


Abstract
Background/Aim: Cisplatin nephrotoxicity includes early activation of the pro-apoptotic p66Shc and disorganization of the actin cytoskeleton, integrity which is regulated by heat-shock protein-27 (Hsp27). Here we determined the potential role of p66Shc in abrogating the Hsp27 function. Materials and Methods: Effects of p66Shc knockdown and Hsp27 overexpression on F-actin stress fibers after cisplatin treatment were visualized by phalloidin staining. Binding of p66Shc to Hsp27 after cisplatin treatment was determined by immunoprecipitation in cell and tissue lysates. The role of p66Shc and its Ser36 phosphorylation in Hsp27 binding was assessed by overexpressing it or mutating its Ser36 residue. Results: Knockdown of p66Shc and overexpression of Hsp27 ameliorated cisplatin-mediated collapse of the actin cytoskeleton. Further studies revealed that p66Shc binds Hsp27 after treatment with cisplatin that requires Ser36 phosphorylation of p66Shc. Conclusion: We propose a novel function of p66Shc that, through interacting with Hsp27, accelerates cisplatin-dependent disruption of the actin cytoskeleton.
Cisplatin is a widely applied antineoplastic drug, use of which is hampered by its nephrotoxicity (1). Cisplatin treatment induces apoptosis in cells of the proximal tubules of the kidney, which is reproducible in vitro using cultured proximal tubule cells (2). An early step in cisplatin-mediated nephrotoxicity is the loss of F-actin stress fibers, which then results in detachment of cells from their basement membrane and ultimately to cell death (3). The small heat-shock protein-27 (Hsp27) is important in the maintenance of an intact actin cytoskeleton and as such provides protection against stress-mediated disruption of the microfilament (4). In renal epithelial cells, Hsp27 ameliorates oxidant-mediated disruption of the actin cytoskeleton in vitro (5) and protects against renal ischemia/reperfusion injury in vivo (6). However, the role of Hsp27 in cisplatin-mediated nephrotoxicity has, to our knowledge, not been studied.
In previous of ours work, we showed that the activated (Ser36 phosphorylated) p66Shc is involved in apoptosis of cisplatin-treated renal proximal tubule cells (7), but its role in cisplatin-mediated disorganization of the actin cytoskeleton is unknown.
In this study we investigated the effects of p66shc and Hsp27 on cytoskeletal organization and their potential interaction in cultured renal proximal tubule cells.
Materials and Methods
Animals and treatment. In a previous experiment, 6-to 8-week-old male 129Sv mice (Harlan Laboratories, Madison, WI, USA) received a single intraperitoneal injection of 20 mg/kg cisplatin. Three untreated and three treated mice were sacrificed one day after cisplatin treatment (2). Kidney lysates obtained in the previous experiments were used in this study (2).
Cell culture and treatment. The immortalized mouse proximal tubule cell line (TKPTS) was a gift from Dr. Bello-Reuss (8). The p66Shc knockdown variant of TKPTS cells was developed by transfecting a p66Shc short-hairpin-RNA construct (9). Cells were treated with 25 μM cisplatin (Sigma-Aldrich, ST. Louis, MO, USA) for 4 h. A fraction of cells were infected with an adenovirus that contains the human HSP27 gene [a gift from Dr. Vander Hide (10)] as described elsewhere (11).
Western blotting and immunoprecipitation. Cell and kidney lysates were separated by sodium dodecylsulphate/polyacrylamide gel electrophoresis (SDS/PAGE), transferred to a polyvinylidene fluoride (PVDF) membrane (Invitrogen, Grand Island, NY, USA) and hybridized with the following primary (mouse anti-p66Shc: Nanotools/Axxora, Farmingdale, NY, USA; rabbit anti-Hsp27: Millipore, Temecula, CA, USA) and secondary (HRP conjugated anti-mouse or anti-rabbit IgG, Cell Signaling Technology, Danvers, MA, USA) antibodies. Bands were visualized by an enhanced chemiluminescence system (Thermo Scientific, Rockford, IL, USA) and analyzed by densitometry (UnScan-It; Silk Scientific Corp., Orem, UT, USA). For immunoprecipitation, 500 μg of lysates were incubated with an Hsp27 antibody overnight at 4°C using the Catch and Release v2.0 reversible immunoprecipitation system (Millipore, Charlottesville, VA, USA). Immunoprecipitated proteins were resolved by SDS/PAGE and immunoblotted as described above.
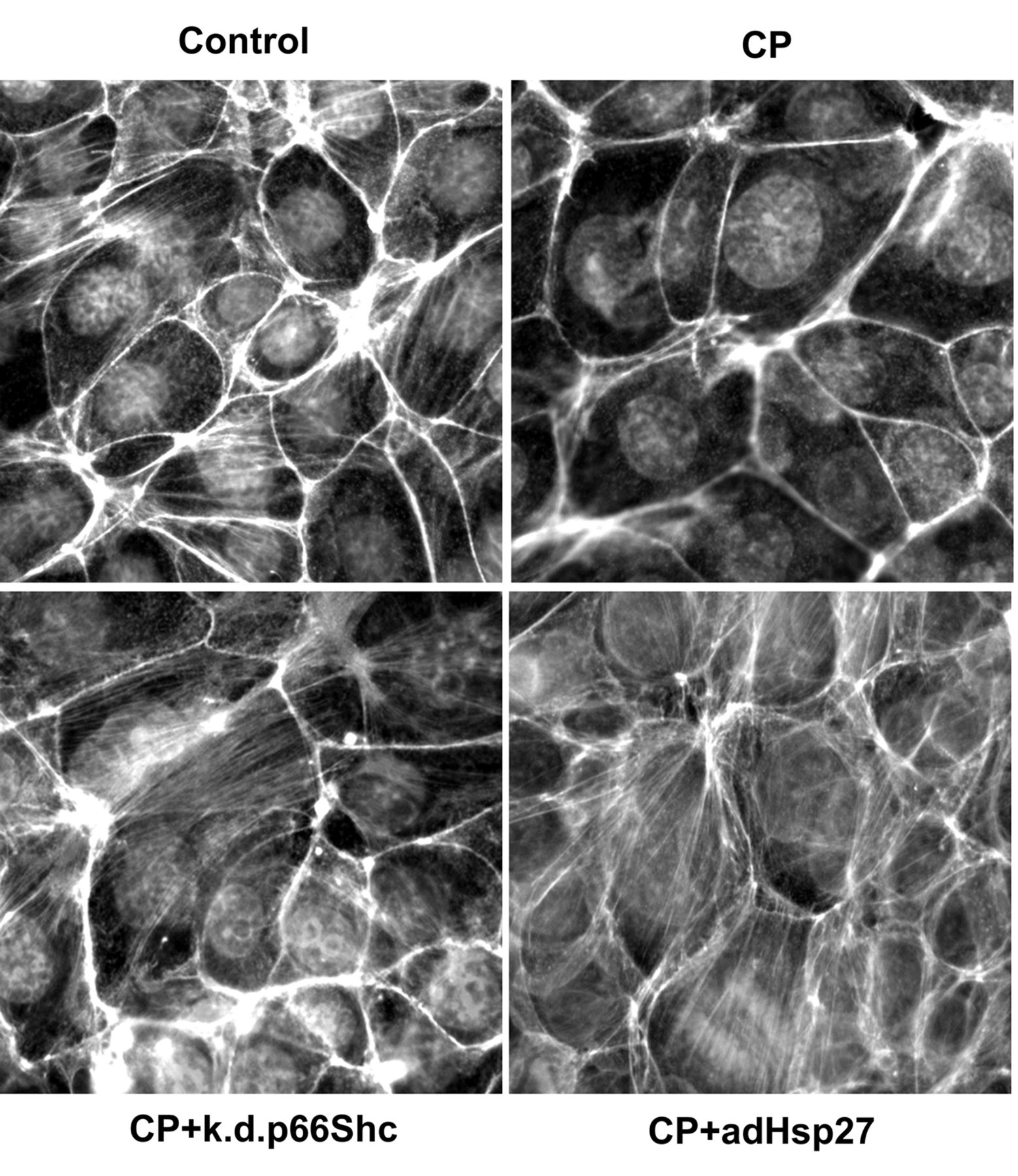
Cisplatin (CP)-dependent collapse of the actin cytoskeleton involves p66Shc and Hsp27 in cultured renal proximal tubule cells. TKPTS cells were treated with 25 μM cisplatin for 4 h and F-actin stress fibers were visualized as described in Materials and Methods. Images shown are representative of three independent experiments at magnification of ×400. k.d.p66Shc: p66Shc-knockdown cell line, adHsp27: adeno-Hsp27.
Visualization of the actin cytoskeleton and fluorescent microscopy. Cells grown on chambered coverglass were fixed and permeabilized, then labeled with Alexafluor 488 phalloidin (Invitrogen) and counterstained with 4’,6-Diamidino-2-Phenylindole, Dihydrochloride (DAPI, Invitrogen), as recommended by the manufacturer. Fluorescence was observed with a Nikon Eclipse TS100F inverted microscope equipped with a DAPI, Fluorescein-isothiocyanate (FITC) and Cyanine 3 (CY3) filter at x400 magnification. Images were captured by a Nikon DS cooled camera and analyzed with the NIS Elements Basic Research 3.0 software (Nikon Instrument Inc., Lewisville, TX, USA).
Statistical analysis. Statistical differences between the treated and control groups were determined by the Student's t-test. Differences between means were considered significant if p<0.05. All analyses were performed using the SigmaStat 3.5 (Systat, San Jose, CA, USA) software package.
Results
Cisplatin-mediated disruption of the actin cytoskeleton is regulated by p66Shc or Hsp27. Semi-confluent TKPTS cells were treated with 25 μM cisplatin for 4 h then fixed and stained with phalloidin to visualize F-actin stress fibers. As seen in Figure 1, cisplatin treatment resulted in loss of stress fibers, which was ameliorated in p66shc knockdown cells. Similarly, overexpression of Hsp27 via adenoviral gene transfer prevented cisplatin-dependent loss of stress fibers.

Cisplatin (CP) enhances binding of p66Shc to Hsp27 in vitro and in vivo. A: TKPTS cells were treated with 25 μM cisplatin for 4 h, cell lysates were prepared, that were immunoprecipitated with an Hsp27 antibody followed by immunoblotting with antibody, against p66Shc. After stripping, the blot was re-hybridized with an Hsp27 antibody to demonstrate equal pull-down. As control, lysates from untreated cells were used. Data shown are representatives of three independent experiments. B: Mice received 20 mg/kg cisplatin i.p. and sacrificed 24 h later. Kidney lysates were immunoprecipitated as described in the Materials and Methods. Data shown are representative of three independent experiments. C: Densitometry of blots shown in (A) and (B). p66Shc binding was calculated as the ratio of p66Shc and Hsp27 and expressed as percentage of the control values. N=3, *p<0.05 compared to control values.

Cisplatin (CP)-mediated binding of p66Shc to Hsp27 depends on serine 36 phosphorylation of p66Shc. A: TKPTS cells were treated with 25 μM cisplatin for 4 h in the presence or absence of a p66shc-expressing or a mutant p66shc plasmid in which the Ser36 residue was mutated to alanine (S36A). Data shown are representative of three independent experiments. B: Densitometry of blots shown in (A). p66Shc binding was calculated as the ratio of p66Shc and Hsp27 and expressed as percentage of the cisplatin-treated values. N=3, *p<0.05 compared to CP values or as indicated.
Cisplatin treatment increases binding of p66Shc to Hsp27 both in vitro and in vivo. To determine whether there is an interaction between p66Shc and Hsp27, TKPTS cells were treated with cisplatin 25 μM CP for 4 h and cell lysates were immunoprecipitated with an antibody to Hsp27 and immunoblotted with an antibody to p66Shc. Figures 2A and C demonstrate that cisplatin-treatment significantly increased binding of p66Shc to Hsp27. Rehybridization of the blots with an Hsp27 antibody demonstrated equal pull-down. Similar findings were noted in the kidneys of mice that were treated with cisplatin for 24 h (Figures 2B and C).
Cisplatin-mediated serine 36 phosphorylation of p66Shc is required for its interaction with Hsp27. TKPTS cells were transfected with a p66Shc-expressing plasmid followed by treatment with 25 μM cisplatin for 4 h. Immuno-precipitation studies revealed highly increased binding of p66Shc to Hsp27 (Figure 3). In contrast, mutation of Ser36 of p66Shc to alanine (S36A mutant) dramatically reduced binding of Hsp27 after treatment with cisplatin.
Discussion
Proximal tubule cells of the kidney are attached to the basement membrane via specific family of integrins, which interact with the F-actin fibers (12). This interaction initiates/sustains signaling that is necessary for cell survival (13). Therefore, any insults that interfere with proper F-actin organization may induce cell death. Indeed, early action of nephrotoxic agents, including cisplatin, involves loss of cytoskeletal F-actin fibers followed by cell detachment and ultimately apoptosis (14). Our data confirmed this observation: cisplatin treatment of cultured renal proximal tubule cells resulted in re-organization of the actin cytoskeleton, i.e. loss of F-actin fibers (Figure 1). Not surprisingly, overexpression of HSP27 prevented this loss (Figure 1). This effect is due to stabilization of actin cytoskeleton by Hsp27 that prevents disruption of cytoskeletal reorganization by stress signals as suggested by others (4). It was more surprising that knockdown of p66Shc also ameliorated re-organization of F-actin fibers (Figure 1). p66Shc is an alternative splicing product of the ShcA gene and is activated (Ser36-phosphorylated) by a wide variety of stress signals including cisplatin, as we reported earlier (7). However, the role of p66Shc in disorganization of the actin cytoskeleton is little known. Recently, Natalicchio et al. demonstrated that lack of p66Shc leads to complete disorganization of the actin cytoskeleton in myoblasts (15). Our results somewhat contradict with those of Natalicchio et al: we showed protective effects of p66Shc knockdown in cisplatin-mediated disruption of the actin cytoskeleton. This discrepancy may be due to the use of different cell types and/or stress stimuli.
Importantly, we found that cisplatin enhances binding of p66Shc to Hsp27 both in vivo and in vitro (Figure 2). This is a completely unknown phenomenon: only an interaction between p66Shc and mitochondrial Hsp70 has been described (16) that modulates mitochondrial transmembrane potential. We determined that overexpression of p66Shc enhances while mutation of its Ser36 residue (S36A) attenuates cisplatin-dependent binding to Hsp27 (Figure 3). These results suggest that activation, i.e. Ser36-phosphorylation is required for this interaction. We postulate that early binding of p66Shc to Hsp27 after treatment with cisplatin may interfere with Hsp27, diminishing its function in the maintenance of the integrity of the actin cytoskeleton.
Conclusion
Based on our results we propose a novel function for p66Shc that, through interacting with Hsp27, accelerates cisplatin-dependent disruption of the actin cytoskeleton and consequently apoptosis. Thus, therapeutic means that interfere with p66shc may ameliorate renal toxicity of CP.
Acknowledgements
The Authors thank Dr. Richard S. Vander Hide for providing the Hsp27 adenovirus and Dr. Judith Megyesi (UAMS) for the help in animal experiments. These studies were supported by an American Heart Association Grant-in-Aid (10GRNT3790019, I.A.) and an Intramural Research Support Program Award from the University of Mississippi Medical Center (I.A.), as well as an NIH grant (DK073401, LAJ).
- Received August 23, 2012.
- Revision received October 11, 2012.
- Accepted October 12, 2012.
- Copyright© 2012 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved





{kind=link}
{kind=link}
{kind=link}