


Abstract
Background: Defects of some DNA polymerases have shown associations with cancer, but data on DNA polymerase ϵ are limited. This study investigated mutations in the 55 kDa subunit gene of DNA polymerase ϵ in colorectal cancer. Materials and Methods: DNA from 16 human colorectal cancer and 9 control samples was studied with polymerase chain reaction–single-strand comformation polymorphism analysis and DNA sequencing. Results: DNA polymerase ϵ gene alterations were identified in 5 out of the 16 cases (31.2%). Two samples showed a T-C transition at exon 17 (potential tyrosine to histidine substitution), and an A-G transition at intron 7; one sample showed an A-G transition at intron 8. An AATT deletion was observed at intron 18 in 3 out of the 16 colon cancer cases (grades 2, 3, and 2, and Dukes' classes C, D, and C, respectively). Conclusion: Because the AATT deletion has also been found in breast cancer, the region may be a mutation hot spot, possibly involved in the carcinogenetic path in advanced colorectal cancer.
The modified Dukes' class (or TNM classification) is the best general prognosticator for colon cancer (1). However, there are still situations in which other prognosticators will be able to offer valuable advice for clinicians, e.g. within individual classes. This especially applies to Dukes' class B cases, and proposals for additional prognosticators have been suggested (2). Many potential and proven prognostic features are negatively or positively associated with proliferation-associated features, such as Ki-67 score (3). Proliferation, on the other hand, is intimately linked with DNA polymerases. This is why we wished to compare the genomic status of the second largest (55 kDa) subunit of DNA polymerase ϵ (POLE2) with several prognostic features to get an idea how genomic abnormalities in DNA polymerase ϵ might be related to clinical presentation or prognosis associated features (4-6).
DNA polymerases have a central role in the maintenance of genome integrity, as they are the enzymes that actually synthesise DNA (7). At least 14 such polymerases have been identified in the mammalian cell, but only three of these, α, δ and ϵ, are considered to perform the bulk of DNA synthesis during replication (7). These members of family B DNA polymerases are structurally related enzymes and are all essential for nuclear DNA replication. They also have additional roles in DNA repair and cell cycle control (8).
Information on the association of DNA polymerase defects with biological phenomena have been slow to emerge. It has been suggested that defects of mitochondrial DNA polymerase are associated with premature aging (9). There are links to cancer with some DNA polymerases, especially with β (POLB). POLB mutations or splice variants of POLB mRNA have been found in human colorectal, prostate and bladder cancer (10-12). Other DNA polymerases have been implicated in tumorigenesis. Mutations in the DNA polymerase η (POLH) gene underlie the cancer-prone xeroderma pigmentosum variant syndrome (XP-V) (13, 14). Point mutations were detected in the DNA polymerase δ (POLD) in colon cancer cell lines, and in primary human colorectal cancer (15). This phenotype was also found in mouse models carrying mutated forms of POLD (16-19). There are no comparable reports of mutation for POLE. This is surprising, since several mutations have been reported for yeast pole that reduce replication fidelity and cause genomic instability. This underlines the importance of POLE in chromosomal replication (20), suggesting the potential pathologic role of human POLE mutations. Human POLE is composed of four subunits, a large catalytic subunit of 261 kDa and three associated subunits of 55, 17 and 12 kDa (21). Our earlier report demonstrated that POLE2 gene variations were observed in human breast cancer (22). In this study, we further investigate the POLE2 gene variations in human colon cancer.
Materials and Methods
Studied samples. Fresh colon tumor tissues from 16 patients were collected at the Turku University Central Hospital and Turku City Hospital (Table I). All samples were immediately transferred to our laboratory in liquid nitrogen, and processed for RNA and DNA isolations. Corresponding normal colon tissues at a distance of 2-5 cm from the tumor were obtained from 8 patients at the same time. A normal human placental sample was collected at Turku University Central Hospital. The histological grades and Dukes' stages are shown in Table I.
PCR amplification. The 19 sets of intronic primers shown in Table II were designed to amplify the entire coding region of the POLE2 DNA sequence reported by Huang et al. (23). The SP6 promoter sequence, GACACTATAGAATAC, and T7 promoter sequence, CGACTCACTATAGGG, were attached to the 5' end of each upstream and downstream primer, respectively. The polymerase chain reaction (PCR) was run for 35 cycles, each consisting of denaturing for 1 min at 94°C, or at 95°C for exon 1, 11 and 12, annealing at different temperatures (Table II) for 1 min, and extension for 1 min at 72°C. PCR was performed in a Perkin Elmer Cetus DNA Thermal Cycler 480 (Perkin Elmer, USA). The second pair of Cy5-labelled primers (SP6 5': TTTAGGTGACACTATAGAATAC and T7 5':GTAATACGACTCACTATAGGG) were used for subsequent secondary PCR and to produce Cy5-labelled PCR products. The 30 cycles included 30 s denaturation at 94°C (except exon 1 at 96°C), 30 s annealing at 55°C and 30 s extension at 72°C. The PCR products were electrophoresed on 2.5% agarose gel.
Single-strand comformation polymorphism (SSCP) analysis. The Cy5-labelled PCR products were mixed with an equal volume (4.5 μl) of denaturing solution containing 100% deionized formamide, and 0.05% bromophenol. The mixture was heated at 95°C for 4 min, thereafter chilled on ice. Two undenatured samples (one from among the cancer cases, one from among controls, no heating at 95°C) along with the denatured test samples were studied with SSCP. A volume of 2.5 μl of the mixture was loaded on a 6% polyacrylamide gel (acrylamide:bisacrylamide, 99:1) in an ALFexpress II with Cool kit (Pharmacia Biotech, Sweden). The running conditions for the gel electrophoresis were 35 W for 10 h at 12°C with external cooling. The SSCP data were collected by ALFwin software and analyzed by fragment analyzer 1.02 (Pharmacia Biotech).
Sequence analysis. Samples displaying variant bands on SSCP were additionally analyzed by sequencing. DNA Samples were amplified by PCR as described above. The amplification products were purified with a GFX™ PCR DNA and Gel Band Purification Kit (Amersham Biosciences, Sweden). Purified PCR products were sequenced using Thermo Sequenase Cy™5 Dye Terminator Kit according to protocols provided by the manufacturer (Amersham Biosciences, Sweden). The sequencing products were electrophoresed on 6% PAGE gel at ALFexpress II DNA sequencer (Pharmacia Biotech). The sequence data were collected by ALFwin software and analyzed by sequence Analyser 2.00 (Pharmacia Biotech). The sequencing results were compared with chromosome 14 genomic sequence data from bacterial artificial chromosome (BAC) clones (24).
The clinical profile of cancer specimens, and the mutationsa of POLE2 found in this study. Histologically, all cases represented adenocarcinomas.
Immunohistochemistry. Her-2 (ErbB2): A monoclonal mouse antibody was used (clone CB11; Novocastra Laboratories Ltd, Newcastle upon Tyne, UK) diluted 1:50, with microwave treatment 600 W, 2×7 mins in citrate buffer at pH 6. Both membranous and cytoplasmic staining were evaluated from highest intensity areas, and in the area representing average staining as judged subjectively. Intensity of staining was scored as 0, 1, 2, or 3, and the immunohistochemical staining index calculated according to the formula:

where f0 – f3 refer to the fractions of cells staining with intensities 0-3 (25). This type of evaluation in our experience has given more meaningful prognostic results than subjective intensity grading alone (26).
Intronic primer sets for amplification of exons 1-19 for the POLE2 gene.
Ki-67: Clone MIB-1 monoclonal mouse antibody was used (DakoCytomation, Denmark), diluted 1:100, with microwave treatment as above. Nuclear staining was evaluated as a fraction of positively stained nuclei (% of all nuclei present). Staining was evaluated in the area of most prominent positive staining, and in an area of average staining as judged subjectively.
Bcl-2: Clone 124 monoclonal mouse antibody was used (DakoCytomation), diluted 1:20, with microwave treatment as above. Cytoplasmic staining was evaluated with an index based on the formula above (25). Four intensity categories were used: 0, 1, 2 and 3.
p53: Clone DO-7 monoclonal mouse antibody was used (DakoCytomation), diluted 1:300, with microwave treatment as above. Nuclear staining was evaluated as in the case of Ki-67 (see above).
DakoCytomation link biotinylated secondary antibodies (AB2) were used. The final stain was produced with ChemMate (Dako) streptavidin peroxidase using 4',6-diamidino-2-phenylindole (DAPI) as the chromogen.
Statistical evaluation. Associations between POLE2 gene status and various immunohistochemical stainings were evaluated by using the non-parametric Kruskal-Wallis test, and Fischer's exact test (27-29).
Results
Genomic DNA from 16 colon tumors and 9 control samples (8 tumor-associated corresponding normal tissue, and 1 placental sample) was amplified by PCR, and subsequent SSCP revealed abnormal electrophoretic patterns in five PCR products, which were derived from cases 4, 6, 10, 14 and 16. All SSCP abnormalities were sequenced.
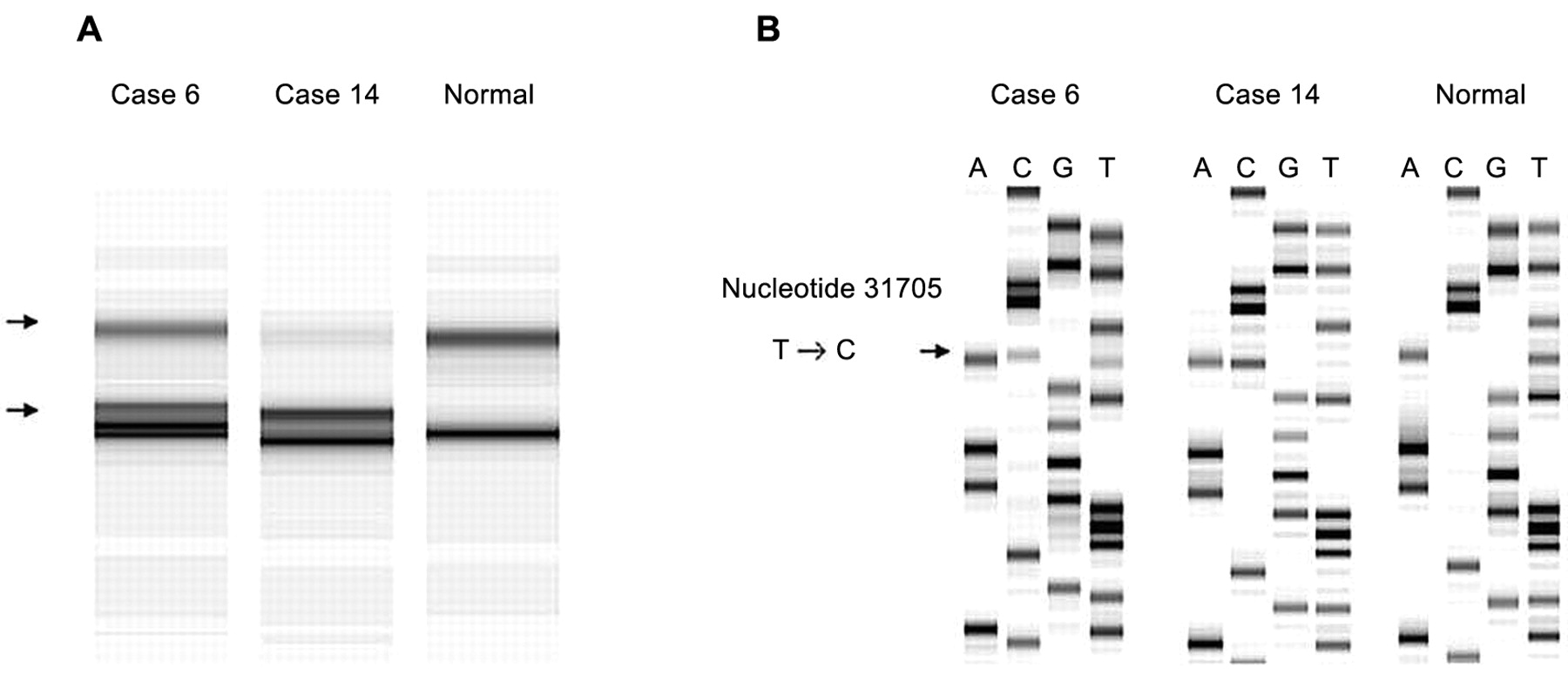
A, SSCP analysis of PCR products derived from primer set 17. Abnormal patterns were observed in cases 6 and 14. B, The DNA sequences of the PCR products of cases 6 and 14, and normal tissue sequence. Arrow, T to C transition at nucleotide 31705.
Cases 6 and 14 showed an abnormal pattern in SSCP from primer set 17 and sequence analysis revealed a T→C transition at nucleotide (nt.) 31705 in exon 17 (Figure 1), which could be expected to result in a tyrosine → histidine substitution at codon 472. Moreover, these two cases showed an A→G transition at nt. 47656 in intron 7 and a G→A transition at nt. 47680 in exon 7 with primer set 7 (Figure 2). The alterations in exon 7 both coded for glutamine, although the wild-type gene read CAG and the mutated tumor gene read CAA at codon 189.
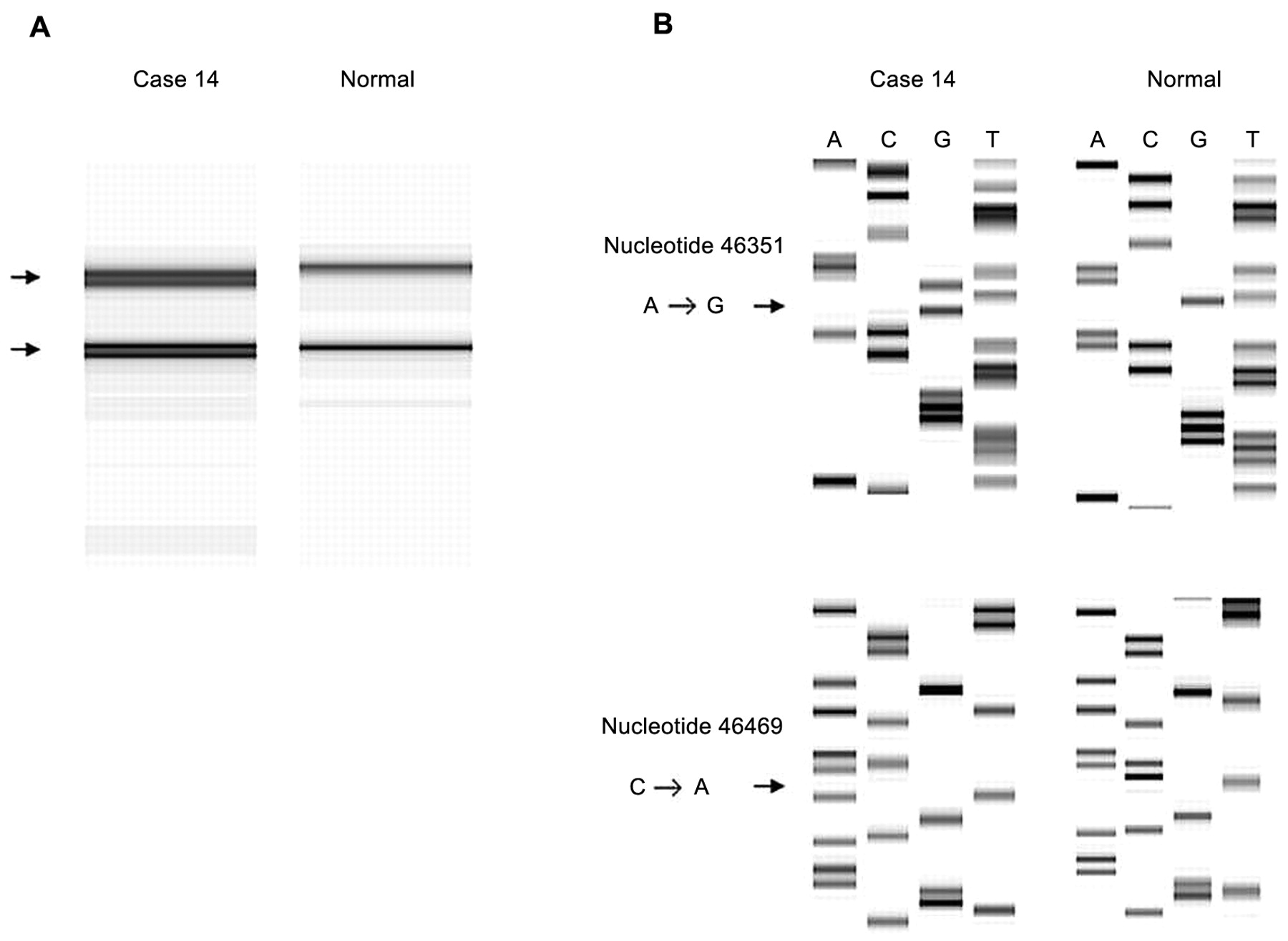
Case 14 also showed abnormal patterns in SSCP associated with primer set 8. The sequencing analysis of PCR product revealed an A→G transition at nt. 46351 in intron 8, and a C→A transition at nt. 46469 in exon 8 (Figure 3), which did not affect the amino acid encoded.
Cases 4, 10 and 16 showed an AATT deletion at nt. 25115-25118 in intron 18 by primer set 19 (Figure 4). This deletion is the same that we previously reported in two cases of breast cancer (22).
In addition to the aforementioned mutations, we found a clearly polymorphic DNA alteration which showed a G→A transition at nt. 35481 with primer set 14.
Interestingly, the T→C, A→G, and G→A transitions in case 6 and the AATT deletion in case 10 were also found in the corresponding normal tissues (Table I). Histologically, the tumors corresponding to the AATT deletion in cases 4, 10 and 16 were of grades 2, 3 and 2, respectively and of Dukes' stages C, D and C, respectively. There was no significant association of the presence of mutations with tumors of the left or right side of the colon, but two samples showing single base changes were from tumors of the left side.
Immunohistochemical results are shown in Table III and some clinical features are intercorrelated with them. Samples with mutations showed higher proliferation rates than the samples with no POLE2 mutations, and these samples on average had a lower p53 index, but the differences are not statistically significant. The data (Table I) also show a trend for tumors with mutations on average being of a higher stage than non-mutated ones (p=0.15).
Discussion
In the study reported here, we found that mutations occurred in POLE2 in 5 out of 16 cases of human colon cancer examined. These results suggest that specific changes can occur in the POLE2 gene in colon cancer. POLE2 is not yet fully characterized, and there is no definite evidence that the mutations found in this study are actually inactivating mutations. It is important to note that the AATT deletion observed at intron 18 in 3 out of the 16 colon cancer cases was also seen in our previous study on human breast cancer (22), which suggests that this region could be a hot spot for mutation in this gene or a rare polymorphism which could increase cancer risk in general.
Jaszczur et al. (30) recently isolated temperature-sensitive mutants in the corresponding yeast gene dpb2. Even at permissive temperature, the mutants increased mutation rates to the level of mutants defective in the 3'→5' proofreading exonuclease (Pol2-4), or mutants defective in mismatch repair (msh6) enzymes. dpb2 mutants can exhibit a mutator phenotype comparable to that of mismatch-repair-defective strains (msh6), indicating that defects in the 55 kDa subunit of the human POLE2 gene may increase genomic instability and the risk of disease and carcinogenesis. Furthermore, Jaszczur et al. (31) reported that in Saccharomyces cerevisiae strains carrying different mutated alleles of the dpb2 gene, the mutated alleles are responsible for the observed mutator phenotypes and controlling spontaneous mutagenesis.
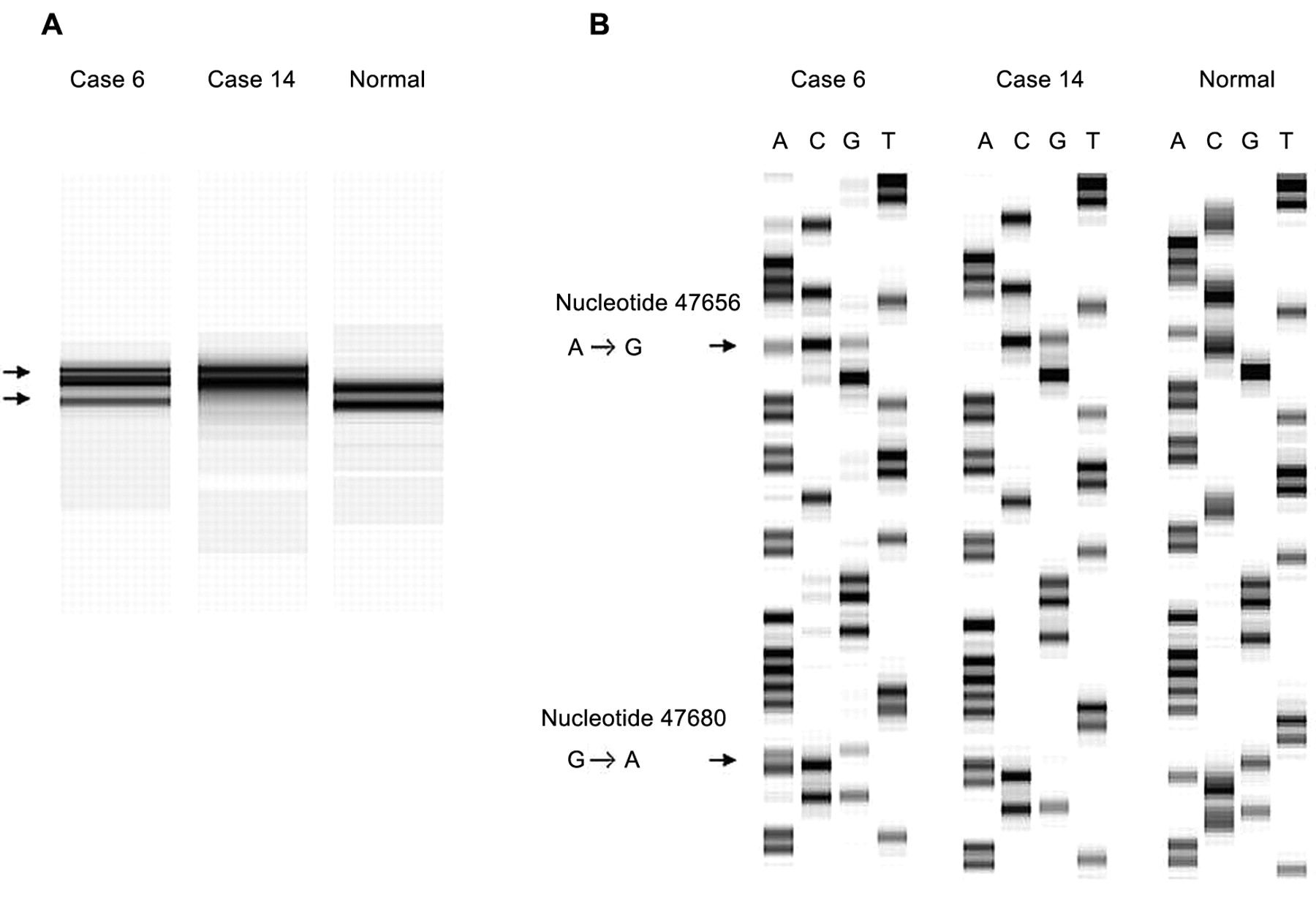
A, SSCP analysis of PCR products produced by primer set 7. Abnormal patterns were observed in cases 6 and 14. B, The DNA sequences of the PCR products of cases 6 and 14, and normal tissue sequence. Arrows: A to G transition at nucleotide 47656; G to A transition at nucleotide 47680.

A, SSCP analysis of the PCR products derived from primer set 8. An abnormal pattern was observed in case 14. B, The DNA sequence of the PCR product of case 14, and normal tissue sequence. Arrows: A to G transition at nucleotide 46351; C to A transition at nucleotide 46469.

A, SSCP analysis of the PCR products derived from primer set 19. Abnormal patterns were observed in cases 4, 10 and 16. B, The DNA sequences of the PCR products of cases 4, 10 and 16, and normal tissue sequence. Arrow, AATT deletion of nucleotides 25115-25118.
Associations of sequence findings of the POLE2 gene, and location of the tumor (right side, left side) with immunohistochemistry results for Her-2, Bcl-2, p53 and Ki-67 of the studied 16 colorectal tumors. Interrelationships of p53 and Ki-67 immunohistochemical results are also shown.
There are certain technical questions that should be discussed in association with the results. One is the presence of the same mutation in the adjacent normal appearing gut tissue and in the tumor in two samples. This may be related to the development of POLE2 mutations in the early stage of colon tumorigenesis and cause large areas of mucosa to be sensitive to further carcinogenesis. Alternatively, these patients may have carried a hereditary genetic change in POLE2, i.e. the changes could actually be polymorphisms. Normal POLE2 sequence was found in all tumor specimens except in case 14, indicating that tumor tissues were carrying both normal and mutated genes. This either suggests that the tumors are heterogeneous, or that the profile is due to there being normal tissue present within the sample. It is probable that the latter is the case for the finding that both normal and mutated DNA were found in these samples.
Table I shows that of the five cases with mutations, four were of histological grade 2 and one was of grade 3. This may suggest that higher grades may be associated with mutations in POLE2, but there is no conclusive evidence because the number of cases is low. On the other hand, there is a tendency for tumors with mutations being linked to advanced Dukes' class: 4 out of 5 tumors with mutation were Dukes' C or D tumors. However, the difference is not statistically significant.
The trend for an increased proliferation rate and a lower p53 index may suggest that the POLE2 alterations observed may affect the checkpoint function of POLE (32) rather than its essential role in DNA replication. Alternative, POLE2 mutations could cause a general mutator phenotype, as described for mutations of POLD.
In many types of cancer, proliferative activity is associated with prognosis, breast cancer being the classical example (33-35). In colorectal carcinoma, the immunohistochemical evaluation of the fraction of Ki-67-staining nuclei (Ki-67 index) has shown association with improved survival (5, 36) after chemotherapy. This may be treatment-associated because in some tissues Ki-67 index and survival showed an inverse relationship (37). The relationship of Ki-67 index and mitotic activity in colon cancer is not as well known as in breast cancer (38). In the work of Zavrides et al. (3), Ki-67 index was inversely related to the intensity of Bcl-2 staining. This is also apparent in our study. The tumors with POLE2 mutations also had a slightly higher Ki-67 index than those without mutations and they were on average of higher stage and grade, although the true significance of this finding could not be defined due to the small number of non-wild-type cases. The results, however, are not contrary to the potential significance of the findings in the origin and/or progression of colorectal carcinoma. On the other hand, cases with intronic AATT deletion had a lower Her-2 index, lower Bcl-2 index, and higher p53 index than other mutations, but the significance of these findings cannot be evaluated at this stage.
In summary, although the relationship between the variations of POLE2 gene and alteration in DNA replication and tumorigenesis remains to be resolved, our results suggest that POLE2 mutations may contribute to the development of certain cases of human colon carcinoma. It seems to us that further studies on this subject are warranted.
Footnotes
-
↵* Present address: Department of Pathology, Al-Arab Medical University, Benghazi, Libya, Finland
- Received September 22, 2009.
- Revision received November 25, 2009.
- Accepted November 27, 2009.
- Copyright© 2009 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved




{kind=link}
{kind=link}
{kind=link}
{kind=link}