


Abstract
In this review, we summarize approaches to treat cancer with genetically engineered fusion proteins. Such proteins can act as decoy receptors for several ligands or as recruiters of immune effector cells to tumor. Examples of interference with growth factor-mediated tumor growth and tumor-related angiogenesis with fusion proteins consisting of the extracellular domains, and in some cases also of entities of one or several receptors and the Fc part of human IgG1, are discussed. In addition, we present strategies for recruitment of immune effector cells to tumor with fusion proteins. This can be achieved with fusion proteins consisting of a tumor-related antibody and a cytokine or major histocompatibilty complex class-I-peptide complexes, by T-cell receptor cytokine fusion proteins or by combination of a T-cell-recruiting antibody with a tumor-related ligand or a defined T-cell receptor.
- Angiogenesis
- antibody cytokine fusion proteins
- chimeric extracellular receptor domains
- decoy receptor
- HER signaling; Fc-based fusion proteins
- immune effector cell recruitment
- treatment resistance
- review
One of the aspects of biologics-based cancer therapy is the inhibition of angiogenesis and of tumor-promoting effects of growth factors. Another approach is based on the recruitment of immune effector cells to the tumor in order to initiate an antitumor immune response. These approaches are based on the identification of the hallmarks of cancer (1-3). Using the first approach, several recombinant receptor decoys are under pre-clinical and clinical investigation. They are based on genetic fusion of the extracellular domain (ECD) of a receptor and the Fc-part of human immunoglobulin IgG1, which improves the pharmacokinetic properties of the fusion protein. Chimeric decoy receptor fusion proteins can be generated by combining entities derived from the ECDs of two, or theoretically more, plasma membrane-associated receptors. The fusion proteins, as outlined, can bind several ligands dedicated to one or several receptors inhibiting, processes such as tumor-associated angiogenesis or human epidermal growth factor receptor (HER)-based signaling. Another approach is the recruitment of immune effector cells to the tumor with genetically engineered fusion proteins. We discuss fusion proteins of tumor-related antibodies and cytokines or MHC-class I peptide complexes, T-cell receptor (TCR) cytokine fusion proteins, and fusions proteins of a T-cell-recruiting antibody and a tumor-cell receptor ligand or a defined TCR.
Technologies for Generation of Genetically Engineered Fusion Proteins
Genetically engineered fusion proteins are composed of domains of antibodies (predominantly IgG), and of additional protein entities that mediate functionality. The entities that are derived from antibodies can either contain binding regions or constant antibody regions.
Binding region modules are applied for targeting approaches, i.e. to direct a fused ‘effector’ payload specifically to those cells that shall be addressed. These binding entities are composed, in most cases, of variable regions (Fv) of antibodies which specifically bind the desired antigen. Fv fragments are heterodimers of variable heavy chain (VH) and variable light chain (VL) domains. They are the smallest modules that retain a complete structure of the binding region of antibodies. Their small size (approx. 25 kDa) enables the generation of small fusion proteins which may penetrate tissues and tumors quite rapidly. Such molecules may be particularly useful in applications that require rapid tissue/tumor penetration, e.g. for targeted cancer therapy. The complete removal of the constant regions of antibodies not only reduces the size of fusion proteins, but it also restricts the functionality of antibody modules to ‘targeting-only’. Removal of the constant domains eliminates any (Fc-receptor mediated) effector functionality of the parent antibody, such as complement-dependent cytotoxicity (CDC) or antibody-dependent cellular cytotoxicity (ADCC). Application of Fvs without attached constant domains generates technical challenges, in particular stability issues: while fragment antigen binding (Fabs), [heterodimers composed of VH first constant domain of the heavy chain (CH1) and VL (CL)] are by themselves quite stable heterodimers, VH-VL heterodimers are instable. The CL and CH1 domains contribute significantly to the formation of the heterodimeric Fabs, and their interactions (including an interchain disulfide at the C-terminal end of CH1 and CL) stabilize Fab fragments. Once these domains are removed, VH and VL have only very limited capability to form functional Fv heterodimers. Furthermore, VH-VL interfaces are too weak to stably retain heterodimeric Fvs without CH1 and CL attached (4, 5). One method that has frequently and successfully been applied to overcome the problem of instable VH-VL heterodimers is the covalent connection of VH and VL by a flexible peptide linker. These linkers are frequently composed of glycine and serine, containing stretches of 15-20 amino acids. The tethering between VH and VL generates single-chain Fv molecules (6, 7), which are more stable compared to VH-VL heterodimers by themselves. Single-chain (sc)Fvs are widely applied as recombinant binding modules and components of fusion proteins.
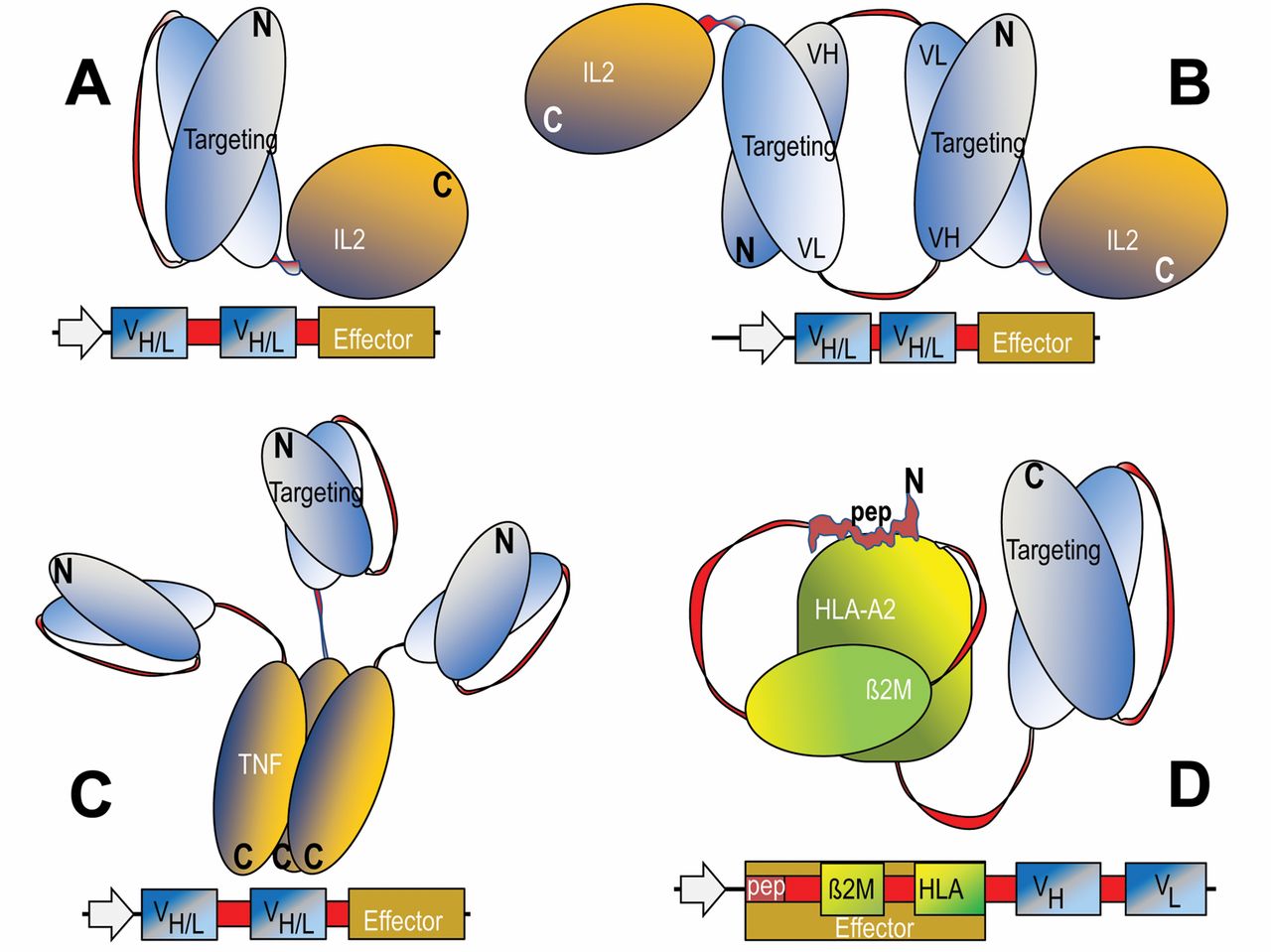
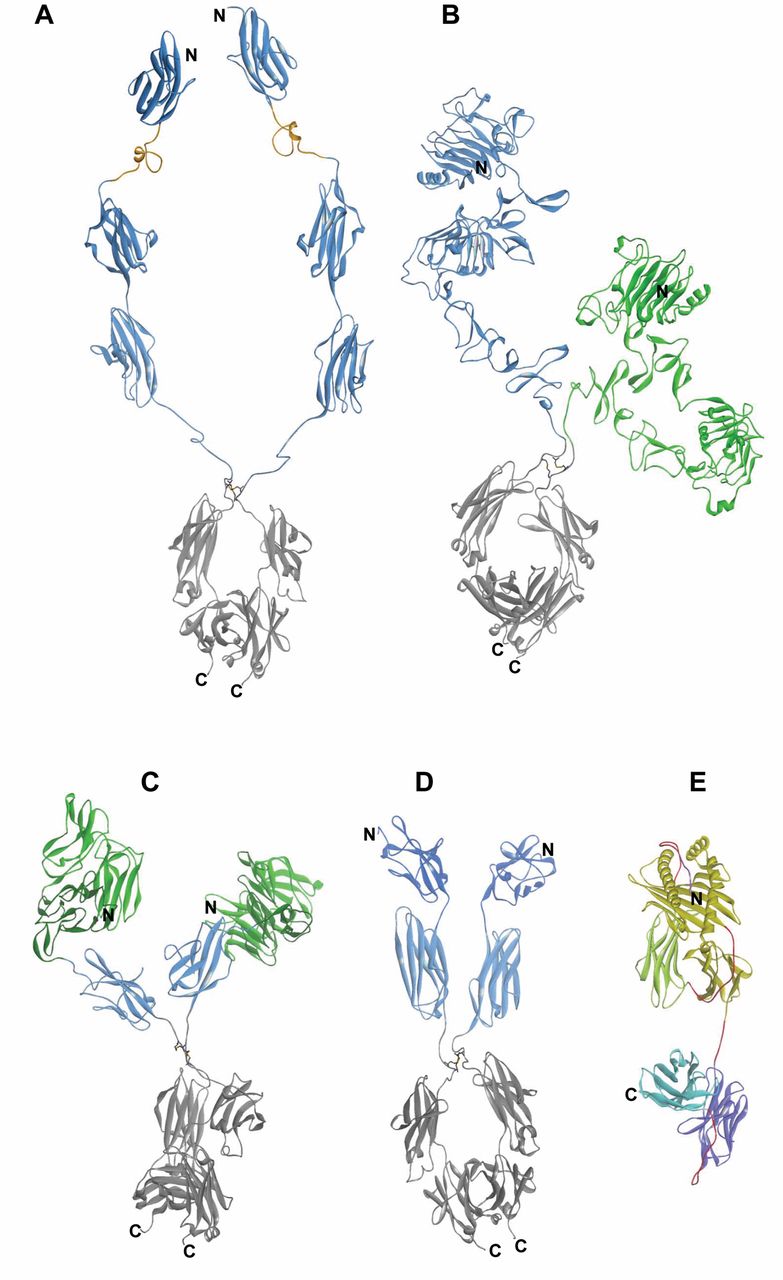
Additional technologies have been applied to further stabilize Fv fragments for therapeutic applications. For some variable regions, linkage of VH and VL by flexible peptide linkers only partially restores its stability. Such scFvs still have a tendency to dissociate and in consequence have a propensity to aggregate. These problems can be overcome by increasing the stability of the VH-VL interface by mutations and/or by introduction of covalent disulfide connections between the VH and VL domains (8-10). The fusion partners of Fv-containing proteins are connected to each other in a modular manner. In most cases, flexible linker or connector sequences are set between the Fv module and the additional entity to prevent undesired interactions (including steric hindrance) between the fusion partners. This modular composition retains the binding specificity of the Fv region as well as the functionality of the targeted-effector entity. Engineered Fv fusion proteins can be efficiently produced in bacteria such as Escherichia coli, either as soluble proteins or as inclusion bodies, followed by re-folding to obtain fully active proteins (11). Examples for genetically engineered fusion proteins that contain variable antibody domains as cell targeting modules are the cytokine fusions that contain interleukin-2 (IL2), or tumor necrosis factor-α (TNFα), or those that display human leukocyte antigen (HLA) peptide complexes (Figure 1, A-D).
Constant region entities of antibodies are also applied for the generation of fusion proteins. In contrast to antibody-binding entities (for specific targeting approaches), the main rationale for using constant region entities as fusion partners is the modulation of size and the pharmacokinetics of the therapeutic fusion proteins. The Fc-fragment of antibodies (IgG) starts with an N-terminal at the flexible hinge region of antibodies, thereafter followed by CH2 and CH3 domains. These entities form stable homodimers, covalently linked by interchain disulfide bonds between cysteine residues in their hinge regions. The fusion partners of Fc-containing therapeutic proteins are connected to each other in a combinatorial manner. In most cases, the fusion to Fc is made at the hinge region, followed thereafter by CH1 and CH2. Hinge regions are flexible by nature, thus fusion at this position minimizes or prevents undesired interactions and steric hindrance. This combinatorial composition retains the functionality of the Fc region, as well as that of the additional entity, as will be shown in this review. Constant regions bind Fc receptors, including FcRn (12, 13). Binding to FcRn, which enables antibody recycling/export from cells, is the key contributor to the long serum half-life of antibodies. This favorable pharmacokinetic property can also be transferred to fusion proteins that contain Fc domains as FcRn-binding modules. A further advantage of having Fc regions as part of recombinant fusion proteins is that they can be applied to facilitate production and downstream processing. For this reason, Fc fusions can be expressed and purified by procedures that are similar to those applied for the production of therapeutic antibodies. This includes expression in mammalian cells, secretion into culture supernatants and subsequent affinity-based (ProteinA) purification steps. Examples of genetically engineered fusion proteins that contain constant regions of antibodies are ‘ligand trap’ molecules such as those containing extracellular domains of fibroblast growth factor (FGF), epidermal growth factor receptor (EGFR), human epidermal growth factor receptor-2 (HER2), and of vascular endothelial growth factor (VEGF) receptors (Figure 2, A-F).
VEGF Trap
The members of VEGF family are involved in processes such as angiogenesis, lymphangiogenesis, vasculogenesis, and are also modulators of host innate and adoptive immunity (14). Clinical studies have validated VEGF as a target for treatment of cancer and vascular diseases affecting the eye. These studies have led to the approval of drugs targeting the VEGF pathway, such as an antibodies directed against VEGF-A (Avastin), and kinase inhibitors which inhibit activation of the respective receptors (VEGFRs) (15, 16). The biological functions of VEGFs (A, B, C, D and E) are mediated by interactions with VEGFRs 1, 2 and 3. VEGFRs 1 and 2, each contain seven different ECDs. A soluble VEGF trap was created by genetic fusion of domain 2 of VEGFR1 to domain 3 of VEGFR2 with the Fc portion of human IgG1, resulting in a composite soluble decoy receptor referred to as Aflibercept (17) (Figure 2F and 3D). It binds to all VEGF-A isoforms generated by differential splicing, such as VEGF121, VEGF165, VEGF189 and VEGF206, in addition to other members of the VEGF family such as VEGF B, C and D, as well as placental growth factor (PLGF), which is also involved in pathological angiogenesis (15). The binding to all isoforms of VEGF-A occurs with high affinity (Kd <0.5 pM) and the Kd for hPLGF is in the range of 25 pM. Several pre-clinical properties are attributed to VEGF Trap, such as normalization of the tumor vasculature, pruning of excess vessels, reduction of interstitial fluid pressure and vascular permeability (18-20). VEGF Trap has shown significant antitumoral and antiangiogenic activity in numerous pre-clinical models (21). A significant impact of the VEGF Trap was documented when administered together with chemotherapy or radiation (21). Similarly to Avastin, the VEGF Trap has a marked effect on pre-existing or newly-generated blood vessels (22). While antibodies form multimeric complexes that are readily cleared from the circulation, VEGF Trap forms an inert 1:1 complex with VEGF that remains in the circulation and can be monitored, thus providing a biomarker for predicting efficacious angiogenic blockade (23). Measurement of the level of free and bound VEGF provides an indicator of the doses of the VEGF Trap required for capturing all available VEGF. VEGF Trap exhibits excellent pharmacological properties and has a circulation half-life of 2-3 weeks, allowing for bi-weekly or even less frequent dosing (23). It should be kept in mind that trapping all members of the VEGF family at first glimpse seems to be a conceptual advantage in comparison to Avastin which binds exclusively to VEGF-A. However, it has been shown that VEGF-B and VEGF-D can have a negative impact on angiogenesis and lymphangiogenesis (24, 25) and in this context controversial effects for PLGF have emerged (26).
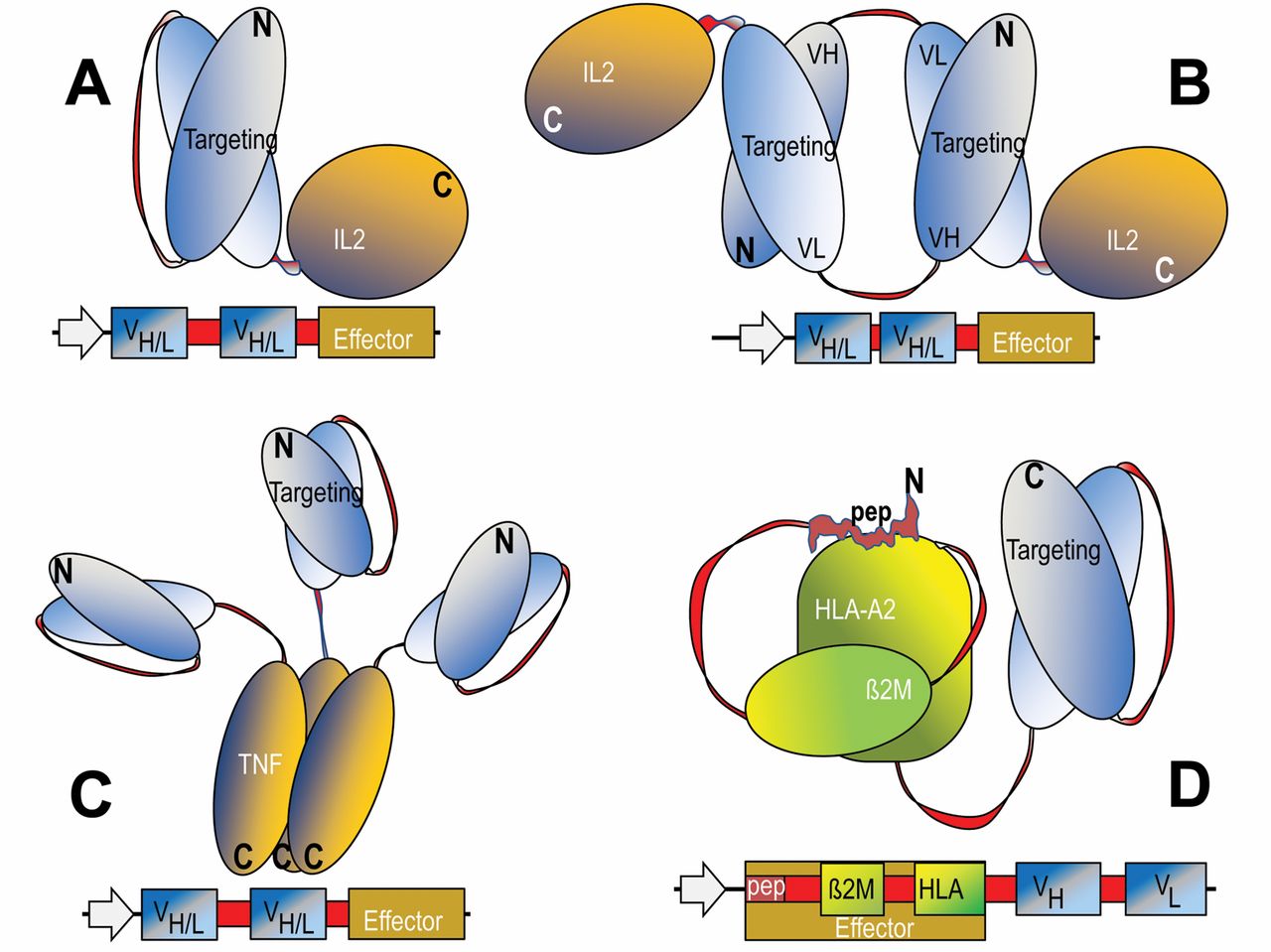
Design and composition of fusion proteins that combine antibody-derived targeting domains with effector modules. The domain compositions of fusion proteins are shown schematically in the upper panels and the sequences that encode these entities and summarized in lower panels of each figure. The general design principle includes recombinant fusion of cell surface, targeting modules (blue) to effector modules (yellow). Flexible linker or connector sequences are applied to combine variable heavy-chain (VH) and variable light-chain (VL) domains, and are also incorporated at the fusion sites of Fvs to effector entities (red). These linker and connector sequence stretches contain in many cases serine and glycine residues to permit flexibility. A and B: Monomeric and di-meric fusion proteins for targeted accumulation of IL2. C: Targeted tri-meric tumor necrosis factor. D: Molecules that are applied for targeted accumulation of human leukocyte antigen (HLA)-presented peptides.
Aflibercept is currently clinically-evaluated in three phase III studies in combination with chemotherapy for second-line metastatic colorectal cancer (mCRC), second-line non-small cell lung carcinoma (NSCLC) and first-line hormone-refractory prostate cancer (27). Aflibercept was approved for wet macular degeneration in the US, and phase III studies are currently being conducted as a first-line treatment in combination with docetaxel and prednisone in patients with androgen-resistant prostate cancer and in patients with mCRC after failure of an oxaliplatin-based regimen together with folinic acid plus fluoruracil and irinotecan (FOLFIRI).
Fibroblast Growth Factor Receptor 1 (FGFR1) Fc
The FGFs are a family of proteins comprising of 22 structurally related members with a wide range of biological activities (28). FGF1 and FGF2 were the first angiogenesis factors identified (29, 30). FGFs activate a family of tyrosine kinase receptors, designated as FGFR1-FGFR4 (31, 32). They are composed of an ECD with three Ig-like domains (D1, D2, D3), a transmembrane domain and a cytoplasmic tyrosine kinase domain.The ligand binding site is located in the Ig-like domains D2 and D3 and the amino acids connecting them (33). FGFRs can be spliced differentially, FGF binding specificity is determined by the third Ig-like domain, the N-terminal half of which by an invariant exon IIIa, with alternative usage of IIIb and IIIc exons for the C-terminal half. FGFRs containing exon IIIb are expressed on mesenchymal cells and those with FGFR IIIc are expressed on epithelial cells, establishing a paracrine signaling relationship between epithelial and mesenchymal tissues. FGFR IIIb binds efficiently to FGF1, FGF3 and FGF10, whereas FGFRIIIc binds to FGF1, FGF2, FGF6, FGF8 and FGF9 (34). The role of FGF-FGFR interactions in the development of vasculature and tumor angiogenesis has been extensively documented (35, 36). An autocrine growth factor role for FGF2, FGF9 and FGFR1, FGFR2 has been reported (37). Selective amplification of the FGFR1 gene in lung squamous cell carcinomas and cell lines emphasizes the role of FGF FGFR signaling in these tumors (38). Amplification of FGFR2 is observed in 4-20% of patients with breast cancer, while FGFR4 is overexpressed in 30% of patients (38). Ligand-dependent mutations of FGFR2 (S252W/P253R) are observed in 7% of endometrial carcinomas (39, 40).
In order to sequester mitogenic and angiogenic FGFs, FP-1039, a fusion protein consisting of the ECD of FGFR1 IIIc and the Fc domain of human IgG1 was experimentally evaluated (41) (Figure 2A and 3A). Surface plasmon-resonance experiments indicated binding to FGF1, FGF2, FGF4 and FGF18 (41). FP-1039 reduced the number of viable Caki-1 and A549 tumor cells in a concentration-dependent manner and displayed potent anti-angiogenic activity in FGF2- and VEGF-induced angiogenesis in matrigel plugs in mice (41). FP-1039 elicited dose-dependent inhibition of growth of the head and neek squamous cell carcinoma (HNSCC) cell lines 584-A2, CCL30 and Detroit 562, but not of UMSCC19, which produces small amounts of FGF2 (42). FP-1039 significantly inhibited tumor growth of a renal carcinoma xenograft model (Caki1) in a dose-dependent manner. Decrease of tumor vascularization and inhibition of phosphorylation of ERK1/2 was observed. Tumor growth of primary human tumor xenografts was also inhibited (43). Single-agent therapeutic efficiency was observed in vitro and in vivo with MFE-280 endometrial carcinoma cells which carry the activating S252W FGFR1 mutation, whereas HEC-1B cells with wild-type FGFR1 status were much less sensitive (43). Phase I studies are currently being performed for this rare population of patients with endometrial carcinoma (43).
The paracrine FGFs bind to their receptors together with heparin sulfate proteoglycan (HSPG), resulting in dimerization and activation. The mode of action of FP 1039 is not yet clearly resolved since paracrine FGFs can be pre-bound to HSPG on the cell surface (28) rather than being present in the circulation. FGFRs have also significant affinity for HSPG and therefore FP-1039 could also be directed to the extracellular matrix. Therefore, competition of FP-1039 with the binding of FGFs to HSPGs might be part of the underlying mode of action.
Activin-like Receptor Kinase 1 (ALK1) Fc
ALK1 is a type I transforming growth factor β (TGFβ) receptor which is preferentially expressed in proliferating vascular endothelial cells (44, 45). Upon ligand binding, the type II serine-threonine kinase receptor phosphorylates and activates ALK1, which by itself phosphorylates a SMAD protein (SMAD 1/5/8). Phosphorylated SMADs dimerize with SMAD-4 and the complexes translocate into the nucleus and mediate transcription of genes involved in endothelial cell function and angiogenesis (46). Involvement of ALK1 in lymphangiogenesis has been also demonstrated (47). TGFβ, bone morphogenetic protein 9 (BMP9) and BMP10 have been identified as ligands of the type II receptor (48) and endoglin, a co-receptor for TGFβ, displays a modulatory function for the ALK receptor complex (46). Genetic studies of ALK1–/– mice and zebrafish with loss-of-function mutations indicated a role for ALK1 in vessel maturation, differentiation of pericytes and in the organization and patency of neo-angiogenic vessels (49, 50). In humans, type 2 hereditary hemorrhagic telangiectasia (HHT2), which exhibits direct arterovenous connections without an intervening capillary bed, is linked to inactivating mutations of ALK1 (51). VEGF and basic fibroblast growth factor (bFGF) can promote endothelial cell attachment, spreading and tubulogenesis in an ALK1-dependent manner (52). Since ALK1 cannot bind to VEGF or bFGF, these findings point to a possible cross-talk between these signaling systems.
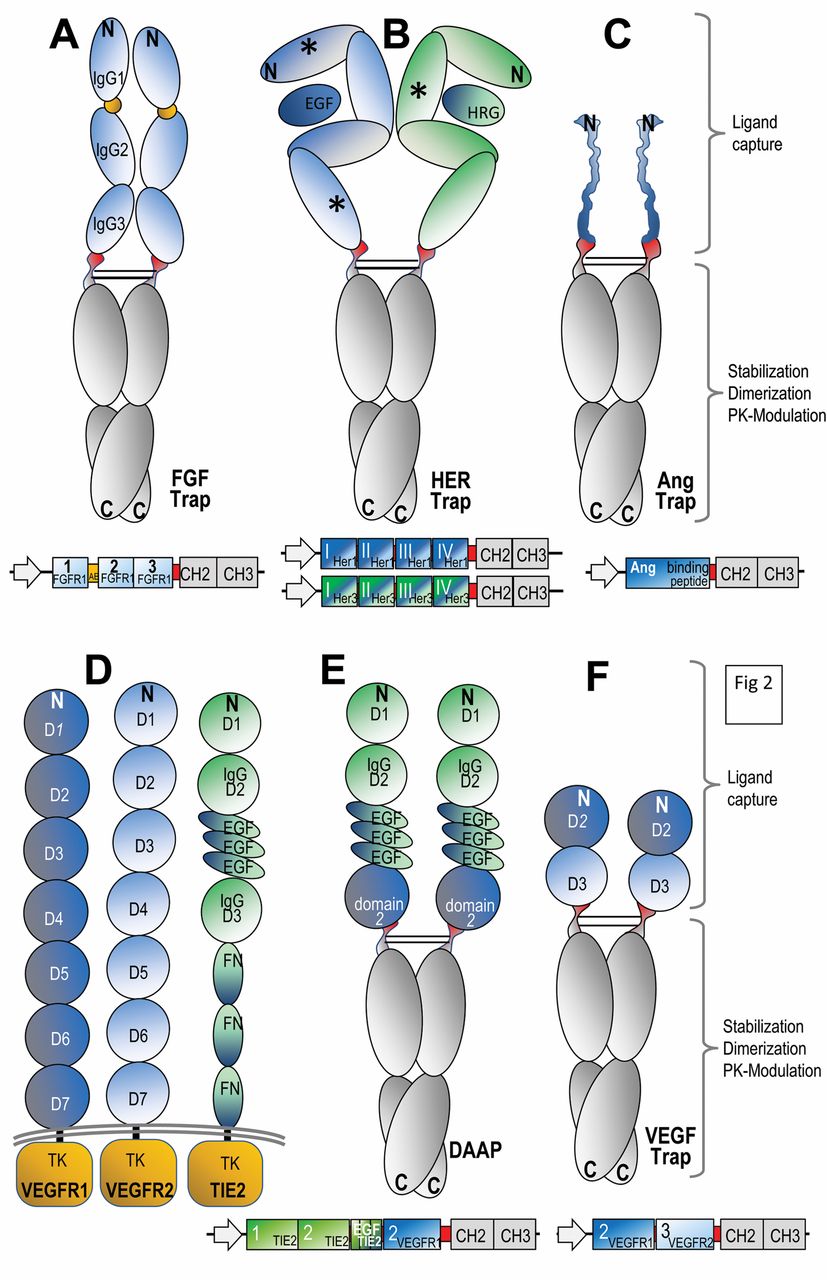
Design and composition of Fc fusion proteins. The domain compositions of Fc fusion proteins are schematically shown in the upper panels and the sequences that encode these entities are summarized in lower panels of each figure. The general principle for design of Fc fusions includes recombinant fusion of effector domains (e.g. extracellular domains of receptor tyrosine kinases or peptides that capture ligands (blue or green) at a hinge region of IgGs (red) to constant regions (CH2 and CH3, grey). These molecules form dimers via their dimerization propensities of CH2-CH3 domains. These dimers are stabilized by disulphide bonds in their hinge regions. A: fibroblast growth factor (FGF) Trap; B: human epidermal growth factor receptor (HER) Trap; C: angiopoeitin (Ang) Trap; D: domain structure of vascular endothelial growth factor receptor 1 (VEGFR1), VEGFR2 and tyrosine kinase receptor in endothelial cells 2 (TIE2); E: double antiangiogenic protein (DAAP); F: vascular endothelial growth factor (VEGF) Trap.
In order to elucidate the biological function of ALK1 in several experimental systems, ALK1 ligand traps have been constructed (53, 54). Murine Alk1 ECD has been fused to mouse Fc (RAP-041), and in huALK1 Fc (ACE-041), the corresponding human equivalents have been fused. The murine Alk1 ligands have been shown to bind to human ALK1 and vice versa (53). Surface plasmon resonance experiments indicated binding of BMP9 and BMP10 with high affinity and not TGFβ by the chimeric fusion proteins (53, 54). ALK1 Fc blocks BMP9 signaling in human umbilical venule endothelial cells (HUVECs) and reduces the expression of transcription factor, encoded by the ID1 gene (Id-1), induces VEGF expression and represses the antiangiogenic factor thrombospondin-1. In a chick chorioallantoic membrane assay, ALK1 Fc reduced VEGF, and FGF and BMP10 induced vessel formation. ALK1 Fc treatment reduced tumor burden in mice receiving orthotopic grafts of MCF-7 mammary adenocarcinoma cells, indicating that ALK1-Fc has a role in mitigating vessel formation. Making use of RAP-041, it was shown that blunting of ALK1 signaling retards the angiogenic switch and tumor growth in the transgenic Rip-1-Tag2 mouse model of endocrine pancreatic tumorigenesis (53) in which BMP9 and TGFβ are increased during the tumor progression pathway. RAP-041 impairs tumor angiogenesis in vivo. It was demonstrated that ALK1 is a haploinsufficient gene in the context of tumor angiogenesis since ALK1+/– Rip1/Tag2 mice displayed a reduction in the number of angiogenic islands, as well as a reduced total tumor burden (54).
In humans, ALK1 is expressed in a broad range of tumor types (46). Acceleron Pharma has started a phase I study of the ALK1 Fc fusion protein ACE-041 in patients with advanced solid tumors or refractory multiple myeloma, assessing safety and tolerability of ACE-041, as well as changes in tumor metabolism by 18F-deoxyglucose positron emission tomography (46). The side-effect profile arising due to dual inhibition of BMP9 and BMP10 will be monitored carefully, since it has been shown that BMP10 ablation in mice leads to embryonic lethality due to impaired cardiac growth and function (46). Taken together, the pre-clinical data available, point to a role of attenuation of ALK1 signaling of this decoy receptor in mouse models of cancer for inhibition of tumor angiogenesis and growth.
Podoplanin Fc
Podoplanin is a transmembrane glycoprotein that is abundantly expressed on lymphatic vessels but not on blood vascular endothelial cells in vitro and in vivo (55, 56). Podoplanin seems to be involved in cytoskeletal organisation because it interacts with proteins of the ezrin/radixin/moesin family, which function as cross-linkers between actin filaments and the plasma membrane (57). Podoplanin-null mice die at birth of respiratory failure and display numerous lymphatic defects, indicating that podoplanin is essential for the function and development of lymphatic vessels (58, 59). The function of the podoplanin ectodomain remains to be investigated in more detail. Interactions with the following proteins have been documented: galectin 8, a modulator of several functions of lymphatic endothelial cells (60); C-type lectin-like receptor (CLEC)-2 on the surface on platelets involved in mediating podoplanin-induced platelet aggregation (61); and the lymphatic-specific chemokine (C-C motif) ligand-21 (CCL21), a chemoattractant for chemokine (C-C motif) receptor-7 (CCR7), expressed on subsets of immune cells (62). To explore the function of podoplanin in more detail, podoplanin Fc was evaluated in several experimental systems. It was shown that podoplanin Fc inhibits adhesion, migration and tube formation of lymphatic endothelial cells, as well as lymphatic vessel formation, in the mouse embroid body assay and impairs lymphatic vessel formation in inflamed mouse cornea in vivo (63). Podoplanin Fc inhibited skin tumor formation in a transgenic skin cancer model, but also induces intravascular coagulation. This property is independent of the Fc portion of the molecule and it was shown that MCF-7 cells expressing transmembrane podoplanin on their surface were able to accelerate coagulation of mouse blood (64, 65). If the platelet aggregation activity of podoplanin can be eliminated, e.g. by mutation of the crucial threonine 52 (63), podoplanin Fc might become an agent interfering with lymphangiogenesis in a therapeutic setting. Interestingly, tumor lymphangiogenesis and metastasis to lymph nodes are also induced by cancer cell-based expression of podoplanin (65). Endothelin-1 (ET-1) might play a crucial role in this context since it was found that ET 1 expression is increased in podoplanin-1 overexpressing tumor xenografts. ET-1 can interact with endothelin receptor B (ENDRB) on lymphatic endothelium and might induce lymphangiogenesis (65).
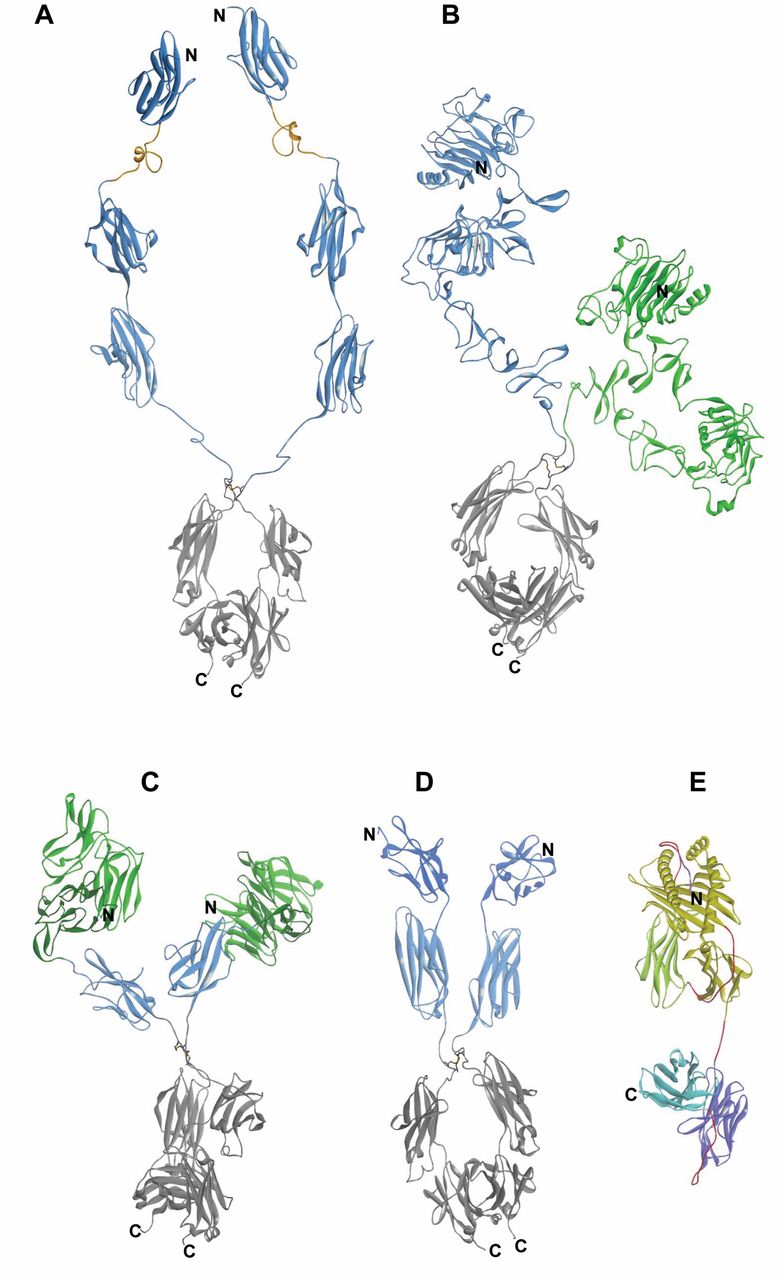
Structure models of cytotoxic fusion proteins. The structure models were generated based on available structure data of domains or entities. Structural data from the Protein DataBank (PDB, Nov. 2011) (134) were assembled and minimized using DiscoveryStudio31 (135). Ribbon representations are shown to facilitate identification of secondary structures where the N-terminal domain is placed on the top and the C terminal domain on the bottom. The color scheme of the models follows that of Figure 1 and 2. A: Model of the of fibroblast growth factor receptor 1 (FGFR1) Trap composed of several different structures starting with the nuclear magnetic resonance (NMR) structure of domain 1 (pdbcode: 2CKN), a model of the acidic box based on the homology to part of coagulation factor VII, in orange (pdcode: 3CDZ), followed by domains 2 and 3, in blue (pdcode: 3OJV), and, at the C-terminal end, the Fc portion, in grey (pdbcode: 1HZH). B: The HER Trap model is composed of the extracellular domain (ECD) domain of epidermal growth factor receptor (EGFR), in blue (pdbcode: 3NJP) and the ECD domain of HER3, in green (pdbcode: 1M6B), fused to the Fc portion, in grey (pdbcode: 1HZH). C: Double antiangiogenic protein (DAAP) model is based on the first two immunoglobulin domains and the three EGF domains of tyrosine kinase receptor in endothelial cells 2 (TIE2), in green (pdbcode: 2GY7), domain-2 of vascular endothelial growth factor receptor-1 (VEGFR1), in blue (pdbcode: 2XAC), and the Fc portion, in grey (pdbcode: 1HZH). D: Model of VEGF Trap (Aflibercept) is based on the crystal structure of VEGFR1 domain-2, in dark blue (pdbcode: 1QSZ), followed by domain-3 of VEGFR2, in light blue (pdcode: 2X1W), and fused to the IgG1 Fc portion, in grey (pdbcode: 1HZH). E: A major histocompatibility complex (MHC) complex structure comprising the α chain of the MHC allele 201, in yellow, the common β2-microglobulin domain, in green, and a peptide in the groove, in magenta (pdbcode: 2VLR), is used to build the model of a fusion with on scFv, in light and dark blue; the linkers comprising G4S motifs are indicated in red.
Angiopoietin Trap
The human angiopoietin (ANGPT) tyrosine kinase receptor in endothelial cell (TIE) systems consists of two tyrosine kinase receptors (TIE1 and TIE2) and three secreted ligands (ANGPT1, ANGPT2 and ANGPT3) (66, 67). ANGPT1 functions as a TIE2 receptor agonist, whereas ANGPT2 normally acts as an ANGPT1 antagonist. ANGPT1 can excert an antiangiogenic function, whereas ANGPT2 mediates a pro-angiogenic function, and the physiological effects of ANGPT3 are poorly resolved (66, 67). ANGPT1 is constitutively expressed in many human tissues, whereas ANGPT2 is predominantly expressed in tissues undergoing vascular remodeling. ANGPT2 can promote the dissociation of pericytes from pre-existing vessels, increase vascular permeability and facilitate the infiltration of proteases, cytokines and angiogenic myeloid cells thus preparing the vasculature for a proliferative response to angiogenic growth factors (67). Angpt2 knock-out mice are born normally, but have defects in lymphatic vasculature, have edema, and the remodeling of the retinal vessels is impaired and many of them die at postnatal day 14 (67). With Angpt2 knock-out mice, it was shown that host-derived Angpt2 affects early steps of tumor development and vessel formation, but is dispensable for later stages (68).
The functional contribution of the ANGPT-TIE2 system in angiogenesis and tumor growth was assessed with AMG 386 [2xCon4(c)], a peptide Fc fusion protein (peptibody) which prevents the interaction of ANGPT1 and ANGPT2 with their receptor TIE2 (69). Phage-display libraries were planned against human ANGPT2, converted into peptide-Fc fusions and expressed in Escherichia coli. AMG 386 potently neutralizes human ANGPT2/TIE2 interaction (IC50=23 pM), as well as human ANGPT1/TIE2 interaction (IC50=900 pM), with similar potency for murine and rhesus monkey Angpt2. AMG 368 is an Fc-fusion protein with 58 amino acids fused at its C-terminus (Figure 2C).
Systemically administered AMG 386 induced regression of large A431 (human epidermoid tumor) and Colo 205 (human colorectal carcinoma) xenografts, with documented suppression of proliferation of endothelial cells in vivo, whereas it had no impact on proliferation of these cells in vitro. AMG 386 also prevented neovascularization in a rat corneal model of angiogenesis. A pharmacological effect on epiphyseal growth plate thickness was observed in rats treated with AMG 386 (69). Epiphyseal thickness is considered to be a consequence of anti-angiogenic therapy in growing rodents treated with VEGF antibodies (70). AMG 368 selectively inhibits ANGPT2, but also displays some inhibitory activity against potentially antiangiogenic ANGPT1. Despite this ANGPT1 neutralizing activity, AMG 368 elicits a similar response profile to the more ANGPT2 selective agents L1-7 (N) and Ab 536 (69). This may indicate that ANGPT2 might be the dominant angiogenic factor and ANGPT1 has a limited function in the context of the experimental settings as described above. In order to elucidate the consequences of ANGPT1 inhibition, ANGPT1-specific inhibitory molecules need to be generated. AMG 386 is currently being evaluated in phase II studies in a variety of cancer types, such as HER2-negative breast cancer, hepatocellular, ovarian, colorectal, gastric and renal cancer (71-73). As the safety profiles of AMG 386 and chemotherapy do not overlap considerably, AMG 386 may be combined with chemotherapy.
ANG VEGF Trap
A chimeric decoy receptor, double antiangiogenic protein (DAAP), which can simultaneously bind VEGF-A and angiopoietins and blocking their actions was designed (74) (Figure 2D, E and 3C). Several reports have indicated that ANGPT-1 and ANGPT-2 are up-regulated in the context of angiogenic rescue when VEGF-A/VEGFR2 signaling is blocked (75-78).
DAAP is composed of the two Ig-like domains and the three EGF-like domains (E1, 2 and 3) of TIE-2, the second IgG domain of VEGFR1 and the Fc portion of IgG1. It was shown that DAAP binds to hVEGF-A165, mouse (m) Vegf-A164, hVEGF-A189, mPLgf, hANGPT2, hANGPT1, mAngpt3 and hANGPT4, but not to hVEGF-C, hVEGF-E, human angiopoietin-like-2 (hANGTPL-2) or mAngptl-4. It should be stressed-out that ANGPT1 has been described as an antiangiogenic factor, and ANGPT2 as a pro-angiogenic factor, whereas the function of ANGPT4 has been poorly resolved (66, 67). For comparison, VEGF Trap was able to bind to all isoforms of VEGF-A but not any of the angiopoietins, whereas TIE2 Fc was able to bind to all ANGPTs tested, but not to VEGF. In vivo evaluation of DAAP in an established Lewis lung carcinoma (LLC) tumor model demonstrated regressions of VEGF-A induced active vessel sprouting and network formation, but also blockage of ANGPT-induced vessel enlargement in tumor angiogenesis. DAAP was shown to inhibit growth and metastasis of implanted B16 melanoma cells and its combination with dacarbazine or cisplatin in the LLC model resulted in improved efficacy in comparison to treatment with the single-agents. Overall, the in vivo experiments indicate superiority of DAAP in comparison to treatment with VEGF Trap and TIE2-Fc and the combination of the latter agents (74). DAAP also reduced growth and metastasis of orthotopically implanted CT-26 colon cancer and in the transgenic MMTV-PyMT breast cancer model. In an ovarian orthotopic cancer model (MDAH-2774), reduction of vascular leakage and induction of tumor vessel normalization was shown. A close correlation between in vivo efficacy and expression of VEGF-A and ANGPT-2 in the models referred above was noted (74). Thus, simultaneous inhibition of VEGF-A and ANGPTs seems to be an effective strategy in blocking angiogenesis, metastasis, vascular leakage and potential treatment of VEGF-treatment-resistant tumors. Whether the potential dual function of ANGPT1 and ANGPT2, with respect to inhibition and promotion of tumor-related angiogenesis, is a critical issue for safety and clinical efficacy remains to be resolved.
HER Trap
The receptor tyrosine kinase (RTK) family of the HER (ERBB) or EGFRs comprises four members: EGFR/HER1, HER2, HER3 and HER4 (79). When bound to a ligand they undergo dimerization and transmit intracellular signals resulting in cell migration, proliferation and differentiation. Two of these receptors, HER1 and HER4 are autonomous, undergoing dimerization and signaling upon ligand binding. The other two receptors form heterodimeric signaling complexes with other ligand-bound HER family members (80). Seven ligands were shown to bind to EGFR/HER1: EGF, heparin-binding EGF (HB-EGF), TGFα, amphiregulin (AR), betacellulin (BTC), epigen (EPG) and epiregulin (EPR). Four neuregulins (NRGs) have been identified. NRG1, NRG2, HRG1 and HRG2 bind to HER3 and NRG1 to 4 bind to HER4. In addition, BTC, HB-EGF and EPR can bind to HER4 (81). The seven ligands which bind to the EGFR are synthesized as transmembrane precursors, cleaved by metalloproteases thus releasing mature growth factors (82, 83).
HER signaling is dysregulated in many types of cancers and based on this observation, drugs for treatment of cancer have been developed, such as Cetuximab, Herceptin, Pertuzumab and trastuzumab-DM1 (TDM1), as well as small-molecule kinase inhibitors, such as erlotinib, gefitinib and lapatinib (84, 85). A common theme for these mono- or dual-specific target inhibitors is the development of resistance during treatment (86, 87), with involvement of HER family members which are not targeted by the specific drug. Therefore, an approach targeting several members of the ERBB family and their ligands might be superior from a conceptual point of view.
Based on this reasoning an HER-ligand binding protein has been designed. RB200 (Hermodulin) is an HER1-HER3 heterodimer dimerized with an Fc fragment of human IgG1 (88-91) (Figure 2B and 3B). Binding studies revealed that RB200 interacts with HER1 ligands such as EGF, TGFα and HB-EGF, as well as HER3 ligands NRG1 and HRG, and inhibits EGF and NRG1 stimulated HER family protein phosphorylation. RB200 was shown to inhibit both growth-factor stimulated and unstimulated cell proliferation in monolayer cultures and synergistic growth inhibition was observed in combination with tyrosine kinase inhibitors (88). Single-amino acids were identified in the HER1 and HER3 part of the ligand binding trap which confer high affinity sequestration of the cognate ligands (91). A single mutation (T15S, EGFR subdomain I) increased affinity for EGF two-fold, TGFα 26-fold and HB-EGF six-fold. Another mutation (Y246A, HER3 subdomain II) enhanced NRG1 binding eight-fold, probably by interfering with subdomain II-IV interactions. Further work revealed that the HER3 subunit of an HER1/HER3 heterodimer suppresses EGFR ligand binding. Reverting this suppression by mutation resulted in enhanced ligand binding (EGF 10 fold, TGFα 34 fold, HB-EGF seven-fold, NRG1 31-fold). Increased improvement of ligand-binding was reflected in improved inhibition of tumor cell proliferation (91). RB200 inhibited xenograft growth of A431 (epidermal carcinoma) and H1437 (NSCLC) (91). Further experiments need to be performed in order to demonstrate differentiation of this HER-ligand trap from other HER-signaling inhibitors and in treatment-resistant settings.
Transmembrane- and Calcium-modulating Cyclophilin Ligand (CAML) Interactor (TACI) Fc
B-lymphocyte stimulator (BLyS) (also known as BAFF, TALL-1, THANK and zTNF4) is a member of the tumor necrosis factor (TNF) family and is functionally involved in survival of B-lymphocytes, regulation of peripheral B-cell populations and is overexpressed in several B-cell malignancies. BLyS binds to three receptors: B-cell maturation antigen (BCMA), TACI and B-cell activating factor of the tumor necrosis factor family receptor (BAFF-R) (92, 93). A proliferation-inducing ligand (APRIL), a factor closely related to BLyS in sequence and function and which is also overexpressed in several B-cell malignancies, has also been identified (92). TACI and BCMA bind to BLyS and APRIL and BAFF-R binds to BLyS. This indicates that BLyS can signal through three receptors, whereas APRIL signals through TACI and BCMA (92). BLyS and APRIL act as survival factors for malignant myeloma cells cultured within the bone marrow microenvironment and for B-cells of patients with Waldenströms macroglobulinemia (WM) (93-96).
Atacicept is a fusion protein composed of the ECD of TACI and the Fc portion of human IgG1, resulting in neutralization of BLyS and APRIL (97). In vitro studies have demonstrated that Atacicept exerts a negative effect on proliferation and a pro-apoptotic effect on B-cells from patients with lymphoma, multiple myeloma and WM and can also block the biological activity of heterodimeric complexes of this receptor family (97, 98). Atacicept is currently being clinically investigated in phase I studies in patients with relapsed and refractory B-cell non-Hodgkin’s lymphoma (NHL), multiple myeloma and WM (99,100). Atacicept is currently being also evaluated in several phase I to III studies in patients with autoimmune diseases (101-103).
Antibody Cytokine Fusion Proteins
A promising strategy for the treatment of cancer is the recruitment of immune effector cells to the tumor for induction of an antitumor response. Systemic administration of IL2, granulocyte-macrophage colony stimulating factor (GM-CSF), or IL12 were shown to increase the immunogenicity of tumors and their elimination, based on the underlying immune response (104). However, severe side-effects have been observed, since systemic administration does not correspond to their physiological mode of action (105). In order to increase the therapeutic index, cytokines were genetically fused to antibody-relating targeting modules directed against tumor antigens (106, 107) (Figure 1 A-C). The half-life of cytokines is significantly improved in antibody cytokine fusion proteins. Pre-clinical in vivo efficacy data are summarized in (106, 107). In addition to their evaluation in a therapeutic setting, some of these fusion proteins were also evaluated in an adjuvant setting. For example, targeted IL2 was shown to enhance immune responses induced by DNA vaccination (108). Antibodies directed against CD20, epithelial cellular adhesion molecule (EpCAM), ganglioside GD2 and single-stranded DNA in conjunction with IL2, IL12, GM-CSF and TNFα have been evaluated pre-clinically (106, 107). The side-effect profile of these fusion proteins is triggered by the tumor specificity of the antibody moiety. The fibronectin B-domain, generated by differential splicing, was detected selectively in the extracellular matrix of newly formed tumor-related blood vessels. An antibody (L19) with specificity for this domain was identified (109). Fusion proteins of this antibody with TNFα, IL2 and IL12 have been evaluated pre-clinically (110). A fusion protein between L19 and IL2 gave rise to an improved therapeutic index for IL2, significant antitumor activity, and infiltration of experimental tumors with macrophages, lymphokine-activated killer (LAK), natural killer (NK) cells and T-lymphocytes (111). This and other antibody cytokine fusion proteins are presently under clinical investigation (112-114).
Antibody-MHC Class I (peptide) Fusion Proteins
Passive immunization of patients with cancer with monoclonal antibodies has resulted in a proven record of therapeutic benefit (107, 115, 116). Another approach is immunization of patients with cancer with peptides recognized by T-lymphocytes and adoptive transfer therapies with tumor-reactive selected T-cells directed against tumor antigens (117). These approaches are based on the elimination of tumor cells presenting MHC/peptide complexes by cytotoxic T-cells. An important pitfall of this approach is the inability to select tumor cell populations with loss of MHC expression, or dysfunctions in the MHC peptide presentation pathway (118).
Therefore, recombinant proteins have been designed, which combine an antibody-based targeting module with a fused β2 microglobulin MHC module. In some cases, the specific peptide binding to MHC has also been fused to the designed proteins, as described above (119-122) (Figure 1D and 3E). The specific peptide can be derived from a tumor or viral antigen. Proof-of-concept was demonstrated with a single HLA-A2 molecule genetically fused to the variable domains of antibody directed against the α subunit of the IL2R. Tumor cells coated with this fusion protein were susceptible to lysis by HLA-A2 restricted melanoma gp100 peptide specific cytotoxic T-cells (CTLs) (121). scab-sc HLA-A2 complexes targeting the α-subunit of the IL2R or human mesothelin with specificity for melanoma differentiation antigen gp100 or an Epstein Barr virus (EBV)-derived peptide epitope were evaluated in vivo after refolding and loading with the corresponding peptide. In vivo efficacy was demonstrated after intratumoral (i.t.) or intravenous (i.v.) injection of human CTLs (121). Another example involved an antibody scFv fragment derived from EGFR antibody C225 fused to monomeric sc-HLA-A2 complexes containing immunodominant tumor-related or viral epitopes (122). The corresponding fusion protein mediated CTL-dependent lysis of EGFR-expressing tumor cells regardless of the expression of self-peptide MHC complexes. In vivo efficacy in corresponding xenografts was shown after i.t. and i.v. administration of EBV-specific CTLs. A potential problem associated with the MHC peptide complexes is the stability of the complex since the peptide can dissociate from the MHC binding groove. Therefore, N-terminal peptides were fused with sc-HLA-A2 molecules and C-terminal scFv of an anti-IL2R α subunit-specific antibody. It was shown that the fusion protein induced HLA-A2-restricted, specific CTL-mediated lysis after binding to antigen-positive tumor cells (122). In comparison to the use of bi-specific antibodies, the approach, as described, induces a monoclonal instead of a polyclonal T-cell response. It should be stressed out that in none of the described fusion proteins is an Fc moiety incorporated. As outlined, Fc regions can impact pharmacokinetic properties and are mediators of antibody-based effector functions.
NK Cell Receptor Ligand Fusion Proteins
NK cells can eliminate tumor cells in the absence of MHC restriction, making use of multiple activating receptors (123). One of the activating receptors is NKG2D (124, 125). Ligands in human are MHC class I chain-related protein A/B (MICA/B) and retinoic acid early-transcript (RAET1), also called ULBPs (UL-16 binding proteins). NK2G2D ligands are preferentially overexpressed in tumor cells and tumor-associated suppressor cells. It was shown that a fusion protein between scFv anti-CD3 and the ECD of NKG2D binds to NKG2D ligand-positive tumor cells and activates T-cells via scFv anti-CD3 (126). Administration of scFv-NKG2D promoted survival in a murine lymphoma model. It was shown that an immunological memory response was generated in these mice.
TCR Fusion Proteins
Many tumor-related antigens are located intracellularly and cannot be targeted with antibodies. Cancer testis antigens are one class of such targets among others (127, 128). Peptides derived from these targets are presented as T-cell epitopes complexed with MHC molecules and induce a cytotoxic T-cell response. The concept of a TCR-derived fusion protein for recruitment of immune effector cells to the tumor has been described (129, 130). A cytokine fusion protein (264 sc TCR/IL2), consisting of IL2 genetically-fused to soluble sc HLA-A2-restricted TCR recognizing a peptide derived from p53, which is overexpressed in tumors, was constructed. In this context, IL2 can be delivered directly to the tumor with minimized toxicity. With human melanoma A375 xenografts (p53+/HLA-2.1+) in nude mice better than equivalent antitumor doses of IL2 was noted. Antitumor activity was mediated by NK cell activation and tumor infiltration. Tumor growth inhibition was observed in p53+/HLA-A2+, not in p53+/HLA-A2– tumors. Furthermore it was shown that the ECD of a TCR can be genetically fused to anti-CD3 antibodies for recruitment of T-cells (131). Soluble high-affinity TCR, specific for HLA-A2-presented melanoma-associated epitope gp100 281-288 was fused to anti-CD3 sc Fv domain. The activation of a polyclonal T-cell response, when targeted to melanoma cell presenting the gp100 derived epitope, was reported. Pico-molar (pM) activity was found in a panel of tumor cells. Inhibition of tumor growth was observed after co-injection of the fusion protein with human peripheral blood monocuclear cells. Phase I studies have been performed (132).
Concluding Remarks
From a technical point of view, genetically engineered fusion proteins confer improved pharmacokinetic characteristics on decoy receptors and cytokines. This allows for less frequent and optimized dosing of these agents in comparison to their non-fused counterparts (134). Potential immunogenicity, due to the newly-generated junctions is a theoretical concern, but does not match the clinical observations. Tumor-targeting is dependent on the tumor specificity of the antigen that the antibody-based molecule is directed against. In the case of cytokines such as IL2 and TNFα, a significant improvement of the therapeutic index was observed upon administration of the fusion proteins.
Sequestration of Fc-based fusion proteins by interaction with Fc receptors also does not seem to be a problem. The maintainance of the cognate binding affinity of the ECD for its ligands must be investigated case-by-case and if necessary, can be improved by mutagenesis (91).
From a conceptional point of view, through creation of ECD-based fusion proteins by incorporation of ligand-binding modules from different receptors, more potent decoy receptors can be generated in comparison to ECD-based fusion proteins derived from a single receptor. The specificity of the antitumoral response mediated by immune effector cells is largely triggered by the specificity of the target molecule, its expression levels on tumor cells, its continuous expression and its tumor penetration. Down-modulation of the corresponding antigens targeted by the appropriate fusion proteins is a general mechanism of resistance. In the case of fusion proteins targeting angiogenesis- or tumor-related growth related receptors switching to alternative pathways after inhibition of the original pathways has been observed.
The recent approval of Aflibercept for the treatment of macular degeneration might be the beginning of a bright future for genetically engineered fusion proteins for the treatment of cancer.
Footnotes
-
This article is freely accessible online.
- Received July 3, 2012.
- Revision received July 27, 2012.
- Accepted July 31, 2012.
- Copyright© 2012 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue




{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- Technologies for Generation of Genetically Engineered Fusion Proteins
- VEGF Trap
- Fibroblast Growth Factor Receptor 1 (FGFR1) Fc
- Activin-like Receptor Kinase 1 (ALK1) Fc
- Podoplanin Fc
- Angiopoietin Trap
- ANG VEGF Trap
- HER Trap
- Transmembrane- and Calcium-modulating Cyclophilin Ligand (CAML) Interactor (TACI) Fc
- Antibody Cytokine Fusion Proteins
- Antibody-MHC Class I (peptide) Fusion Proteins
- NK Cell Receptor Ligand Fusion Proteins
- TCR Fusion Proteins
- Concluding Remarks
- Footnotes
- References
- Figures & Data
- Info & Metrics