Article Text

Abstract
Objective Pancreatic cancer is characterised by invasive tumour spread and early metastasis formation. During epithelial–mesenchymal transition, loss of the cell adhesion molecule E-cadherin is frequent and can be caused by genetic or epigenetic modifications, recruitment of transcriptional activators/repressors or post-translational modifications. A study was undertaken to investigate how E-cadherin expression in human pancreatic adenocarcinoma and pancreatic cancer cell lines is regulated.
Methods In 25 human pancreatic cancer resection specimens, the coding region of the E-cadherin gene (CDH1) was sequenced for somatic mutations. The tumour samples and 11 established human pancreatic cancer cell lines were analysed by immunohistochemistry, western blot analysis, chromatin immunoprecipitation and methylation-specific PCR. The role of specific histone deacetylase inhibitors (HDACi) on pancreatic tumour cell migration and proliferation was studied in vitro.
Results Neither somatic mutations nor CDH1 promoter hypermethylation were found to be responsible for downregulation of E-cadherin in pancreatic cancer. In the transcriptionally active CDH1 promoter, acetylation of histones H3 and H4 was detected whereas HDAC1 and HDAC2 were found attached only to a silent promoter. Expression of ZEB1, a transcription factor known to recruit HDACs, was seen in E-cadherin-deficient cell lines in which ZEB1/HDAC complexes were found attached to the CDH1 promoter. Moreover, knockdown of ZEB1 prevented HDAC from binding to the CDH1 promoter, resulting in histone acetylation and expression of E-cadherin. HDACi treatment attenuated tumour cell migration and proliferation.
Conclusions These findings imply an important role for histone deacetylation in the downregulation of E-cadherin in human pancreatic cancer. Recruitment of HDACs to the CDH1 promoter is regulated by the transcription factor ZEB1, and inhibition of HDACs may be a promising antitumour therapy for pancreatic cancer.
- Cancer genetics
- epithelial cell adhesion
- pancreatic cancer
- E-cadherin
- RNA expression
- tumour markers
- epigenetics
- liver cirrhosis
- molecular oncology
- pancreatic tumours
- pancreatic disease
- pancreatic pseudocyst
- cadherins
- ACINI
- acute pancreatitis
- adhesion molecules
- pancreatic surgery
- liver metabolism
- pancreatic enzymes
- pancreatitis
- pancreatic disorders
- chronic pancreatitis
- trypsinogen activation peptide
Statistics from Altmetric.com
- Cancer genetics
- epithelial cell adhesion
- pancreatic cancer
- E-cadherin
- RNA expression
- tumour markers
- epigenetics
- liver cirrhosis
- molecular oncology
- pancreatic tumours
- pancreatic disease
- pancreatic pseudocyst
- cadherins
- ACINI
- acute pancreatitis
- adhesion molecules
- pancreatic surgery
- liver metabolism
- pancreatic enzymes
- pancreatitis
- pancreatic disorders
- chronic pancreatitis
- trypsinogen activation peptide
Significance of this study
What is already known about this subject?
E-cadherin is an important mediator of intercellular adhesion and tissue integrity. Loss of E-cadherin expression advances epithelial–mesenchymal transition and promotes metastasis formation in some tumours.
Different regulatory mechanisms can contribute to the inactivation of E-cadherin in human cancers. These include functional inactivation by somatic mutation or transcriptional repression by epigenetic modification.
Transcription of specific genes can be controlled by multiprotein complexes involving transcriptional repressors and histone deacetylases (HDACs).
What are the new findings?
Reduced E-cadherin expression correlates with poor survival of patients with pancreatic cancer.
Neither somatic mutations nor promoter hypermethylation were found to play a major role in E-cadherin regulation in pancreatic cancer specimens.
The transcriptional repressor ZEB1 binds to the E-cadherin promoter and was found to associate with HDAC1 and HDAC2 which emerges as a critical regulatory mechanism for E-cadherin expression.
HDAC inhibition and knockdown of ZEB1 were shown to induce re-expression of E-cadherin in pancreatic carcinoma cell lines.
How might it impact on clinical practice in the foreseeable future?
Reduced E-cadherin expression is associated with poor survival in patients after successful pancreatic cancer resection. Histone modification is a critical regulatory mechanism of E-cadherin inactivation in pancreatic cancer. Owing to the reversible nature of histone modification, specific inhibitors of histone deacetylation should be considered for targeted pancreatic cancer therapy.
Introduction
Pancreatic cancer is characterised by aggressive tumour spread, early metastasis and a poor prognosis. Despite recent advances, the 5-year survival remains below 5%.1 Tumour invasion and metastasis formation are multistep processes characterised by epithelial–mesenchymal transition and the disorganisation of cell–cell adhesions, thus destabilising tissue integrity.2–5 Intercellular contacts formed by cadherin/catenin complex proteins provide strong mechanical attachment between adjacent cells. The extracellular domains of transmembrane-spanning cadherins associate in a Ca2+-dependent manner and their intracellular domains provide attachment to actin filaments via associating catenins.6 In epithelial cells the major cadherin is E-cadherin, and its degradation correlates with increased invasion and metastasis and is a sign of poor prognosis in many types of human carcinomas.7 8 Downregulation mechanisms of E-cadherin expression have been investigated in vitro and multiple repressor proteins and regulatory mechanisms were found to be involved. However, their specific role and contribution to the tumorigenesis of different tumour types remains unknown.
Inactivation of E-cadherin via somatic or germline mutations has been reported for gastric, endometrial and breast carcinoma.9–11 Downregulation of E-cadherin expression can also occur at the transcriptional level. So-called E-boxes within the CDH1 promoter region are recognition sites for transcriptional repressors of the basic helix-loop-helix (bHLH) and the zinc finger protein family, such as Snail (SNAI1), Slug (SNAI2), ZEB1, ZEB2 or E12/E47.12–17 As a third possibility, epigenetic modification of DNA and histones plays a role in the control of gene expression and has been linked to the initiation and progression of cancer.18 Promoter hypermethylation of CpG islands by a specific group of DNA methyltransferases (DNMTs) downregulates transcription whereas gene promoter hypomethylation induces transcription and may cause genomic instability.18–21 Other epigenetic changes involve the reversible acetylation of histones by antagonistic acetyltransferases (HATs) and members of the deacetylase family (HDACs).22 Nucleosomes consist of four core histones (H2A, H2B, H3 and H4) which can all be modified by acetylation.23 Acetylation of nucleosomal lysine residues is central to the switch from repressive chromatin to a conformational decondensation and transcriptional activation. On the other side, deacetylation by HDACs restores chromatin condensation and induces transcriptional silencing.24
We examined whether E-cadherin expression is downregulated in pancreatic cancer specimens and cell lines, whether it correlates with survival and whether E-cadherin loss is caused by mutations in the CDH1 gene, hypermethylation or histone acetylation of the E-cadherin promoter. We identified histone deacetylation to be the major repression mechanism of E-cadherin expression and found that ZEB1-dependent repression of CDH1 occurs by recruitment of HDAC1 and HDAC2.
Materials and methods
Cells, tissues and reagents
Pancreatic cancer cell lines were obtained from the DSMZ (Braunschweig, Germany; German resource centre for biological material) and cultured at 37°C and 5% CO2 in Dulbecco's modified Eagle's medium (Invitrogen, Carlsbad, California, USA) supplemented with 10% fetal calf serum (PAA, Pasching, Austria). Pancreatic resection specimens were obtained from consenting patients after approval of the local ethics committee. Haematoxylin and eosin (H&E)-stained sections of paraffin embedded tissues were reviewed by a senior pathologist and tumour staging was assessed according to the American Joint Committee on Cancer classification system (TNM). Only tissue with a tumour cell area of >50% was used for subsequent experiments. Pancreatic tissue free of tumour and specimens from patients with chronic pancreatitis served as controls for measurements of HDAC activity. Trichostatin A (TSA), sodium butyrate and MS-275 were obtained from Sigma, dissolved in dimethyl sulfoxide (DMSO) and added to the media in final concentrations of 1–10 μM (TSA), 1–10 mM (sodium butyrate) and 0.5–5 μM (MS-275). Cells including DMSO-treated controls (<0.1% DMSO final) were incubated for up to 48 h. Mouse monoclonal antibodies against E-cadherin or E47 (BD Biosciences, Bedford, Massachusetts, USA) were used. Detection of ZEB1 and Snail was done with rabbit polyclonal antibodies (H-102 and H-130; Santa Cruz Biotechnology, California, USA). Rabbit polyclonal antibodies were also used against Slug (Abcam, Cambridge, UK), acetyl histones H3 and H4 (Upstate, Temecula, California, USA) as well as HDAC1 or HDAC2 (GeneTex, Irvine, California, USA). Monoclonal anti-GAPDH clone 6C5 was from Meridian Life Science (Saco, Maine, USA).
Bisulfite modification and methylation-specific PCR
Bisulfite modification of DNA was done according to the modified protocol of Herman et al.25 Purification of DNA was performed using QIAEX II Gel Extraction Kit (Qiagen, Hilden, Germany) and eluted in 160 μl water. Modification was continued by desulfonation with sodium hydroxide (final concentration 0.3 M) for 10 min at room temperature. DNA was precipitated by ethanol and dissolved in water. PCR amplification was done using primers for the human E-cadherin promoter (gene bank L34545). Primers for methylated (5′-TTAGGTTAGAGGGTTATCGCGT-3′ (sense), 5′-TAACTAAAAATTCACCTACCGAC-3′ (antisense)) and unmethylated (5′-TAATTTTAGGTTAGAGGGTTATTGT-3′ (sense), 5′-CACAACCAATCAACAACACA-3′ (antisense)) CDH1 promoter were purchased from Invitrogen (Carlsbad, California, USA). The PCR mixture contained 2× FastStart Master Mix (Roche, Basel, Switzerland), primers (200 nM each), bisulfite modified DNA (approximately 100 ng) in a final volume of 50 μl. Amplification was carried out in an Eppendorf cycler for 35 cycles, annealing temperatures were 57°C for methylated primers and 53°C for the unmethylated pair. Negative controls without DNA were set up for each PCR reaction. PCR products were visualised under ultraviolet light on a 2.5% agarose gel containing ethidium bromide.
CDH1 sequence analysis
Genomic DNA from tumour tissues (n=25) was screened for CDH1 mutations by PCR amplification of all 16 exons and genomic sequencing. Primer and PCR conditions were used as described by Brooks-Wilson et al.9 PCR products were purified using Agencourt AMPure purification kit (Beckman Coulter, Beverly, Massachusetts, USA) and the sequencing reaction was done on 2 μl DNA template in a total volume of 10 μl containing 3 nM sequencing primer and 0.25 μl BigDye Terminator Mix V.3.1 (Applied Biosystems, Foster City, California, USA). Forward and reverse directions were sequenced and reactions were purified using Agencourt Clean SEQ kit according to the manufacturer's instructions. The products were loaded on a ABI capillary sequencer (3130xl Genetic Analyzer, Hitachi, Japan) and the results were processed using software Sequencing Analysis V.5.2 and Variant Reporter.
Knockdown of ZEB1 and western blot analysis
PaTu-8988T cells were transfected with 25 nM and 100 nM of ON-TARGETplus SMARTpool siRNA against human ZEB1 or siRNA control using Dharmafect transfection reagent (Therma Scientific Dharmacon, Lafayette, Colorado, USA). Cells were harvested 24, 48, 72 and 96 h after transfection and 200 μg of lysate were used for western blot analysis. SU.86.86 cells were transfected by electroporation with AMAXA according to the manufacturer's protocol using solution V and 1–5 μM siRNA. Immunoblotting was performed as previously reported.4 7 Incubation with specific primary antibodies was followed by antibody/enhanced chemiluminescence (ECL) detection using ECL peroxidase (HRP)-linked secondary antibodies (GE Healthcare, Buckinghamshire, UK). Equal protein loading was confirmed by reprobing with GAPDH.
Immunostaining of E-cadherin, ZEB1 and SNAI1
Paraffin sections (5 μm) were deparaffinised and incubated with primary antibodies following antigenic recovery using a pressure cooker. Non-specific binding was blocked by 30 min incubation in 1% bovine serum albumin. Incubation with primary antibody (against E-cadherin, ZEB1 or SNAI1) was done overnight at 4°C. After rinsing with phosphate buffer solution (PBS), cells were incubated with FITC- or Cy3-conjugated secondary antibody (1:200) (Jackson ImmunoResearch, Newmarket, UK) at room temperature. 4',6-diamidino-2-phenylindol (DAPI) was used for nuclei staining and slides were mounted in Vectashield Mounting Medium (Vector Labs, Burlingame, California, USA).
Measurement of HDAC activity
Determination of HDAC activity was performed using the HDAC Fluorescent Activity Assay/Drug Discovery Kit (Biomol, Plymouth Meeting, Pennsylvania, USA). Briefly, Fluor de Lys substrate (50 μM) was added to 5 μg protein lysates in buffer (50 mM Tris HCl, pH 8.0, 137 mM NaCl, 2.7 mM KCl, 1 mM MgCl2) and incubated for 45 min at room temperature. Developer solution including TSA (0.1 μM) was added and analysed in a spectrofluorometer (Molecular Devices, Sunnyvale, California, USA, Ex: 360 nm, Em: 460 nm). A standard curve (1–5 μM deacetylated standard) was used to calculate enzymatic activity in nkatal (nmol/s)/mg protein. Experiments were done in triplicate.
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) assays were performed using reagents and a protocol from Upstate Biotechnology with some modifications. PaTu-8988S and -T cells were treated with TSA (10 μM) for 24 h or remained untreated and were cross-linked by incubation for 10 min in 1% formaldehyde. Cells were lysed in SDS lysis buffer (50 mM Tris HCl, pH 8.1, 10 mM EDTA, 1% SDS including protease inhibitors) and sonicated with seven 10 s bursts of a Bandelin Sonicator UW 2200 (Bandelin Electronic, Berlin, Germany). After 10 min centrifugation at 14 000 rpm (4°C), supernatant was saved and an aliquot of 200 μl was saved for ‘input control’. Lysates were diluted in buffer (16.7 mM Tris HCl, pH 8.1, 167 mM NaCl, 1.2 mM EDTA, 1.1% Triton X-100, 0.01% SDS), precleared with protein A agarose/salmon sperm DNA and immunoprecipitation was performed by addition of antibodies against acetyl histones H3 and H4, ZEB1, HDAC1 or HDAC2 and incubated overnight at 4°C under rotation. Precipitated complexes were collected with protein A agarose/salmon sperm DNA (1 h) and washed with low salt, high salt and LiCl wash buffer. DNA complexes were eluted in 500 μl elution buffer (0.1 M sodium hydrogen carbonate, 1% SDS), heated at 65°C for 4 h and treated with proteinase K. DNA was recovered using the QIAEX-II Gel Extraction Kit (Qiagen, Hilden, Germany) and finally dissolved in 60 μl water. Immunoprecipitated and input DNA were subjected to PCR analysis. CDH1 primer sequences were: 5′-GGGCTGGGATTCGAACCCAGTG-3′ (s1), 5′-CCAATCAGCAGCGCGGACCC-3′ (as1) and 5′-GGGCTGGAGTCTGAACTGAC-3′ (as2) (see figure 2 in online supplement). Control ChIP primers sequences in CDH1 exon 13 (3′UTR) were: 5′-AGACTTCTTGCCCCAGATGA-3′ (s) and 5′-AACCACCAGCAACGTGATTT-3′ (as). ChIP was also done from approximately 40 mg of tumour tissue that was dounced and cross-linked in 1% formaldehyde for 20 min. Cell pellets were washed with PBS, dissolved in 200 μl SDS lysis buffer containing protease inhibitors and chromatin was sheared by sonication. Amplified DNA was separated on 2.5% agarose gel and visualised with ethidium bromide.
Co-immunoprecipitation
PaTu-8988S, PaTu-8988T and MIAPaCa-2 cells were lysed and lysates were precleared with protein A-sepharose beads (GE Healthcare). Supernatant was incubated with 25 μg HDAC1 or HDAC2 antibody overnight at 4°C and protein A-sepharose was added for 1 h. The beads were washed four times with wash buffer and immune complexes were subjected to immunoblot analysis with anti-ZEB1 or anti-SNAI1 antibody. ECL detection was done using a horseradish peroxidase-conjugated secondary antibody. Membranes were stripped and reblotted with HDAC1 and HDAC2 antibody.
Determination of cell proliferation and in vitro wounding assay
104 cells/well were seeded into a 96-well plate and allowed to adhere overnight. After treatment with sodium butyrate, TSA or MS-275 for 24–48 h, 10 μl thiazolyl blue tetrazolium bromide (MTT, Sigma, St Louis, Missouri, USA, 0.4 mg/ml) were added, cells were incubated at 37°C for 2 h and the reaction was terminated by addition of 150 μl HCl/isopropanol. Colorimetric measurement was done at 570 nm in a microplate reader (Spectra Max 190, Molecular Devices). In vitro wounding assay was performed by drawing a scratch with a 20 μl tip on the surface of a confluent dish of PaTu-8988T and SU.86.86 cells. After incubation with TSA, MS-275 or AZA for 24 h, pictures were taken and evaluated using tscratch software (see http://www.cse-lab.ethz.ch).
Statistical analysis
Experiments were done at least in triplicate and mean±SEM values were calculated. Differences were analysed by unpaired Student t test and p values <0.05 were considered statistically significant. Survival analysis of patients with pancreatic cancer was calculated by log rank test.
Results
Downregulation of E-cadherin expression in pancreatic tumour correlates with poor survival
E-cadherin expression was assessed by immunofluorescence staining of sections of human pancreatic tumour resection specimens. Only samples with a tumour content of ≥50% (verified by H&E staining) were analysed. E-cadherin expression was present in early (T1–2) and advanced tumour stages (T3–4) but, in 10 of 25 tumour specimens (40%), E-cadherin signals were not detected (table 1 and representative micrographs in figure 1A). Most tumours were moderately to poorly differentiated with no significant correlation between grade of differentiation and E-cadherin expression. Kaplan–Meier analysis of patient survival after surgery over an observation period of 30 months showed a mean survival benefit of >16 months for patients with E-cadherin-positive tumours compared with E-cadherin-negative tumours (figure 1B).
Clinical and pathological characteristics of patients with pancreatic cancer with E-cadherin-expressing or E-cadherin-deficient tumours

(A) H&E-stained sections of representative pancreatic tumour samples and immunofluorescence labelling for E-cadherin (red) and nuclei (DAPI, blue). Tumour cell content in the resection specimens always exceeded 50%. (B) Kaplan–Meier analysis of successfully resected patients with pancreatic cancer showing prolonged survival for patients with E-cadherin-positive tumours.
In the 11 pancreatic carcinoma cell lines the levels of E-cadherin expression varied from absent to strong, with a tendency for poorly differentiated cell lines to be deficient in E-cadherin expression (PaTu-8988T, MIAPaCa-2 and SU.86.86; table 2). E-cadherin expression was predominantly localised at the cell membrane and resulted in maintenance of intact cell–cell contacts in culture, whereas loss of E-cadherin was associated with growth in small clusters or as dispersed single cells. Interestingly, differential E-cadherin expression was found in PaTu-8988S and PaTu-8988T cells which were isolated from the same liver metastasis of a patient with pancreatic cancer. In contrast to E-cadherin-positive PaTu-8988S cells, PaTu-8988T cells are poorly differentiated and exhibit no E-cadherin expression.
Characterisation of human pancreatic tumour cell lines
Methylation status of the CDH1 promoter in pancreatic tumour samples
We investigated whether aberrant hypermethylation of CpG islands in the CDH1 promoter region is involved in the downregulation of E-cadherin expression in pancreatic tumours. Using methylation-specific PCR, we found no aberrant hypermethylation of the CDH1 promoter in all 10 E-cadherin-deficient human tumour samples (see representative examples in figure 2). MIAPaCa-2 was the only carcinoma cell line in which we were able to detect promoter hypermethylation, whereas in two other cell lines (PaTu-8988S and SU.86.86) the absence of E-cadherin expression was not associated with promoter hypermethylation (figure 2). The demethylating agent AZA induced E-cadherin expression in MIAPaCa-2 cells but not in PaTu-8988T and SU.86.86 cells (data not shown). From these results we conclude that promoter hypermethylation is not the principal regulatory mechanism for the downregulation of E-cadherin in pancreatic cancer.

Methylation-specific PCR was performed on bisulphite converted DNA of E-cadherin-deficient pancreatic carcinoma cell lines and five representative resection specimens from E-cadherin-deficient pancreatic tumour samples. 1 μg of DNA was converted according to a modified protocol of Herman et al and specific primers were used for the detection of methylated (M) and unmethylated (U) CDH1 promoter sequences. In MIAPaCa-2 cells the presence of a 115 bp fragment indicated methylated cytosines; all other cell lines and all human pancreatic cancer tissues were found to have an unmethylated CDH1 promoter.
Somatic mutations are not present in E-cadherin-deficient tumours
Inactivating somatic mutations have been reported as a possible cause of E-cadherin deficiency in some types of human cancer. From the 25 pancreatic resection specimens, we isolated genomic DNA and sequenced all 16 exons of the CDH1 gene. In exons 12, 13 and 14 we detected sequence variations which correspond to known SNPs rs1801025, rs1801552 and rs33964119 (table 3). As these SNPs were equally frequent in tumour samples with or without expression of E-cadherin and caused no changes in the amino acid sequence of E-cadherin nor affected intron/exon splicing, we could exclude somatic mutations as the cause of E-cadherin deficiency in our tumour samples.
Sequence variations in human tumour samples
HDAC inhibitors induce E-cadherin expression in pancreatic cancer cells
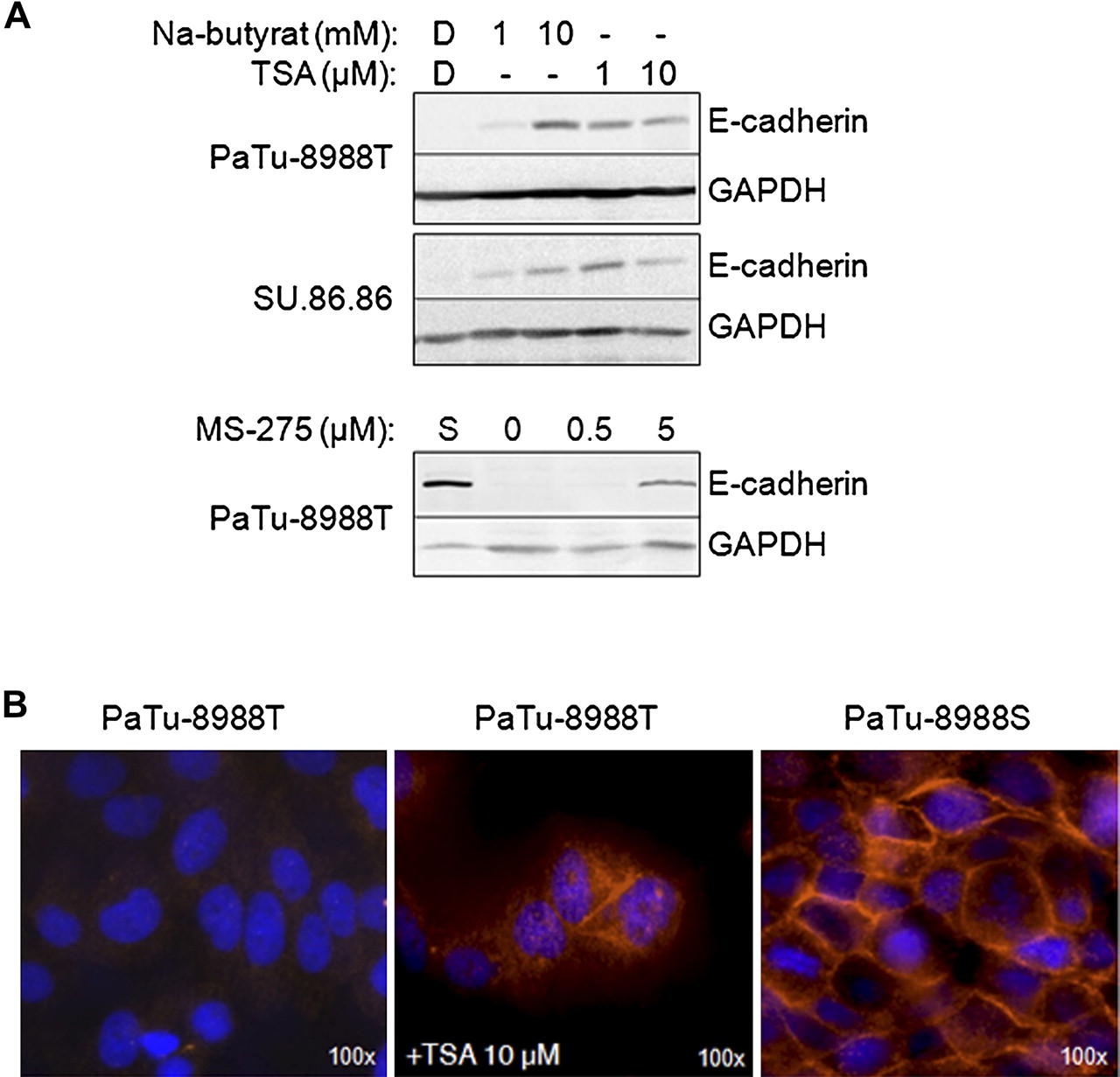
We next investigated whether HDAC inhibitors (HDACi) can induce E-cadherin expression in the E-cadherin-deficient cell lines PaTu-8988T and SU.86.86. HDAC class I and II inhibitors TSA and sodium butyrate, as well as MS-275 (a more selective inhibitor of HDAC1 and HDAC2) were tested in different concentrations. In both cell lines E-cadherin was expressed following 48 h HDACi treatment (figure 3A) but not in DMSO-treated controls. Restored E-cadherin expression following MS-275 treatment suggests that class I HDACs—in particular HDAC1 (and possibly HDAC2)—play a role in CDH1 inactivation. By immunofluorescence staining we confirmed re-expression of E-cadherin in the cytosol and at the cell membrane of TSA-treated PaTu-8988T cells (figure 3B), comparable to its localisation in PaTu-8988S cells.
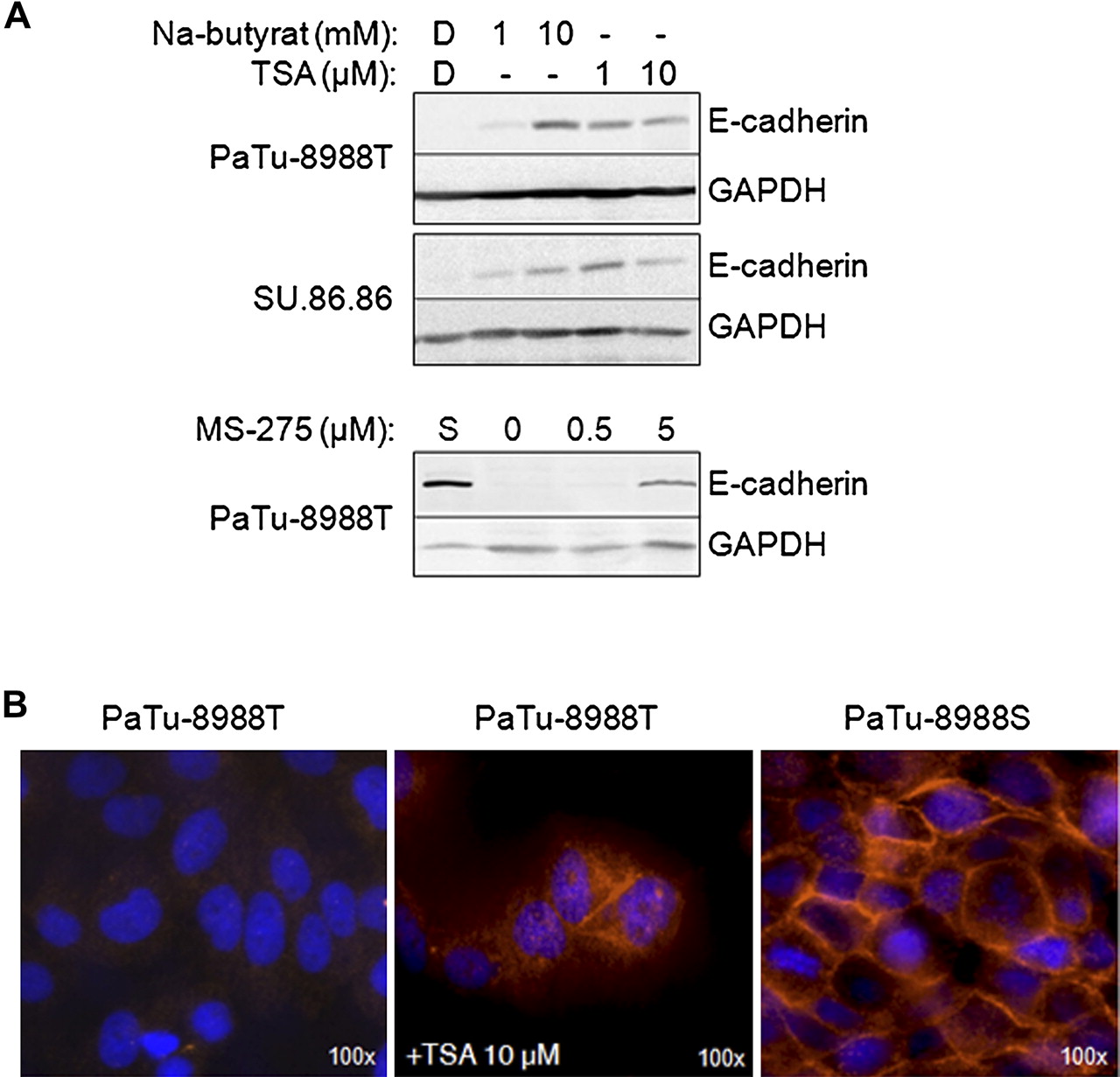
E-cadherin-deficient pancreatic cancer cell lines PaTu-8988T and SU.86.86 were incubated for 48 h with histone deacetylase inhibitors followed by immunoblot analysis for E-cadherin expression. (A) Indicated concentrations of trichostatin A (TSA), sodium butyrate or MS-275, but not the solvent dimethyl sulfoxide (DMSO, D), induced E-cadherin expression in these cell lines. GAPDH immunoblot analysis confirmed equal protein loading and a lysate from E-cadherin-positive PaTu-8988S (S) cells served as a positive control. (B) Immunofluorescence labelling of TSA-treated PaTu-8988T cells also showed expression of E-cadherin which was localised intracellularly and partly at the membrane, comparable to the control labelling of PaTu-8988S cells.
HDAC1 and HDAC2 mediate deacetylation of histones H3 and H4 within the CDH1 promoter
Histone acetylation is known to modify N-terminal lysine residues of histones, transforming the condensed chromatin into a more relaxed structure which is associated with greater levels of gene transcription. By western blotting we detected increased acetylation of histones H3 and H4 in PaTu-8988T, MIAPaCa-2 and SU.86.86 cells 12–24 h after treatment with TSA (figure 4A). ChIP analysis in PaTu-8988T cells further confirmed increased acetylation of histones H3 and H4 within the CDH1 promoter, which may be responsible for reactivation of E-cadherin expression (figure 4B). Comparable results were obtained with the specific inhibitor MS-275, suggesting a specific involvement of HDAC1 in the transcriptional regulation of CDH1 in these tumour cells (figure 4C).

E-cadherin expression is influenced by histone acetylation. (A) Western blot analysis of trichostatin A (TSA)-treated PaTu-8988T cells shows induced acetylation of histones H3 and H4. (B,C) Specific immunoprecipitation of acetylated histones H3 (acH3) and H4 (acH4) followed by PCR with CDH1-specific primers showed that treatment with TSA or MS-275 (a more specific inhibitor of histone deacetylases HDAC1 and HDAC2) increases the histone acetylation status of H3 and H4 within the CDH1 promoter (Ø=negative control without antibody). (D) Chromatin immunoprecipitation further shows that HDAC1 and HDAC2 bind to the CDH1 promoter in PaTu-8988T cells but not in PaTu-8988S cells. HDAC inhibitor treatment detaches HDACs from the CDH1 promoter. A negative control without antibody (no Ab) and a positive control PCR from the original cell lysate after sonication (input DNA) are also shown. (E) HDAC binding to the E-cadherin promoter is seen in most E-cadherin-deficient pancreatic tumour samples (#1–4) but not in E-cadherin-expressing tumours (#6–10). (F) Immunoprecipitation of HDAC1 or HDAC2 followed by immunoblot analysis for ZEB1 and SNAI indicates binding differences in MIAPaCa-2 and PaTu-8988T cells. In PaTu-8988T cells, HDAC1 and HDAC2 form complexes with ZEB1 and SNAI1 (snail) whereas, in MIAPaCa-2 cells, more SNAI1 precipitated with HDAC1 and only a small amount of ZEB1 bound to HDAC1. Additional factors may influence the molecular affinities in different cells. Membranes were reprobed for HDAC1 and HDAC2 to confirm equal loading.
We therefore investigated whether HDAC1 and HDAC2 were directly associated with the CDH1 promoter. As shown by ChIP analysis, both HDACs were found to be associated with CDH1 in untreated PaTu-8988T cells. However, this association was resolved following incubation with TSA (figure 4D). These results indicate that HDAC1 and HDAC2 have a role in E-cadherin downregulation in pancreatic cancer cells.
We next analysed samples of pancreatic tumour resection specimen by ChIP assays. E-cadherin-deficient carcinoma samples showed selective association of HDAC1 or HDAC2 to the CDH1 promoter (4/5), whereas such an association was not seen in E-cadherin-expressing carcinoma tissue (0/5) (figure 4E). One sample of the E-cadherin-deficient tumours did not show HDAC binding to the CDH1 promoter, suggesting the existence of another CDH1 repressor mechanism in this tumour. A possible explanation for the observed selective binding of HDAC1 or HDAC2 to the promoter in E-cadherin-deficient tumour tissue might be a different affinity of HDACs to known E-cadherin repressors such as SNAI1 and ZEB1. E-cadherin-deficient tumour cell lines MIAPaCa-2 and PaTu-8988T also showed different binding of transcriptional repressors with HDACs (figure 4F). In PaTu-8988T cells, HDAC1 and HDAC2 were both found to bind to ZEB1 and SNAI1. However, in MIAPaCa-2 cells, more SNAI1 precipitated with HDAC1 than with HDAC2 and only a small amount of ZEB1 bound to HDAC1 in contrast to HDAC2. Additional factors may therefore influence the molecular affinities between HDACs and transcription factors in different cells.
ZEB1 is inversely correlated with E-cadherin expression in pancreatic cancer and attaches to the CDH1 promoter in a complex with HDAC1 and HDAC2
ZEB1, SNAI1, SNAI2 and E12/E47 have been reported as repressors of E-cadherin transcription. We investigated their co-expression with E-cadherin in pancreatic carcinoma cell lines by western blot analysis. A reverse correlation of expression with E-cadherin was only detectable for ZEB1 and, to a lesser extent, with SNAI1. In Capan-1 cells, a weak expression of both E-cadherin and ZEB1 was still detectable (figure 5A). In immunofluorescence labelling of pancreatic tumour sections (n=5), we found that tumour cells were either positive for E-cadherin or ZEB1 but there was hardly any co-expression in the same cell (figure 5B). We also found that the ratios of ZEB1-positive cells were higher in E-cadherin-negative tumour sections (see figure 1 in online supplement). In ChIP assays we demonstrated ZEB1 binding to the CDH1 promoter in PaTu-8988T and SU.86.86 cells but not in PaTu-8988S cells (figure 5C). Co-immunoprecipitation experiments further demonstrated that ZEB1 can recruit HDAC1 as well as HDAC2 in PaTu-8988T cells (figure 5D). After knockdown of ZEB1 by siRNAs in PaTu-8988T and SU.86.86 cells, the binding of HDAC1 and HDAC2 to the E-cadherin promoter was lost or attenuated (figure 5E, upper panel) and acetylation of histones was increased (figure 5E, lower panel), indicating that ZEB1 mediates the specific interaction of HDACs with the CDH1 promoter (but not, for example, with the 3′ UTR of the CDH1 gene). The knockdown of ZEB1 was also sufficient to induce re-expression of E-cadherin in PaTu-8988T and SU.86.86 cells within 48 h (figure 5F). Neither ZEB1 knockdown nor induction of E-cadherin expression were seen when the control siRNA was used (data not shown). Taken together, these results indicate cooperation of ZEB1 with HDAC1 and HDAC2 in the binding and modification of the CDH1 promoter, which affects E-cadherin expression.
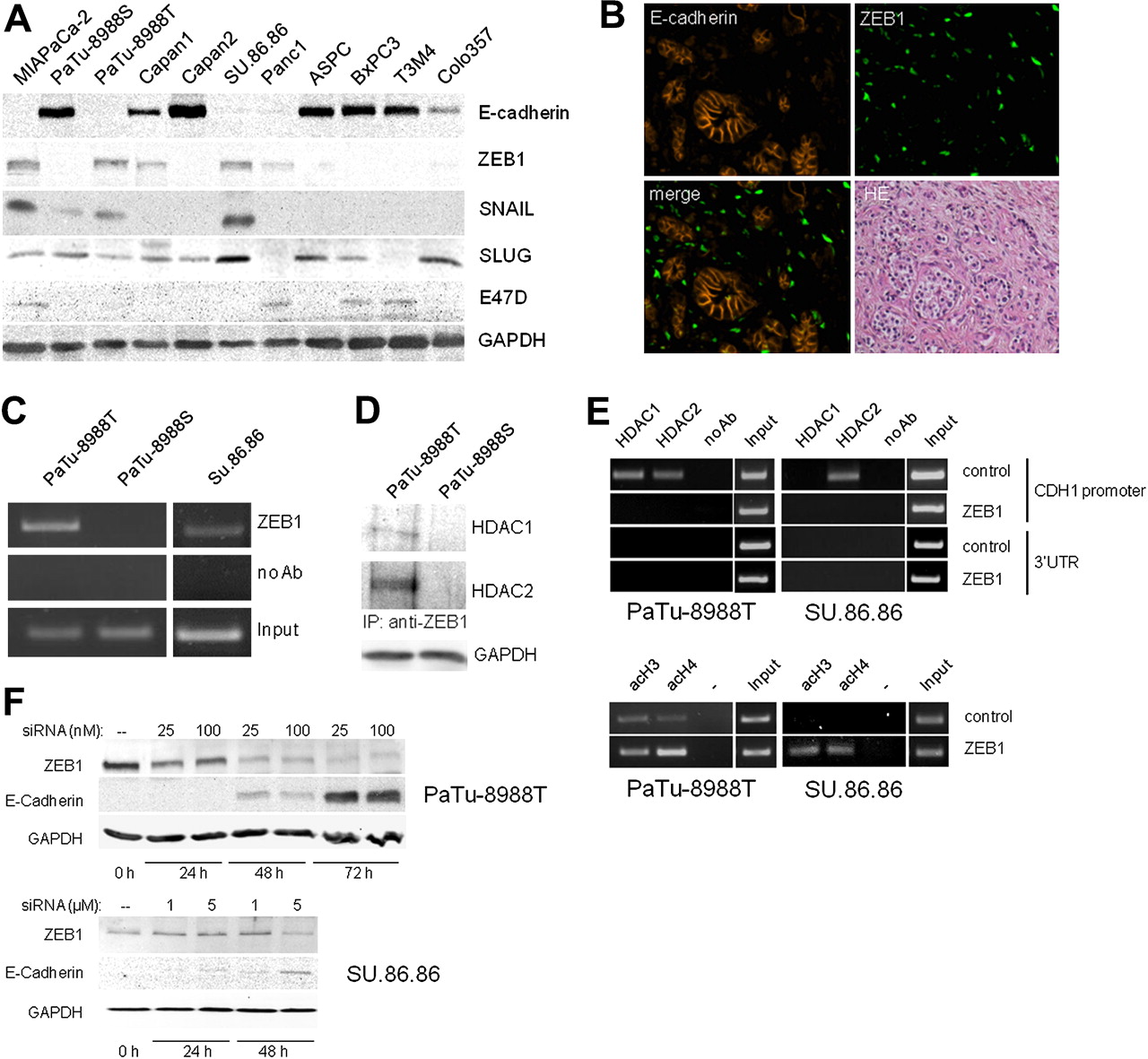
Inverse correlation of ZEB1 and E-cadherin expression in pancreatic tumour cells. (A) Expression of E-cadherin and transcriptional repressor proteins ZEB1, SNAI1 (Snail), SNAI2 (Slug) and E47D was analysed from 100 μg lysate of indicated human pancreatic carcinoma cell lines by immunoblot analysis using specific antibodies. (B) A representative pancreatic tumour section was co-immunolabelled for E-cadherin (red) and ZEB1 (green). The merged picture indicates that ZEB1 expression is incompatible with co-expression of E-cadherin. HE represents the H&E-stained section from the same sample. (C) Chromatin immunoprecipitation demonstrates ZEB1 binding to the E-cadherin promoter in PaTu-8988T cells and SU.86.86 cells but not in PaTu-8988S cells. (D) Immunoprecipitation of ZEB1 revealed complex formation with histone deacetylases HDAC1 and HDAC2 in PaTu-8988T cells but not in PaTu-8988S cells. (E) PaTu-8988T and SU.86.86 cells were transfected with control or ZEB1 siRNA. Chromatin immumoprecipation shows specific binding of HDACs with the promoter but not with the 3′UTR region of the CDH1 gene (upper panel). After knockdown of ZEB1, no HDAC binding is detectable. In the lower panel, specific immunoprecipitation of acetylated histones H3 (acH3) and H4 (acH4) followed by PCR with CDH1-specific primers shows increased H3 and H4 histone acetylation within the CDH1 promoter after ZEB1 knockdown. (F) Proof of ZEB1 knockdown in PaTu-8988T and SU.86.86 cells was obtained by immunoblot analysis at the indicated time points after transfection. Inverse correlation of ZEB1 and E-cadherin expression is demonstrated.
With regard to SNAI1 expression, an inverse correlation with E-cadherin expression was seen in most pancreatic tumour cell lines (figure 5A). However, immunostaining of tumour sections showed sparse SNAI1 expression in only six of the 10 E-cadherin-deficient cancer tissues (not shown). In addition, SNAI1 expression was also present in nine of 13 E-cadherin-expressing tumour tissues, suggesting that SNAI1 expression alone does not determine the downregulation of E-cadherin in pancreatic cancer.
HDAC activity is increased in pancreatic cancer tissues
Recruitment of HDACs to a definite gene locus may be achieved by binding to specifically attached repressor proteins such as ZEB1. We investigated whether deregulated activity of HDACs, as a consequence of either altered expression or due to other regulation mechanisms, could also contribute to differential transcriptional activities of the CDH1 promoter in pancreatic tumour cells. By using a fluorogenic HDAC substrate (BioMol), we found that the total HDAC enzymatic activity was significantly increased in PaTu-8988T and SU.86.86 cells compared with PaTu-8988S cells, but only marginally increased in MIAPaCa-2 cells (figure 6A). E-cadherin-deficient human pancreatic tumour explants (n=10) showed considerable variation in HDAC activity which was not significantly different from E-cadherin-positive tissues (n=15; figure 6B). Tumour samples still showed significantly more HDAC activity than normal pancreatic tissue (n=8) and control tissue from patients with chronic pancreatitis (n=8). While the total cellular HDAC activity appeared to be upregulated in tumour cells, the upregulation itself does not explain the loss of E-cadherin expression.

Histone deacetylase (HDAC) activity in PaTu-8988S/T cells and pancreatic tissue. HDAC activity was analysed from 5 μg protein lysate with the HDAC Fluorescent Activity Assay/Drug Discovery Kit and measured spectrofluorometrically (Ex: 360 nm, Em: 460 nm). Enzymatic activity in μkatal/mg protein was calculated by comparison with a standard curve (1–5 μM deacetylated standard). (A) HDAC activity is significantly increased in PaTu-8988T cells compared with the sister cell line PaTu-8988S. (B) HDAC activity is higher in resection specimens from pancreatic cancer (PaCa) than in normal pancreas (nP) or chronic pancreatitis (CP). E-cadherin-deficient tissues have slightly higher HDAC activity than E-cadherin-expressing tumours. All experiments were done in triplicate. *p<0.05.
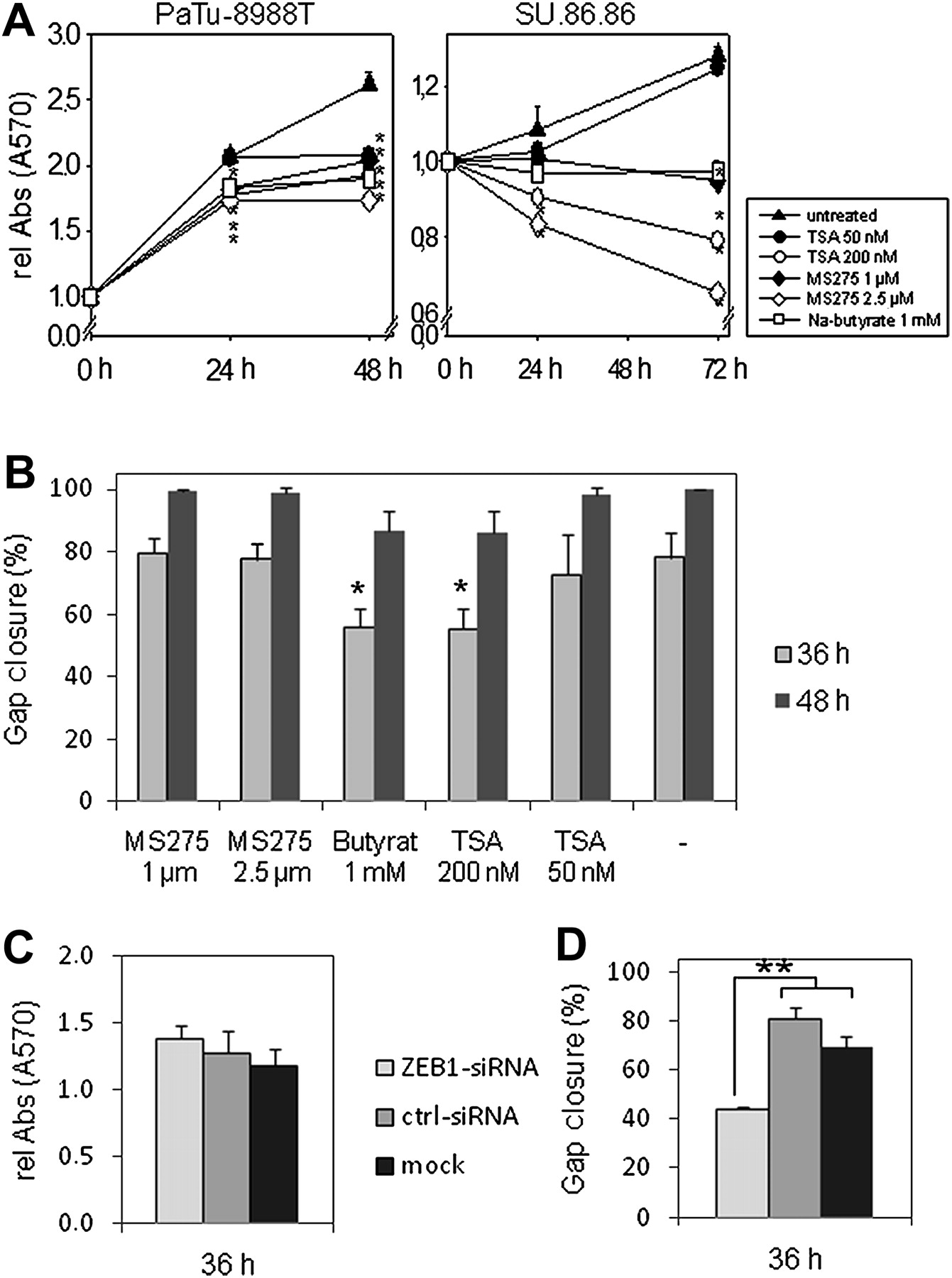
HDACi and ZEB1 knockdown affect cell proliferation and tumour migration in pancreatic carcinoma cells
We further analysed whether HDAC inhibition has an influence on cell proliferation or migration of tumour cells. Treatment with 1 mM sodium butyrate, micromolar concentrations of MS-275 or nanomolar concentrations of TSA clearly attenuated proliferation of PaTu-8988T cells (figure 7A) or induced cell death in SU.86.86 cells. When cell migration of HDACi-treated PaTu-8988T cells was analysed in an in vitro wounding assay (figure 7B), we found delayed gap closure at 200 nM TSA and 1 mM sodium butyrate. Inhibition of cell proliferation and tumour migration by HDACi may partly be ascribed to the tumour-suppressive function of re-expressed E-cadherin. However, we also observed increased caspase-3 cleavage and lactate dehydrogenase release in PaTu-8988T cells with high concentrations of HDACi (see figure 3A,B in online supplement). The antitumour activity of HDACi may therefore reflect concentration-dependent combined effects on different genes that are associated with proliferation, migration, cell cycle and cell death. In addition, we analysed the role of ZEB1 on cell proliferation and migration. After transfection of PaTU-8988T cells with ZEB1 siRNA, marginal inhibition of cell proliferation (figure 7C) but significantly reduced cell migration was seen in comparison with control siRNA-treated cells (figure 7D).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Treatment with histone deacetylase inhibitor (HDACi) affects cell proliferation and migration. (A) 104 PaTu-8988T or SU.86.86 cells were seeded in 96-well plates and MTT assays were performed in the presence or absence of the indicated concentrations of sodium butyrate, trichostatin A (TSA) or MS-275. Relative absorbance (570 nm) was calculated by comparison with time point 0 h and indicates attenuated proliferation in the presence of HDACi. (B) PaTu-8988T cells were cultured for 24 h in medium containing 1 or 2.5 μM MS-275, 1 mM sodium butyrate or 50 or 200 nM TSA. After drawing a scratch on the confluent cell layer with a pipette tip, the gap closure was calculated (using tscratch software) by comparison of gap areas at 0 h, 36 h and 48 h. Experiments were repeated three times in triplicate. (C) ON-TARGETplus SMARTpool siRNA (and control siRNA) were used to knock down ZEB1 in PaTu-8988T cells and MTT assays were performed in 96-well plates. Relative absorbance (570 nm) was calculated by comparison with time point 0 h. (D) PaTu-8988T cells were transfected with ZEB1 or control siRNA. After drawing a scratch on the confluent cell layer with a pipette tip, the gap closure was calculated by comparison of gap areas of three pictures each at 0 h and 36 h (using tscratch software). *p<0.05; **p<0.01.
Discussion
There are several regulatory processes linked to a loss or reduction of E-cadherin expression and a less favourable prognosis in various forms of cancer. Among them are repressor proteins such as SNAI1, SNAI2, ZEB1, E12/E47 and SIP-1 as well as post-transcriptional mechanisms.26–28 In this study we analysed the role of genetic and epigenetic factors in human pancreatic cancer resection specimens for the loss of E-cadherin expression. Neither somatic mutations nor promoter hypermethylation were responsible for the absence of E-cadherin expression, and ZEB1 was found to be the most relevant repressor for E-cadherin in pancreatic cancer which, by recruitment of HDAC1 and HDAC2, attenuates transcription from the CDH1 locus.
To date, 18 different HDACs have been reported and there is a large variation in HDAC expression between different tumours.23 In our study we found increased levels of HDACs in pancreatic cancer samples compared with normal pancreas or chronic pancreatitis tissue. Overall HDAC activity varied considerably in our cohort of pancreatic cancer specimens, which is in accordance with several other studies performed in both haematological and solid tumours.29–31 In pancreatic carcinoma tissue, upregulation of HDAC1, HDAC2, HDAC3 and HDAC7 has recently been reported.32–35 Upregulation of HDAC alone is not associated with downregulation of E-cadherin, as HDAC activity was not significantly different between E-cadherin-positive and E-cadherin-negative pancreatic tumour samples. Substrates for HDACs comprise a number of proteins including histones and transcription factors, making the precise recruitment to a gene locus a prerequisite for its specific transcriptional regulation.
To study the effects of histone deacetylation on E-cadherin repression more closely, we used a cellular model of PaTu-8988S and PaTu-8988T cells which, while being derived from the same pancreatic cancer patient, differ considerably in morphology and the expression of functional cadherin/catenin complexes.36 E-cadherin-deficient PaTu-8988T-cells could be stimulated to express E-cadherin by HDAC inhibition, and HDAC1 and HDAC2 were found attached to the silent CDH1 promoter of PaTu-8988T cells as well as in other E-cadherin-deficient tumour cells. Class I HDACs, especially HDAC1 and HDAC2, therefore seem to play an important role in E-cadherin repression in pancreatic carcinoma. Recruitment of HDACs to a specific gene locus is achieved by binding to locally bound transcription factors and, in our pancreatic tumour samples, the zinc finger protein ZEB1 was identified as an important repressor for the downregulation of E-cadherin. In knockdown experiments in PaTu-8988T cells, we confirmed that, in the absence of ZEB1, HDACs can no longer bind to the CDH1 promoter, which seems to be sufficient for the induction of E-cadherin expression in these carcinoma cells.
The role of HDACs and histone acetylation has previously been examined in various carcinoma types. In Snail overexpressing Madin-Darby canine kidney cells, Peinado et al showed a specific interaction of HDAC1 and HDAC2 with the CDH1 promoter and a repressive effect of Snail by recruitment of HDAC1/HDAC2 in a multiprotein complex.37 In colon cancer cells, acetylation of histone H3 was associated with E-cadherin expression. Furthermore, the authors could not detect promoter hypermethylation as a prominent event in colorectal cancer.38 This is apparently also true for pancreatic carcinoma where we found no evidence for CDH1 promoter hypermethylation in any of our tumour samples and in only one of 11 PaCa cell lines tested (MIAPaCa-2). It was shown recently in murine pancreatic cancer cells that silencing of E-cadherin is dependent on a repressor complex containing Snail and HDAC1/HDAC2.39 In our study, SNAI1 (snail homologue) was expressed in about 65% of tumour tissues and expression was also seen in E-cadherin-positive tumour tissues. These results are in accordance with observations by Hotz et al who found concomitant E-cadherin in about 50% of SNAI1-expressing pancreatic cancer tissues.40
We also confirmed that HDACi can affect cell proliferation and tumour migration, which can be partly ascribed to the tumour suppressive function of re-expressed E-cadherin. However, the antitumour activity of HDACi may reflect effects on different genes that are associated with proliferation, apoptosis and the cell cycle.41 42 HDACs also act on non-histone proteins, and we observed increased caspase-3 levels in PaTu-8988T cells after HDACi treatment which can affect cell proliferation by increased apoptosis. Besides E-cadherin, other anti-oncogenic factors such as retinoic acid receptor α (RARα), p21/WAF1 and the transcription factor C/EBPα can be involved in HDACi-induced growth inhibition in pancreatic cancer cells.43 44
The analysis of an E-cadherin loss in pancreatic cancer has several limitations. First, E-cadherin-deficient tumour material is limited and only few of the established pancreatic carcinoma cell lines are E-cadherin-negative. Histones are modified by acetylation and also by phosphorylation, sumoylation, ubiquitination or methylation, and Cao et al recently reported that an enhancer of zeste homologue 2 (EZH2), which is part of the multiprotein complex of the polycomb-repressive complex (PRC2), can inhibit E-cadherin expression through histone H3K27 trimethylation at the CDH1 promoter.45 Polycomb proteins are known to prevent the recruitment of transcriptional activators and form a repressive chromatin structure.46 The specific role of histone modification in the context of the biology of pancreatic cancer therefore deserves further study.
In conclusion, we have identified a major role for ZEB1 and HDACs in the regulation of E-cadherin expression in pancreatic cancer. Our study shows antiproliferative and antimigratory effects of HDAC inhibitors or after ZEB1 knockdown in pancreatic cancer cells. The reversibility of epigenetic effects and their susceptibility to pharmacological manipulation of inhibition of histone deacetylation, this may allow new therapeutic options in the treatment of pancreatic cancer.
Acknowledgments
The authors thank N Loth and K Gladrow for technical assistance.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Download Supplementary Data (PDF) - Manuscript file of format pdf
- Download Supplementary Data (PDF) - Manuscript file of format pdf
- Download Supplementary Data (PDF) - Manuscript file of format pdf
Footnotes
See Commentary, p 329
Linked articles 301576.
Funding This study was supported by grants from the Alfried-Krupp-von-Bohlen-und-Hahlbach-Foundation (Graduate Schools Tumour Biology and Free Radical Biology), University Medicine Greifswald (Forschungsverbund Molekulare Medizin), the Deutsche Krebshilfe/Dr Mildred-Scheel-Stiftung (102031-Le and 109102), the Deutsche Forschungsgemeinschaft (DFG GRK840-E3/E4, MA 4115/1-2/3, LE 625/8-1 and 9-1), the Federal Ministry of Education and Research (BMBF GANI-MED 03152061A and BMBF 0314107) and the European Union (EU-FP-7: EPC-TM No. 256974 and EU-FP7-REGPOT-2010-1). AA was supported by a Gerhard-Domagk-Stipendium of Greifswald University.
Competing interests None.
Patient consent Obtained.
Ethics approval Ethics approval was provided by the local ethics committee.
Provenance and peer review Not commissioned; externally peer reviewed.