether the encoded protein keeps the original open reading frame (ORF)

{kind=link}
Structures of the BRCA1 mRNA variants that keep the original open reading frame and hence are capable of encoding functional protein species (see table 1). Numbers correspond to different exons, and the missing exons are shown by connecting lines in each variant. Exons are not drawn to scale, although as indicated exon 11 is much longer than the others. Note that exons 1a and 1b are shown with the full length mRNA species only, because it is currently unknown whether the splicing profiles of the further 3′ exons are different for these two mutually exclusive exons.
TISSUE SPECIFIC BRCA1 mRNA PROFILES
The variability of BRCA1 messages begins with the very first exon of the gene—Xu and colleagues57 first provided evidence for the presence of two possible exon 1 sequences at the 5′ end of the mRNA molecule (fig 1). These exclusive exons, 1a and 1b, differ in their lengths and their sequences, but neither of them encodes a part of the normal brca1 protein because the translational start site is located in exon 2. Both transcripts are expressed in various tissues, including testes and thymus, although their relative expression levels are different. In addition, only transcripts with exon 1a are expressed in mammary glands, whereas in placenta only transcripts starting with exon 1b are found.57 Two different promoters at the 5′ region of the gene account for the different exon 1 sequences,67 and the two promoters have different transcription factor binding sites; therefore, the tissue specific distribution of certain regulatory factors could explain the increase in certain transcripts found in different tissues. But what could be the reason for the existence of different 5′ sequences if they do not encode the major protein species? One explanation could be that the two sequences differ in their ability to initiate translation, hence providing a subtle regulatory mechanism for brca1 protein production. Indeed, there is support for this idea, because Sobczak and colleagues68 showed that the two different transcripts have different efficiencies of translation initiation, possibly as a result of the presence of three small upstream open reading frames in exon 1b, which would mean tissue specific brca1 translation profiles. However, it is not known whether there are differences in the splicing profiles of the further 3′ exons of the mRNAs starting with either exon 1a or exon 1b.
“What could be the reason for the existence of different 5′ sequences if they do not encode the major protein species? ”
Despite the existence of the numerous BRCA1 splice variants (table 1), because of methodological difficulties, most of the studies discuss the relative abundance of only a limited number of variants in the total mRNA pool. In earlier studies, some authors did not characterise the observed mRNA species, they just assumed that they were smaller variants such as Δ(11q) or Δ(11) BRCA1.10 Later investigations focused mainly on those variants that were described in most tissues examined, and hence are called predominant mRNA variants.46,49 The relative expression levels of the full length, the Δ(9,10), the Δ(11q), and the Δ(9,10,11q) BRCA1 variants show a tissue specific pattern, with extreme distributions of Δ(11q) being the only variant in some cells of ovarian and thyroid origin, or being absent completely from the BRCA1 pool in pancreatic and liver cells.47 In certain normal and malignant breast epithelial cells, the Δ(9,10) and the Δ(9,10,11q) variants were present in significantly lower amounts than the full length and the Δ(11q) BRCA1 variants,46 whereas in other breast and ovarian tumours, lower amounts of the Δ(11q) variant were present, with the Δ(9,10) variant being highly expressed compared with other BRCA1 isoforms.48 Apart from the use of different methodologies, these clearly conflicting results could also be explained by the heterogeneity of different tumour samples. Some authors concentrated on comparing the expression levels of only two parts of the BRCA1 mRNA pool, such as those either containing or lacking the 3′ region of exon 11, and they found a strong cell type specificity in the BRCA1 expression profile.69 In a more detailed investigation, by systematically measuring the relative expression levels of the four predominant splice variants, we provided evidence that when increased BRCA1 transcription occurs at the G1–S transition of the cell cycle, the ratio of the full length BRCA1 compared with the other four variants increased in ovarian and breast cancer cell lines, but decreased in a leukaemia cell line examined. The relative amount of the Δ(11q) variant decreased as a result of synchronisation in all cell lines examined, whereas the other two species—the Δ(9,10) and the Δ(9,10,11q) variants—showed more of a cell line dependent expression pattern. However, this study indicated that breast and the ovarian cells might share some common regulatory pathways in alternative splicing as compared with the leukaemia cell line, and the disturbance of such pathways might be associated with breast and ovarian tumorigenesis.12
Despite the conflicting results of the experiments presented above, the main message of these studies is that the alternative splicing of BRCA1 plays an important role in certain cellular functions, and perhaps in tumour suppression. However, the question remained as to the possible functions of the different variants and also to what extent they contributed to the overall functions of the gene.
EVOLUTIONARY CONSERVATION OF BRCA1 SPLICE VARIANTS
Perhaps one of the best indications for the functionality of a certain mRNA species is its evolutionary conservation. Concerning BRCA1, it was shown that more than one mRNA variant is transcribed from the gene homologue in various mammalian species, including mouse, rat, sheep, and pig.1,70 The Δ(11) variant has been demonstrated in mice,71,72 and it was also shown that the transcription profiles of the full length and the Δ(11) Brca1 variants during the cell cycle are similar to that of the human orthologue.62 In addition, an intriguing evolutionary conservation underlined the importance of BRCA1 alternative splicing: when comparing the BRCA1 orthologues in rat, mouse, dog, and human, a strong purifying selection was found on a certain region of the mRNA.73 By examining the corresponding part of the gene, we identified two putative exonic splicing enhancer elements and hypothesised that the presence of these and perhaps similar regulatory sequences could account for the strong evolutionary conservation of that region.74 In addition, this conserved region may contain regulatory elements for the alternative splicing of the four predominant splice variants, at least in humans. Further experimental support is needed to show that homologues of all of the variants exist in all mammals examined, but the fact that some variants do exist in different species indicates their presumably conserved functions.
POSSIBLE CELLULAR ROLES OF THE VARIANTS
One drawback to the functional studies of the variants at the protein level is the lack of specific antibodies recognising certain variants exclusively, which would be extremely useful for delineating their overlapping and distinct functions. Earlier studies described the presence of protein species with a molecular weight lower than the full length brca1, which were possibly the proteins encoded by the Δ(9,10), the Δ(9,10,11q), the Δ(11q), or the Δ(11) mRNA variants.46,61 The variants that would be expected to differ greatest at the functional level from the full length species are those lacking most of exon 11, as a result of their remarkable size difference and the absence of many functional domains involved in protein–protein interactions. The Δ(11) variant was used to identify two nuclear localisation sequences (NLS) in exon 11 and it was found that, in contrast to the full length protein variant, the Δ(11) protein (and possibly the Δ(11q) also) is localised in the cytoplasm.47 However, its role in the cytoplasm remains controversial, despite various attempts to decipher its cellular function. It was first shown that NIH3T3 fibroblast cells or MCF-7 tumour cells transfected with the Δ(11q) protein showed increased levels of apoptosis.61 In contrast, others found that exogenous Δ(11q) brca1 protein was less toxic to the cells than the exogenous full length protein species.48 This last group also showed that the cellular localisation of the full length species was not without ambiguity, because it localised partly in the cytoplasm.48 In an attempt to resolve these conflicting observations, Wang and colleagues75 provided evidence that the full length brca1 protein is present in the nucleus in the absence of serum; however, they were able to prove that the two protein variants encoded by the Δ(11q) and the Δ(9,10,11q) mRNAs could also enter the nucleus, despite the fact that they lack the two NLS. Similar problems arose with the localisation of the mouse homologue: one group showed that the Δ(11) species is located in the cytoplasm,76 whereas others could detect both the full length and the Δ(11) proteins in the nucleus.62 At this time, the picture was very confusing and it was thought that another approach was needed to resolve these contradictory results.
The possible explanation to this problem arose when two separate groups showed that, in the presence of wild-type p53, the full length brca1 and the Δ(11q) and Δ(9,10,11q) variants could transactivate the p21 promoter.77,78 In addition, this effect was synergistic if the cells were transfected with the combination of any two of the variants, but was completely abolished in a mutant p53 background.78 Further characterising this phenomenon, Lu and Arrick78 showed that mutations disturbing one of the BRCT domains at the C-terminal of the protein abolished the ability of the shorter proteins to transactivate the p21 promoter, but not that of the full length brca1. They explained this by suggesting that the shorter variants can also enter the nucleus by associating with other proteins through the BRCT domains, and that is why a mutation affecting that region would result in their inability to transactivate. Such mutations do not affect the full length species because it has its own NLS, and is therefore able to enter the nucleus by itself. For the possible identity of the shuttle protein, the authors suggested more potential binding partners, including p53, CtIP, BAP1, or perhaps the full length BRCA1 itself. Although the exact mechanism needs to be deciphered, this hypothesis could explain the contradictory results concerning the cellular localisation of the shorter brca1 variants.
“Some cellular roles are common among at least the four predominant variants”
The above results undoubtedly raised further speculation as to whether proteins interacting with the evolutionarily conserved RING finger motif at the N-terminal region of brca1 are also able to act as shuttle proteins. Fabbro and colleagues79 provide evidence for that hypothesis by proving that the bard1 protein can carry both the shorter and the full length brca1 variants into the nucleus. They proposed that the bard1 protein is in fact a molecular chaperone masking a putative nuclear export signal at the N-terminal of BRCA1, and could also help retain brca1 in the nucleus at the site of DNA damage. This notion would also explain some previous conflicting observations concerning the subcellular localisation of the full length brca1.
From studies directed towards understanding the specific functions of the variants, it became apparent that some cellular roles are common among at least the four predominant variants. They are all phosphoproteins and are able to interact with E2F proteins and various cyclins and cyclin dependent kinases through the N-terminal RING finger motif,75 which is in agreement with the proposed function of the gene as a possible cell cycle regulator.2–5 In addition, the Δ(11q) and the Δ(9,10,11q) brca1 variants may both interact with the ELK-1 transcription factor, and this dimer could downregulate the expression of the FOS oncogene, which might explain the tumour suppressor behaviour of these brca1 variants in the various breast tumour cells examined.80 However, it still needs to be confirmed that this also applies to the full length species, but both structural and sequence data suggest that it probably does. Nevertheless, the obvious sequence differences between the predominant variants indicate possible functional differences.
Many potential protein partners of brca1 interact with the amino acid region encoded by the large exon 11, and variants lacking this part of the protein could not perform roles associated with those interactions. A good example of this would be the rad51 protein, which seems to be a crucial partner of brca1.25,81 The Δ(11) homologue in the mouse is unable to interact with the rad51 homologue counterpart and, moreover, this variant is not phosphorylated after γ ray induced DNA damage, as would be the full length species.62 In addition, in functional knockout mice, where only the Δ(11) brca1 species is expressed, proliferating cells have a normal G1–S transition, but are stopped at the G2–M checkpoint of the cell cycle and also develop abnormally amplified centrosomes.71 Although results in mouse models should be extrapolated to humans with care, they do provide a good indication for the differing functions of the brca1 variants. Indeed, recent studies in mouse models were useful for elucidating common and distinct functions of the full length and Δ(11) brca1 proteins. One group showed that in functional knockout mice, the expressed Δ(11) variant is sufficient to maintain the normal development of lymphocytes, but cannot suppress tumorigenesis, because a significant number of such mice develop T cell lymphomas.82 Others supported this by showing that the absence of the full length, but not the Δ(11) brca1, protein causes senescence in mutant embryos and cultured cells, in addition to premature aging and tumorigenesis in adult mice.83 A third group provided evidence that in mouse strains where only the Δ(11) variant was functional, the testicular size of animals is reduced, and impaired meiotic DNA damage repair and lack of crossing over could be observed during spermatogenesis.84 Recently, such overlapping and distinct functions of these two BRCA1 variants were also seen in human cell cultures, although these experiments concentrated on transcriptional transactivation activities.85 One intriguing result of these last experiments was that BRCA1 mutations implicated in cancer formation did not destroy all the transcriptional functions that were investigated.
“Although results in mouse models should be extrapolated to humans with care, they do provide a good indication for the differing functions of the brca1 variants”
In addition to the distinct functions described above, one study in human cells did provide a clue for functional differences between variants containing or lacking the amino acid region encoded by exons 9 and 10. Cui and colleagues86 showed that the N-terminal region of the Δ(11q) protein has a transactivating effect in GAL4 systems, whereas the Δ(9,10,11q) protein does not. This might be a useful fact to know when designing further studies to compare the functions of these variants. Nonetheless, in spite of the numerous experimental data, the exact cellular roles of brca1 variants are still unclear and need to be elucidated further.
DISTURBED EXPRESSION PROFILE: A POSSIBLE DIAGNOSTIC TOOL?
As more authors described changes in the expression pattern of the variants associated with malignant transformation, especially in breast and ovarian tissues, the question arose as to whether it could be a useful tool to be applied in cancer diagnosis.12,72 The disturbed splicing profile is already an accepted indicator of tumour formation in the case of other genes, such as WT1 or CD44,87,88 and the use of alternative BRCA1 splicing as a tumour marker might be useful in the diagnosis and prevention of breast and ovarian cancers. Certain mutations were shown to influence the alternative splicing pattern of BRCA1,59,89 and the phenotypic differences caused by different disease predisposing germline mutations should be reconsidered in the light of BRCA1 alternative splicing. There are some indications that different expression profiles might not correlate with specific types of breast tumours (TI Orban and E Olah, unpublished observation); however, further studies are needed to investigate this question further.
CONCLUSIONS
Although much information has been gathered on the regulation and the possible cellular functions of BRCA1 alternative splicing, this area of genetics is still under intense investigation. Our understanding of the functions and the regulation of the numerous splicing variants is far from clear; however, their tissue specific distribution and evolutionary conservation clearly indicate that they play important cellular roles. Functional studies at the protein level are hindered by the lack of variant specific antibodies, and developing such molecular tools would help in the search for the functions of individual splicing variants. Nevertheless, based on mouse models and experiments carried out on human cell cultures, many overlapping and distinct functions of certain variants have been elucidated, and the role of the BRCA1 gene in transcriptional regulation, in DNA repair pathways, and in cell cycle checkpoints is now better understood. One promising implication of these studies is the possibility of using the BRCA1 splicing profile in cancer diagnostics, even if it still awaits further experimental testing. At present, our knowledge in this field is by no means complete, but we are gaining a better understanding of the underlying mechanisms that regulate BRCA1 alternative splicing. Further studies may elucidate further subtleties in the genetic regulation of this tumour suppressor gene, and could perhaps help resolve the paradox of tissue specific tumorigenesis caused by mutations of a gene with such general cellular functions.
Take home messages
-
The tissue specific distribution and evolutionary conservation of the BCRA1 splice variants suggest that they play important cellular roles
-
Mouse models and experiments carried out on human cell cultures have revealed many overlapping and distinct functions of certain variants
-
These studies have shown that the BRCA1 gene plays a role in transcriptional regulation, DNA repair pathways, and cell cycle checkpoints
-
It is possible that the BRCA1 splicing profile may be used in cancer diagnostics in the future
Acknowledgments
The authors are indebted to Dr J Papp for helpful comments on the manuscript. The work in our laboratory was supported by the Hungarian Research Grant OTKA T-030039 and the Ministry of Education Szechenyi Project NKFP1/48/2001.