
HDAC8 Prevents Anthrax Lethal Toxin-induced Cell Cycle Arrest through Silencing PTEN in Human Monocytic THP-1 Cells


Department of Microbiology and Immunology, The University of Western Ontario, London, ON N6G 2V4, Canada
*
Author to whom correspondence should be addressed.
Toxins 2017, 9(5), 162; https://doi.org/10.3390/toxins9050162
Submission received: 19 April 2017
/
Revised: 9 May 2017
/
Accepted: 10 May 2017
/
Published: 16 May 2017
(This article belongs to the Collection Anthrax Toxins)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Anthrax lethal toxin (LeTx) is a cytotoxic virulence factor that causes cell cycle arrest and cell death in various cell types. However, susceptibility to the cytotoxic effects varies depending on cell types. In proliferating monocytes, LeTx has only transient cytotoxic effects due to activation of the phosphoinositide 3-kinase (PI3K)-AKT-mediated adaptive responses. To date, the mechanism of LeTx in activating PI3K-AKT signaling axis is unknown. This study shows that the histone deacetylase 8 (HDAC8) is involved in activating PI3K-AKT signaling axis through down-regulating the phosphatase and tensin homolog 1 (PTEN) in human monocytic THP-1 cells. The HDAC8-specific activator TM-2-51 and inhibitor PCI-34051 enhanced and prevented, respectively, AKT activation and cell cycle progression in LeTx-treated cells. Furthermore, HDAC8 induced tri-methylation of histone H3 lysine 27 (H3K27me3), which is known to suppress PTEN expression, through at least in part down-regulating the H3K27me3 eraser Jumonji Domain Containing (JMJD) 3. Importantly, the JMJD3-specific inhibitor GSK-J4 induced AKT activation and protected cell cycle arrest in LeTx-treated cells, regardless the presence of HDAC8 activity. Collectively, this study for the first time demonstrated that HDAC8 activity determines susceptibility to cell cycle arrest induced by LeTx, through regulating the PI3K-PTEN-AKT signaling axis.
1. Introduction
Anthrax lethal toxin (LeTx) is a key virulence factor of Bacillus anthracis, the causative agent of anthrax [1]. It comprises the intracellular transporter protective antigen (PA) and the metalloprotease lethal factor (LF), where LF cleaves and inactivates the mitogen-activated protein kinase (MAPK) kinases (MEK) 1 to 6, except 5 [2,3]. Inactivation of MEKs results in an almost complete inactivation of the extracellular signal-regulated kinases (ERKs) and p38 MAPKs, but partial or no effects on c-Jun N-terminal kinases [3,4,5,6]. Inactivation of ERKs and p38 MAPKs leads to cell cycle arrest and cell death in both immune and non-immune cells [4,7,8,9,10,11,12,13,14,15,16]. However, the extent of cytotoxicity elicited by LeTx is dependent on cell types, differentiation stages and their capacity in activating adaptive responses [4,14,15,16]. In human macrophages, inhibition of ERKs by LeTx leads to cell cycle arrest at G0/1 phase due to depletion of cyclin D1 [15], which is required for cell cycle progress from G1 to S phase [17]. In contrast, cells with constitutively active or adaptively activating the PI3K-AKT (also known as protein kinase B) signaling axis are resistant to LeTx- or ERK inhibition-induced cell cycle arrest through protecting degradation and/or inducing expression of cyclin D1 [15,18,19]. However, the mechanism by which LeTx activates the adaptive PI3K-AKT signaling axis is yet to be elucidated.
In certain murine macrophages, LeTx rapidly induces pyroptosis, which is a programmed necrotic cell death mediated by the NACHT-leucine-rich repeat and pyrin domain-containing protein 1b and inflammasome [20]. These macrophages also undergo a similar adaptive response as in human monocytes and become resistant to LeTx-induced pyroptosis [5,21,22]. Our previous studies elucidated the adaptive response as an epigenetic phenomenon that silences mitochondrial cell death genes through the histone deacetylase (HDAC) 8 [23,24]. This study examined the role and mechanism of HDAC8 in adaptive responses of the human monocytic THP-1 cells to LeTx-induced cell cycle arrest. We found that HDAC8 played a pivotal role in activating the PI3K-AKT signaling axis in LeTx-exposed cells through inducing histone H3 lysine 27 tri-methylation (H3K27me3) and subsequent inhibition of PTEN expression.
2. Results
2.1. HDAC8 Decreases Susceptibility to LeTx-Induced Cytotoxicity
To examine the role of HDAC8 in cytotoxic effects of LeTx, THP-1 cells were treated with LeTx in the presence or absence of the HDAC8-specific activator TM-2-51 (TM) [25] or inhibitor PCI-34051 (PCI) [26], and total live cell numbers were counted each day for four days (Figure 1A). Cell numbers of LeTx-treated cells remained constant for 2–3 days and then started increasing on Day 3 (Figure 1A). Numbers of cells treated with both LeTx and PCI were steadily decreased throughout the experimental period (four days). However, cells treated with both LeTx and TM were continuously increased, albeit with a slower rate than that of non-treated cells. The cytotoxic and cytoprotective effects of PCI and TM, respectively, were similarly observed in two different doses of LeTx (Figure 1B). Significant cytoprotective effects of TM were detected, starting at 13 µM and maximum at 25 µM (Figure 1C). In contrast, PCI gradually increased cytotoxicity of LeTx, becoming statistically significant at 0.5 µM and maximally at 5 µM (Figure 1D).
2.2. HDAC8 Prevents Cell Cycle Arrest Induced by LeTx
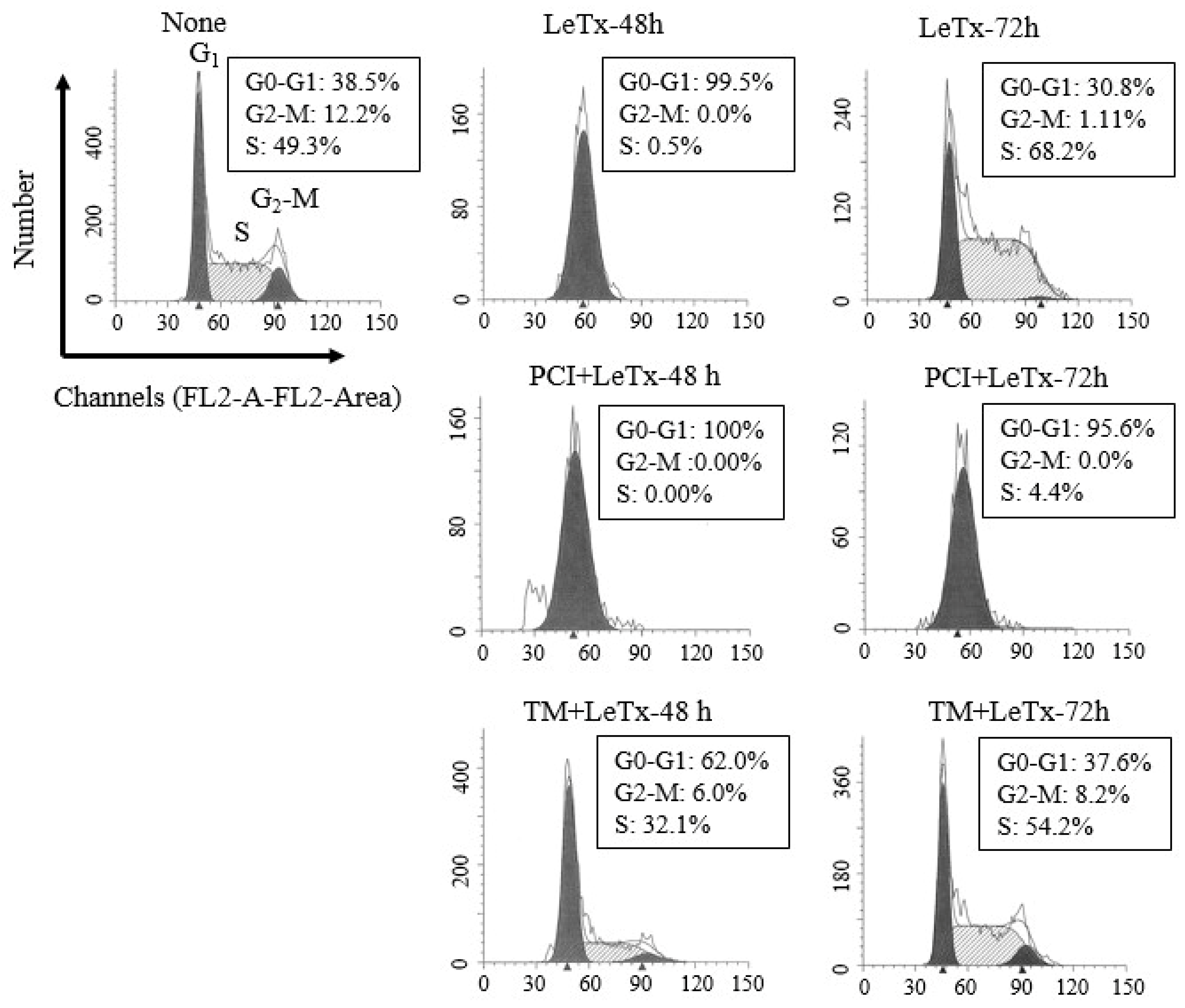
LeTx causes a transient cell cycle arrest in THP-1 cells [15]. Similarly, we detected a complete cell cycle arrest at Go-G1 stage 48 h post LeTx treatment, which was then spontaneously recovered 72 h post LeTx treatment (Figure 2; top lane). However, when cells were treated together with PCI, they failed to recover from the arrest (middle lane). Cell cycle progression of cells treated together with TM was slightly altered in 48 h, but became almost normal in 72 h of LeTx treatments (lower lane). These results indicate that HDAC8 protects cells from LeTx-induced cell cycle arrest.
2.3. HDAC8 Activity is Required for Activating AKT in LeTx-Treated Cells
Previously, we showed that activation of PI3K-AKT signaling axis prevents LeTx-induced cell cycle arrest [15]. To examine if HDAC8 is involved in the activation of AKT in LeTx-treated cells, we analyzed phosphorylation of AKT at the Serine 473 residue. AKT phosphorylation was increased in 24 h and peaked in 72 h after LeTx treatments (Figure 3A). In line with the previous results (Figure 1 and Figure 2), AKT phosphorylation was inhibited by PCI in 48 h post LeTx treatment; whereas, TM further enhanced AKT activation in 24 h after LeTx-treatment. The differences in AKT activation were not due to the levels of LeTx present in cells, since almost complete degradation of MEK3 was detected in all samples throughout the experimental time frame (Figure 3A, the 2nd lane). In addition, the AKT phosphorylation was inhibited by the PI3K inhibitors LY294002 (LY) and wortmannin (Wort; Figure 3B, upper panel), and LY further enhanced cytotoxic effects of LeTx (Figure 3B, lower panel). These results suggest that both HDAC8 and PI3K were involved in LeTx-induced AKT activation.
2.4. LeTx Suppresses PTEN Expression through HDAC8
HDAC8 is an epigenetic modifier that regulates gene transcription. Among several signaling molecules known to regulate PI3K and AKT activation, expression levels of PTEN have shown to be inversely correlated with AKT activation in many cell types [27]. In THP-1 cells, LeTx suppressed PTEN expression in both mRNA and protein levels in 48 h after treatments (Figure 4A). PCI inhibited PTEN down-regulation and subsequently reduced AKT phosphorylation in LeTx-treated cells (Figure 4B). In line with these results, the PTEN-specific inhibitor VO-OHpic enhanced AKT phosphorylation (Figure 4C, upper panel) and cell proliferation (Figure 4C, lower panel) even in the presence of PCI in LeTx-treated cells. These results suggest that AKT activation in LeTx-treated cells was likely due to HDAC8-mediated PTEN down-regulation.
2.5. HDAC8 Induces Histone H3K27me3 by Silencing JMJD3
We previously showed that LeTx or HDAC8 overexpression leads to H3K27Ac deacetylation in murine macrophages [22,28]. Since the levels of H3K27Ac and H3K27me3 are inversely correlated, and an increase of H3K27me3 leads to down-regulation of PTEN expression [29,30,31,32], we examined both acetylation and methylation levels of H3K27. As expected, LeTx reduced H3K27Ac but induced H3K27me3 (Figure 5A). However, H3K27 methylation peaked in 24 h, which was faster than the rate of deacetylation that gradually occurred over 72 h of LeTx treatments. We then examined the expression levels of HDAC8, and the H3K27-specific methyltransferase enhancer of zeste 2 (EZH2)[33] and demethylase JMJD3 [34,35] in response to LeTx. LeTx had no effects on the expression levels of EZH2, but induced and inhibited, respectively, the expression of HDAC8 and JMJD3 (Figure 5B). To further examine if JMJD3 and EZH2 regulate HDAC8 expression or vice versa, THP-1 cells were treated with the JMJD3 inhibitor GSK-J4 and EZH2 inhibitor EPZ-6438. As expected, H3K27me3 levels were increased by GSK-J4 and decreased by EPZ-6438; however, these inhibitors had no effects on HDAC8 expression (Figure 5C). In addition, none of these inhibitors affected the expression levels of HDAC8 in LeTx-treated cells (Figure 5D, left panel). In contrast, PCI was able to prevent down-regulation of JMJD3 expression (Figure 5D, right panel) and subsequently decreased H3K27me3 levels (Figure 5E). These results suggest that HDAC8 regulates JMJD3 expression and H3K27me3 levels.
2.6. GSK-J4 Induces AKT Activation and Prevents Cell Cycle Arrest even in the Presence of PCI in LeTx-Treated Cells
Since down-regulation of JMJD3 was a key step for increasing H3K27me3 levels in LeTx-treated cells, we examined if direct inhibition of JMJD3 influenced cell cycle arrest, PTEN expression and AKT activation in LeTx-treated cells. As expected, the JMJD3 inhibitor GSK-J4 attenuated cell cycle arrest in LeTx-treated cells (Figure 6A). GSK-J4 enhanced H3K27me3 (Figure 6B) and even outcompeted the PCI effects, resulting in decrease of PTEN expression and increase of AKT phosphorylation (Figure 6C). Importantly, GSK-J4 protected cell cycle arrest induced by LeTx even in the presence of PCI (Figure 6D).
3. Discussion
Here, we demonstrated that HDAC8 played a key role in determining susceptibility to cell cycle arrest induced by LeTx. TM, which increases HDAC8 activity up to 12-fold at 10 µM concentration [25], significantly prevented cell cycle arrest induced by LeTx at 13 µM and maximally at 25 µM concentrations. In contrast, PCI further enhanced the duration and extent of cell cycle arrest elicited by LeTx in a dose-dependent manner. In line with these observations, AKT phosphorylation in LeTx-exposed cells was further enhanced by TM, but was inhibited by PCI. Phosphorylation of AKT at Ser-473 is essential for its full activation and stabilization of active conformation [36,37], and mediated by the mammalian target of rapamycin complex 2 and other PI3K-independent kinases [38,39,40]. However, LeTx likely induced AKT phosphorylation through a PI3K-dependent pathway, since the PI3K inhibitors LY and wortmannin inhibited AKT phosphorylation and cell proliferation.
PTEN is a tumor suppressor gene that counteracts PI3K activity through dephosphorylating PI(3,4,5)P3 to produce PI(4,5)P2 [41]. PTEN activity is regulated by multiple post-translational modifications such as phosphorylation, ubiquitylation, acetylation, SUMOylation, oxidation and protein–protein interactions, but transcriptional regulation of PTEN plays a key role in determining PI3K-AKT dependent cell proliferation, survival and metabolic phenotypes [27,42,43,44]. Various cellular stresses and growth factors also regulate expression of PTEN [45,46,47,48]. Activation of MAPKs is involved in the regulation of PTEN expression; however, its role in PTEN expression varies depending on cell types [48,49,50]. We detected PTEN mRNA and protein expression levels were down-regulated by LeTx (Figure 4A) in a PCI-sensitive manner (Figure 4B). Consistently, the PTEN-specific inhibitor VO-OHpic [51] further enhanced AKT phosphorylation and cell survival in LeTx-treated cells. Collectively, these results suggest that LeTx induces AKT activation at least in part through HDAC8-dependent down-regulation of PTEN expression.
HDAC8 has shown to be involved in regulating cell cycle progression of normal and tumor cells [52]. Overexpression of HDAC8 has been associated with cell proliferation of multiple cancer cells [53]; whereas, defective mutations of HDAC8 are linked to Cornelia de Lange syndrome, which is a rare genetic disease manifesting congenital malformations of multiple organs due to defects in cohesin [52]. Cohesin is a multi-protein complex which forms a clutch to hold the sister chromatids together during S phase [54]. HDAC8 deacetylates the cohesin subunit Structural Maintenance of Chromosomes 3 (SMC3) at lysines 105 and 106, resulting in segregation of cohesion from the sister chromatids and replenishes SMC3 for another cell cycle. Inhibition of HDAC8 leads to accumulation of acetylated SMC3, and consequently cell cycle arrest [55]. For example, in breast tumor MCF7 cells and neuronal fibroblasts, inhibition or defects in HDAC8 prolongs G1 phase and delays entering to S phase [56,57]. Similarly, PCI further enhanced cell cycle arrest at G0/1 phase in LeTx-treated cells, which could be an additional cell cycle arrest mechanism induced by PCI. However, PCI alone had no effects on cell cycle progress within our experimental time frame and TM was able to prevent cell cycle arrest in LeTx-treated cells, suggesting that HDAC8 regulates cell proliferation through regulating PI3K-AKT signaling axis induced by LeTx, independent of SMC3 acetylation. However, further studies are required to rule out the involvement of SMC3 in HDAC8-mediated cell cycle regulation in LeTx-treated cells.
Previously, several studies have shown that the Polycomb Repressive Complex 2 (PRC2), which writes and reads H3K27me3, suppresses gene expression including PTEN in various cell types [32,58,59,60,61,62,63]. It is also known that H3K27 acetylation is mutually exclusive to methylation of the residue and H3K27 deacetylation is a prerequisite chromatin context for PRC2 to be active on histones [29,30,31]. In murine macrophages, we showed that LeTx suppresses gene transcription in part through up-regulating HDAC8 that leads to H3K27 deacetylation [23,28]. As expected, LeTx also induced HDAC8 mRNA expression, decreased H3K27Ac levels and increased H3K27me3 levels over 3 days in THP-1 cells. Apparently, increase of H3K27me3 levels was at least in part due to down-regulation of the H3K27 demethylase JMJD3. LeTx decreased JMJD3 expression but not EZH2 which is a subunit of PRC2 with H3K27 methyltransferase activity. PCI was able to prevent down-regulation of JMJD3 and prevent H3K27me3; whereas, the JMJD3- and EZH2-specific inhibitors GSK-J4 and EPZ-6438 had no effects on HDAC8 expression in LeTx-treated cells. However, we could not detect a direct inverse-correlation in the levels of H3K27Ac and H3K27me3. H3K27Ac levels were gradually down-regulated over 3 days, but H3K27me3 levels were rapidly and maximally increased in 24 h after LeTx treatments. At this moment, we could not rule out the involvement of other HDAC8-dependent signaling events in rapidly inducing H3K27me3. However, GSK-J4 suppressed PTEN expression, activated AKT phosphorylation and prevented cell cycle arrest even in the presence of PCI in LeTx-treated cells, supporting a crucial role of JMJD3 in inducing H3K27me3 and PTEN suppression. HDAC8 has also been shown to regulate transcription through targeting non-histone proteins, such as adenoviral E1A-12 protein [64], the inversion-16 fusion gene products in acute myeloid leukemia cells [65], p53 [66], protein phosphatase (PP) 1 [67], heat shock proteins [68], α-actin [69], human ever-shorter telomeres 1B [70], estrogen-related receptor-α [71] and possibly multiple other proteins [72,73]. Of interest, HDAC8 deacetylates and activates p53 [66]. Since p53 binds to the PTEN promoter and enhances PTEN gene transcription [74], it is possible that HDAC8 induces PTEN expression by activating p53. To date, it is unknown whether LeTx induces cell cycle arrest and/or cell death through activating p53. Therefore, the involvement of p53 in preventing LeTx-induced cell cycle arrest by HDAC8 warrants further studies.
Overall, this study showed that the HDAC8-JMJD3-H3K27me3-PTEN-AKT signaling axis played a key role in determining susceptibility to cell cycle arrest in LeTx-intoxicated cells (Figure 7). Activation of the signaling axis by either the HDAC8 activator TM, JMJD3 inhibitor GSK-J4 or PTEN inhibitor VO-OHpic induced AKT activation and prevented cell cycle arrest. In contrast, the HDAC8 inhibitor PCI suppressed AKT activation and potentiated LeTx-induced cytotoxicity. Macrophages play a complex role in the pathogenesis of anthrax, involved in both systemic dissemination of and innate immunity to B. anthracis [75,76]. LeTx is released by germinating spores within macrophage phagosomes at an early stage of infection [77] and systemic vegetative bacilli at a later stage, facilitating bacterial dissemination [75,78] and immune paralysis, respectively [75,76]. Particularly, generation and survival of macrophages were suggested to be important for preventing anthrax at a later stage [4,79]. Therefore, we speculate that therapeutic approaches either inhibiting HDAC8 before systemic dissemination or activating HDAC8 and inhibiting JMJD3 at a later stage could be novel strategies for inhibiting dissemination or maintaining macrophage populations during B. anthracis infection. Interestingly, modified anthrax lethal toxins, which cleave and inactivate MEKs selectively in tumors, is an emerging anti-tumor biomolecule with promising results [80]. However, using LeTx is expected to be limited in its efficacy due to adaptive responses often observed in various tumors. Therefore, targeting HDAC8 could also be a novel strategy for developing combinatory anti-tumor therapies.
4. Materials and Methods
4.1. Reagents
PA and LF were purchased from the List Biological Laboratories (Campbell, CA, USA). Chemical inhibitors used in this study are the following: PCI-34051(Cayman chemical, Ann Arbor, MI, USA), TM-2-51(1-Benzoyl-3-phenyl-2-thiourea, Sigma-Aldrich, St.Louis, MO, USA), JMJD3 inhibitor GSK-J4 (Sigma-Aldrich), EZH2 inhibitor EPZ-6438 (MedChemexpress CO., Ltd; Princeton, NJ, USA, through CEDARLANE), PI3K inhibitors LY294002 (ApexBio Technology, Houston, TX, USA) and wortmannin (Calbiochem, La Jolla, CA, USA), PTEN inhibitor VO-OHpic trihydrate (BioVision, Milpitas, CA, USA). Propidium iodide and RNase A were obtained from Calbiochem and Sigma-Aldrich.
Antibodies raised against the NH2-terminus of MEK1 (MEK1-NT) and MEK3 were obtained from Stressgen Bioreagents (Ann Arbor, MI, USA) and Santa Cruz Biotechnology (Dallas, TX, USA), respectively. The phospho-AKT (Ser-473) and PTEN antibodies were purchased from Cell Signaling and Cedarlane (Danvers, MA, USA and Burlington, NC, USA). Anti- H3K27Ac and anti-H3K27me3 antibodies were from Active Motif (Carlsbad, CA, USA); pan-histone H3 antibodies from Bio Vision; β-actin antibodies from Rockland Inc (Gilbertsville, PA, USA).
4.2. Cell Culture
The human monocytic cell line THP-1 cells were purchased from the American Type Culture Collection and cell cultures were maintained in complete RPMI 1640 medium containing 10% heated-inactivated fetal bovine serum (Sigma), 10 mM MEM non-essential amino acids solution, 100 U/mL penicillin G sodium, 100 μg/mL streptomycin sulfate and 1 mM sodium pyruvate as previously described [24].
4.3. Cell Viability Assay
Living cells in Figure 1A were counted using a hemocytometer after trypan blue staining. MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay was also used for cell viability assay as described in previous studies [24]. Briefly, THP-1 cells were cultured in 96 well plates and treated with LeTx with or without subsequent treatments with chemical reagents for the time indicated. MTT was then added at a final concentration of 0.5 mg/mL, and incubated at 37 °C for an additional 2 h. Culture media was aspirated and 100 µL of dimethyl sulfoxide was added to dissolve crystals. Optical densities of each well were analyzed at OD570 nm using a microplate reader (Synergy H4 Hybrid Reader; BioTek Instruments Inc., Winooski, VT, USA). The percentage of cell survival was estimated based on OD570 nm of wells by comparing those from non-treated cells.
4.4. Western Blot
Preparation of total cell lysates and immunoblotting were performed as previously reported [23]. Briefly, cells were lysed in ice-cold lysis buffer containing 20 mM MOPS, 2 mM EGTA, 5 mM EDTA, 1 mM Na3VO4, 40 mM β-glycerophosphate, 30 mM sodium fluoride, 20 mM sodium pyrophosphate, 0.1% SDS, 1% Triton X-100, pH 7.2, and a protease inhibitor cocktail (Roche, Werk Penzberg, Germany). Cell lysates were collected after centrifugation at 12,500 rpm for 15 minutes at 4 °C. Proteins were then separated by SDS-polyacrylamide gels and transferred onto nitrocellulose membranes (Bio-Rad). The membranes were blocked with 5% (w/v) skim milk for 1 h and incubated overnight at room temperature with primary antibodies. After washing three times with 1 × TTBS (20 mM Tris, 150 mM NaCl, pH 7.5) containing 0.07% tween 20, the membranes were incubated with secondary antibodies for 1 hour at room temperature and films were developed using an enhanced chemiluminescence detection system (ECL; Thermo Scientific, Waltham, MA, USA).
4.5. Quantitative Real-Time PCR
mRNA expression was quantified by quantitative real-time PCR (qPCR) as previously described [23]. Briefly, total cellular RNAs were isolated using TRIzol (Ambion by Life Technologies, Carlsbad, CA, USA) according to the manufacturer's instructions and mRNAs were reverse transcribed using Moloney murine leukemia virus (M-MuLV) reverse transcriptase (New England Biotechnology, Ipswich, MA, USA) and oligo (dT) as a primer. Quantitative real-time PCR (qPCR) analyses were performed with a Rotor-Gene RG3000 quantitative multiplex PCR instrument (Montreal Biotech Inc, Dorval, QC, Canada) using Power UPTM SYBRR Green Master Mix (Applied Biosystems life technologies, Foster City, CA, USA). The data were normalized by expression of the GAPDH housekeeping gene. Primers used for qPCR are listed in the following; for GAPDH, 5’-ACCCACTCCTCCACCTTTG-3’ (5’ primer) and 5’-CTCTTGTGCTCTTGCTGGG-3’ (3’ primer); for PTEN, 5’-ACTTGCAATCCTCAGTTTGTGG-3’ (5’ primer) and 5’-GAAGAATGTATTTACCCAAAAGTG-3’ (3’ primer); for HDAC8, 5’-ATTCTCTACGTGGATTTGGATC-3’ (5’ primer) and 5’-ATGCCATCCTGAATGGGCACA -3’ (3’ primer); for JMJD3, 5’-TCTGATGCTAAGCGGTGGAAG-3’ (5’ primer) and 5’-GCCAATGTTGATGTTGACGGAG-3’ (3’ primer); for EZH2, 5’-GTGCCATTGCTAGGTTAATTGG-3’ (5’ primer) and 5’-AGGGTTGATAGTTGTAAACATGG-3’ (3’ primer).
4.6. Cell Cycle Analysis
Analyses of DNA content were performed using propidium iodide (PI) and Cell Quest software on a FACS calibur flow cytometer (Becton Dickinson Biosciences, San Jose, CA, USA) as previously reported [15]. Briefly, 1.0 × 106 cells were harvested and fixed by drop-wise an addition of ice-cold 70% ethanol after washing three times with 1 × PBS containing 0.1% glucose, and cells were then stored at 4 °C until processing PI staining. Subsequently, cells were pelleted by centrifugation and re-suspended in PI staining solution (0.1% glucose in 1 × PBS) containing 50 µg of PI/mL and 100 unit of RNase A/mL. After two hours of incubation at room temperature, the cell was loaded onto a FACS calibur flow cytometer. Data were acquired and analyzed using Cell Quest and ModFit LT 3.0 software (Becton Dickinson, San Jose, CA, USA).
4.7. Statistical Analysis
Data were analyzed using GraphPad Prism 4.0 (GraphPad Software Inc., San Diego, CA, USA). The results are presented as the means ± SD of three independent repeats. Statistical significance was defined as p < 0.05 (*). Otherwise, it was mentioned in the figure legends.
Acknowledgments
This study was supported by the Natural Sciences & Engineering Research of Canada (No. RGPIN 312482-2013) to S.O.K.
Author Contributions
S.O.K conceived and coordinated the study, designed experiments and wrote the paper. S.-D.H. designed, performed, and analyzed the experiments, and wrote the paper. W.C. performed experiments and wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| ERK | extracellular signal-regulated kinase |
| EZH2 | enhancer of zeste 2 |
| HDAC | histone deacetylase |
| H3K27Ac | histone H3 lysine 27 acetylation |
| H3K27me3 | histone H3 lysine 27 tri-methylation |
| JMJD3 | Jumonji Domain Containing 3 |
| LeTx | anthrax lethal toxin |
| LF | lethal factor |
| LY | LY942006 |
| MAPK | mitogen-activated protein kinase |
| MEK | MAPK kinase |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| PA | protective antigen |
| PI3K | phosphoinositide 3-kinase |
| SMC3 | structural maintenance of chromosomes 3 |
| PCI | HDAC8 inhibitor PCI34051 |
| PTEN | phosphatase and tensin homolog 1 |
| TM | HDAC8 activator TM-2-51 |
References
- Moayeri, M.; Leppla, S.H.; Vrentas, C.; Pomerantsev, A.P.; Liu, S. Anthrax pathogenesis. Annu. Rev. Microbiol. 2015, 69, 185–208. [Google Scholar] [CrossRef] [PubMed]
- Vitale, G.; Bernardi, L.; Napolitani, G.; Mock, M.; Montecucco, C. Susceptibility of mitogen-activated protein kinase kinase family members to proteolysis by anthrax lethal factor. Biochem. J. 2000, 352, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Fang, H.; Frucht, D.M. Anthrax lethal toxin increases superoxide production in murine neutrophils via differential effects on Mapk signaling pathways. J. Immunol. 2008, 180, 4139–4147. [Google Scholar] [CrossRef] [PubMed]
- Kassam, A.; Der, S.D.; Mogridge, J. Differentiation of human monocytic cell lines confers susceptibility to Bacillus anthracis lethal toxin. Cell Microbiol. 2005, 7, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Ha, S.D.; Ng, D.; Lamothe, J.; Valvano, M.A.; Han, J.; Kim, S.O. Mitochondrial proteins Bnip3 and Bnip3L are involved in anthrax lethal toxin-induced macrophage cell death. J. Biol. Chem. 2007, 282, 26275–26283. [Google Scholar] [CrossRef] [PubMed]
- Raymond, B.; Batsche, E.; Boutillon, F.; Wu, Y.Z.; Leduc, D.; Balloy, V.; Raoust, E.; Muchardt, C.; Goossens, P.L.; Touqui, L. Anthrax lethal toxin impairs IL-8 expression in epithelial cells through inhibition of histone H3 modification. PLoS Pathog. 2009, 5, e1000359. [Google Scholar] [CrossRef] [PubMed]
- Park, J.M.; Greten, F.R.; Li, Z.W.; Karin, M. Macrophage apoptosis by anthrax lethal factor through p38 Map kinase inhibition. Science 2002, 297, 2048–2051. [Google Scholar] [CrossRef] [PubMed]
- Comer, J.E.; Chopra, A.K.; Peterson, J.W.; Konig, R. Direct inhibition of T-lymphocyte activation by anthrax toxins in vivo. Infect. Immun. 2005, 73, 8275–8281. [Google Scholar] [CrossRef] [PubMed]
- Koo, H.M.; VanBrocklin, M.; McWilliams, M.J.; Leppla, S.H.; Duesbery, N.S.; Woude, G.F. Apoptosis and melanogenesis in human melanoma cells induced by anthrax lethal factor inactivation of mitogen-activated protein kinase kinase. Proc. Natl. Acad. Sci. USA 2002, 99, 3052–3057. [Google Scholar] [CrossRef] [PubMed]
- Popov, S.G.; Villasmil, R.; Bernardi, J.; Grene, E.; Cardwell, J.; Wu, A.; Alibek, D.; Bailey, C.; Alibek, K. Lethal toxin of Bacillus anthracis causes apoptosis of macrophages. Biochem. Biophys. Res. Commun. 2002, 293, 349–355. [Google Scholar] [CrossRef]
- Kirby, J.E. Anthrax lethal toxin induces human endothelial cell apoptosis. Infect. Immun. 2004, 72, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Xu, L.; Chen, T.Y.; Cyr, J.M.; Frucht, D.M. Anthrax lethal toxin has direct and potent inhibitory effects on B cell proliferation and immunoglobulin production. J. Immunol. 2006, 176, 6155–6161. [Google Scholar] [CrossRef] [PubMed]
- Paccani, S.R.; Tonello, F.; Ghittoni, R.; Natale, M.; Muraro, L.; D'Elios, M.M.; Tang, W.J.; Montecucco, C.; Baldari, C.T. Anthrax toxins suppress T lymphocyte activation by disrupting antigen receptor signaling. J. Exp. Med. 2005, 201, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.Y.; Kang, C.I.; Hur, G.H.; Yang, J.M.; Shin, S. Bacillus anthracis lethal toxin induces cell-type-specific cytotoxicity in human lung cell lines. J. Appl. Microbiol. 2014, 116, 1334–1343. [Google Scholar] [CrossRef] [PubMed]
- Ha, S.D.; Ng, D.; Pelech, S.L.; Kim, S.O. Critical role of the phosphatidylinositol 3-kinase/Akt/glycogen synthase kinase-3 signaling pathway in recovery from anthrax lethal toxin-induced cell cycle arrest and Mek cleavage in macrophages. J. Biol. Chem. 2007, 282, 36230–36239. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Mehta, H.; Chakrabarty, K.; Booth, J.L.; Duggan, E.S.; Patel, K.B.; Ballard, J.D.; Coggeshall, K.M.; Metcalf, J.P. Resistance of human alveolar macrophages to Bacillus anthracis lethal toxin. J. Immunol. 2009, 183, 5799–5806. [Google Scholar] [CrossRef] [PubMed]
- Meloche, S.; Pouyssegur, J. The Erk1/2 mitogen-activated protein kinase pathway as a master regulator of the G1- to S-phase transition. Oncogene 2007, 26, 3227–3239. [Google Scholar] [CrossRef] [PubMed]
- Takuwa, N.; Fukui, Y.; Takuwa, Y. Cyclin D1 expression mediated by phosphatidylinositol 3-kinase through mTor-p70(s6k)-independent signaling in growth factor-stimulated NIH 3T3 fibroblasts. Mol. Cell. Biol. 1999, 19, 1346–1358. [Google Scholar] [CrossRef] [PubMed]
- Radeff-Huang, J.; Seasholtz, T.M.; Chang, J.W.; Smith, J.M.; Walsh, C.T.; Brown, J.H. Tumor necrosis factor-alpha-stimulated cell proliferation is mediated through sphingosine kinase-dependent Akt activation and cyclin D expression. J. Biol. Chem. 2007, 282, 863–870. [Google Scholar] [CrossRef] [PubMed]
- Boyden, E.D.; Dietrich, W.F. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat. Genet. 2006, 38, 240–244. [Google Scholar] [CrossRef] [PubMed]
- Salles, I.I.; Tucker, A.E.; Voth, D.E.; Ballard, J.D. Toxin-induced resistance in Bacillus anthracis lethal toxin-treated macrophages. Proc. Natl. Acad. Sci. USA 2003, 100, 12426–12431. [Google Scholar] [CrossRef] [PubMed]
- Das, N.D.; Jung, K.H.; Chai, Y.G. The role of NF-kappaB and H3K27me3 demethylase, JMJD3, on the anthrax lethal toxin tolerance of Raw 264.7 cells. PLoS ONE 2010, 5, e9913. [Google Scholar] [CrossRef] [PubMed]
- Ha, S.D.; Han, C.Y.; Reid, C.; Kim, S.O. HDAC8-mediated epigenetic reprogramming plays a key role in resistance to anthrax lethal toxin-induced pyroptosis in macrophages. J. Immunol. 2014, 193, 1333–1343. [Google Scholar] [CrossRef] [PubMed]
- Ha, S.D.; Park, S.; Han, C.Y.; Nguyen, M.L.; Kim, S.O. Cellular adaptation to anthrax lethal toxin-induced mitochondrial cholesterol enrichment, hyperpolarization, and reactive oxygen species generation through downregulating MLN64 in macrophages. Mol. Cell. Biol. 2012, 32, 4846–4860. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.K.; Mandal, T.; Balsubramanian, N.; Viaene, T.; Leedahl, T.; Sule, N.; Cook, G.; Srivastava, D.K. Histone deacetylase activators: N-acetylthioureas serve as highly potent and isozyme selective activators for human histone deacetylase-8 on a fluorescent substrate. Bioorg. Med. Chem. Lett. 2011, 21, 5920–5923. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, S.; Ramos, J.; Luo, W.; Sirisawad, M.; Verner, E.; Buggy, J.J. A novel histone deacetylase 8 (HDAC8)-specific inhibitor PCI-34051 induces apoptosis in T-cell lymphomas. Leukemia 2008, 22, 1026–1034. [Google Scholar] [CrossRef] [PubMed]
- Worby, C.A.; Dixon, J.E. Pten. Annu. Rev. Biochem. 2014, 83, 641–669. [Google Scholar] [CrossRef] [PubMed]
- Ha, S.D.; Reid, C.; Meshkibaf, S.; Kim, S.O. Inhibition of IL-1β expression by anthrax lethal toxin is reversed by HDAC8 inhibition in murine macrophages. J. Biol. Chem. 2016, 291. [Google Scholar] [CrossRef]
- Pasini, D.; Malatesta, M.; Jung, H.R.; Walfridsson, J.; Willer, A.; Olsson, L.; Skotte, J.; Wutz, A.; Porse, B.; Jensen, O.N.; et al. Characterization of an antagonistic switch between histone H3 lysine 27 methylation and acetylation in the transcriptional regulation of polycomb group target genes. Nucleic. Acids. Res. 2010, 38, 4958–4969. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, N.; Salmon-Divon, M.; Dvinge, H.; Hynes-Allen, A.; Balasooriya, G.; Leaford, D.; Behrens, A.; Bertone, P.; Hendrich, B. Nurd-mediated deacetylation of H3K27 facilitates recruitment of polycomb repressive complex 2 to direct gene repression. EMBO J. 2012, 31, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Tie, F.; Banerjee, R.; Stratton, C.A.; Prasad-Sinha, J.; Stepanik, V.; Zlobin, A.; Diaz, M.O.; Scacheri, P.C.; Harte, P.J. Cbp-mediated acetylation of histone H3 lysine 27 antagonizes drosophila polycomb silencing. Development 2009, 136, 3131–3141. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Xu, J.F.; Chang, R.M.; Fang, F.; Zuo, C.H.; Yang, L.Y. Jarid2 promotes invasion and metastasis of hepatocellular carcinoma by facilitating epithelial-mesenchymal transition through PTEN/Akt signaling. Oncotarget 2016, 7, 40266–40284. [Google Scholar] [CrossRef] [PubMed]
- Gall Troselj, K.; Novak Kujundzic, R.; Ugarkovic, D. Polycomb repressive complex's evolutionary conserved function: The role of EZH2 status and cellular background. Clin. Epigenetics 2016, 8, 55. [Google Scholar] [CrossRef] [PubMed]
- De Santa, F.; Totaro, M.G.; Prosperini, E.; Notarbartolo, S.; Testa, G.; Natoli, G. The histone H3 lysine-27 demethylase JMJD3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell 2007, 130, 1083–1094. [Google Scholar] [CrossRef] [PubMed]
- Agger, K.; Cloos, P.A.; Christensen, J.; Pasini, D.; Rose, S.; Rappsilber, J.; Issaeva, I.; Canaani, E.; Salcini, A.E.; Helin, K. UTX and JMJD3 are histone H3K27 demethylases involved in hox gene regulation and development. Nature 2007, 449, 731–734. [Google Scholar] [CrossRef] [PubMed]
- Risso, G.; Blaustein, M.; Pozzi, B.; Mammi, P.; Srebrow, A. Akt/PKB: One kinase, many modifications. Biochem. J. 2015, 468, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Hart, J.R.; Vogt, P.K. Phosphorylation of Akt: A mutational analysis. Oncotarget 2011, 2, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Fayard, E.; Xue, G.; Parcellier, A.; Bozulic, L.; Hemmings, B.A. Protein kinase b (Pkb/Akt), a key mediator of the pi3k signaling pathway. Curr. Top. Microbiol. Immunol. 2010, 346, 31–56. [Google Scholar] [PubMed]
- Mahajan, K.; Mahajan, N.P. Pi3k-independent AKT activation in cancers: A treasure trove for novel therapeutics. J. Cell. Physiol. 2012, 227, 3178–3184. [Google Scholar] [CrossRef] [PubMed]
- Faes, S.; Dormond, O. PI3K and AKT: Unfaithful partners in cancer. Int. J. Mol. Sci. 2015, 16, 21138–21152. [Google Scholar] [CrossRef] [PubMed]
- Maehama, T.; Dixon, J.E. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 1998, 273, 13375–13378. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Molina, A.; Efeyan, A.; Lopez-Guadamillas, E.; Munoz-Martin, M.; Gomez-Lopez, G.; Canamero, M.; Mulero, F.; Pastor, J.; Martinez, S.; Romanos, E.; et al. Pten positively regulates brown adipose function, energy expenditure, and longevity. Cell Metab. 2012, 15, 382–394. [Google Scholar] [CrossRef] [PubMed]
- Correia, N.C.; Girio, A.; Antunes, I.; Martins, L.R.; Barata, J.T. The multiple layers of non-genetic regulation of pten tumour suppressor activity. Eur. J. Cancer 2014, 50, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Lei, C.; Fan, J.; Wang, J. Mir-18a promotes cell proliferation of esophageal squamous cell carcinoma cells by increasing cylin D1 via regulating PTEN-PI3K-ALT-mTor signaling axis. Biochem. Biophys. Res. Commun. 2016, 477, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Virolle, T.; Adamson, E.D.; Baron, V.; Birle, D.; Mercola, D.; Mustelin, T.; de Belle, I. The Egr-1 transcription factor directly activates PTEN during irradiation-induced signalling. Nat. Cell Biol. 2001, 3, 1124–1128. [Google Scholar] [CrossRef] [PubMed]
- Patel, L.; Pass, I.; Coxon, P.; Downes, C.P.; Smith, S.A.; Macphee, C.H. Tumor suppressor and anti-inflammatory actions of ppargamma agonists are mediated via upregulation of pten. Curr. Biol. 2001, 11, 764–768. [Google Scholar] [CrossRef]
- Moorehead, R.A.; Hojilla, C.V.; De Belle, I.; Wood, G.A.; Fata, J.E.; Adamson, E.D.; Watson, K.L.; Edwards, D.R.; Khokha, R. Insulin-like growth factor-II regulates PTEN expression in the mammary gland. J. Biol. Chem. 2003, 278, 50422–50427. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.H.; Zhang, L.; Gan, Y.; Wang, X.; Wang, J.; LeMaire, S.A.; Coselli, J.S.; Wang, X.L. Up-regulation of PTEN (phosphatase and tensin homolog deleted on chromosome ten) mediates p38 MAPK stress signal-induced inhibition of insulin signaling. A cross-talk between stress signaling and insulin signaling in resistin-treated human endothelial cells. J. Biol. Chem. 2006, 281, 7727–7736. [Google Scholar] [PubMed]
- Jiang, X.; Li, H. Overexpression of LRG1 regulates PTEN via mapk/mek signaling pathway in esophageal squamous cell carcinoma. Exp. Ther. Med. 2016, 12, 2045–2052. [Google Scholar] [PubMed]
- Wu, Q.X.; Yuan, S.X.; Ren, C.M.; Yu, Y.; Sun, W.J.; He, B.C.; Wu, K. Oridonin upregulates PTEN through activating p38 MAPK and inhibits proliferation in human colon cancer cells. Oncol. Rep. 2016, 35, 3341–3348. [Google Scholar] [PubMed]
- Mak, L.H.; Vilar, R.; Woscholski, R. Characterisation of the pten inhibitor Vo-Ohpic. J. Chem. Biol. 2010, 3, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, A.; Oehme, I.; Witt, O.; Oliveira, G.; Sippl, W.; Romier, C.; Pierce, R.J.; Jung, M. Hdac8: A multifaceted target for therapeutic interventions. Trends Pharmacol. Sci. 2015, 36, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Delcuve, G.P.; Khan, D.H.; Davie, J.R. Targeting class i histone deacetylases in cancer therapy. Expert Opin. Ther. Targets 2013, 17, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Gligoris, T.G.; Scheinost, J.C.; Burmann, F.; Petela, N.; Chan, K.L.; Uluocak, P.; Beckouet, F.; Gruber, S.; Nasmyth, K.; Lowe, J. Closing the cohesin ring: Structure and function of its SMC3-kleisin interface. Science 2014, 346, 963–967. [Google Scholar] [CrossRef] [PubMed]
- Mannini, L.; Lamaze, F.C.; Cucco, F.; Amato, C.; Quarantotti, V.; Rizzo, I.M.; Krantz, I.D.; Bilodeau, S.; Musio, A. Mutant cohesin affects rna polymerase II regulation in Cornelia de Lange syndrome. Sci. Rep. 2015, 5, 16803. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, T.; Antony, J.; Braithwaite, A.W.; Horsfield, J.A. HDAC8 inhibition blocks SMC3 deacetylation and delays cell cycle progression without affecting cohesin-dependent transcription in MCF7 cancer cells. J. Biol. Chem. 2016, 291, 12761–12770. [Google Scholar] [CrossRef] [PubMed]
- Fazio, G.; Gaston-Massuet, C.; Bettini, L.R.; Graziola, F.; Scagliotti, V.; Cereda, A.; Ferrari, L.; Mazzola, M.; Cazzaniga, G.; Giordano, A.; et al. Cyclin D1 down-regulation and increased apoptosis are common features of cohesinopathies. J. Cell. Physiol. 2016, 231, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, J.; Han, Q.; Sun, Z.; Wang, J.; Wang, S.; Zhao, R.C. Enhancer of zeste homolog 2 is overexpressed and contributes to epigenetic inactivation of p21 and phosphatase and tensin homolog in B-cell acute lymphoblastic leukemia. Exp. Biol. Med. (Maywood) 2012, 237, 1110–1116. [Google Scholar] [CrossRef] [PubMed]
- Kaur, M.; Cole, M.D. MYC acts via the PTEN tumor suppressor to elicit autoregulation and genome-wide gene repression by activation of the Ezh2 methyltransferase. Cancer Res. 2013, 73, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.C.; Yang, Z.L. Overexpression of EZH2 and loss of expression of PTEN is associated with invasion, metastasis, and poor progression of gallbladder adenocarcinoma. Pathol. Res. Pract. 2011, 207, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, C.; Ikezoe, T.; Yang, J.; Udaka, K.; Yokoyama, A. Imatinib causes epigenetic alterations of PTEN gene via upregulation of DNA methyltransferases and polycomb group proteins. Blood Cancer J. 2011, 1, e48. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ji, F.; Liu, Y.; Lei, X.; Li, H.; Ji, G.; Yuan, Z.; Jiao, J. Ezh2 regulates adult hippocampal neurogenesis and memory. J. Neurosci. 2014, 34, 5184–5199. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Zang, X.; Ponnusamy, M.; Masucci, M.V.; Tolbert, E.; Gong, R.; Zhao, T.C.; Liu, N.; Bayliss, G.; Dworkin, L.D.; et al. Enhancer of Zeste Homolog 2 inhibition attenuates renal fibrosis by maintaining Smad7 and phosphatase and tensin homolog expression. J. Am. Soc. Nephrol. 2016, 27, 2092–2108. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Ricciardi, R.P. E1A is the component of the MHC class І enhancer complex that mediates HDAC chromatin repression in adenovirus-12 tumorigenic cells. Virology. 2006, 352, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Durst, K.L.; Lutterbach, B.; Kummalue, T.; Friedman, A.D.; Hiebert, S.W. The inv(16) fusion protein associates with corepressors via a smooth muscle myosin heavy-chain domain. Mol. Cell. Biol. 2003, 23, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Singh, S.; Hua, W.K.; Cai, Q.; Chao, S.W.; Li, L.; Liu, H.; Ho, Y.; McDonald, T.; Lin, A.; et al. HDAC8 inhibition specifically targets inv(16) acute myeloid leukemic stem cells by restoring p53 acetylation. Cell Stem Cell 2015, 17, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Canettieri, G.; Morantte, I.; Guzman, E.; Asahara, H.; Herzig, S.; Anderson, S.D.; Yates, J.R., 3rd; Montminy, M. Attenuation of a phosphorylation-dependent activator by an HDAC-PP1 complex. Nat. Struct. Biol. 2003, 10, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Karolczak-Bayatti, M.; Sweeney, M.; Cheng, J.; Edey, L.; Robson, S.C.; Ulrich, S.M.; Treumann, A.; Taggart, M.J.; Europe-Finner, G.N. Acetylation of heat shock protein 20 (hsp20) regulates human myometrial activity. J. Biol. Chem. 2011, 286, 34346–34355. [Google Scholar] [CrossRef] [PubMed]
- Waltregny, D.; Glenisson, W.; Tran, S.L.; North, B.J.; Verdin, E.; Colige, A.; Castronovo, V. Histone deacetylase HDAC8 associates with smooth muscle alpha-actin and is essential for smooth muscle cell contractility. FASEB J. 2005, 19, 966–968. [Google Scholar] [PubMed]
- Lee, H.; Sengupta, N.; Villagra, A.; Rezai-Zadeh, N.; Seto, E. Histone deacetylase 8 safeguards the human ever-shorter telomeres 1B (HEST1b) protein from ubiquitin-mediated degradation. Mol. Cell. Biol. 2006, 26, 5259–5269. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.J.; Tremblay, A.M.; Deblois, G.; Sylvain-Drolet, G.; Giguere, V. An acetylation switch modulates the transcriptional activity of estrogen-related receptor alpha. Mol. Endocrinol. 2010, 24, 1349–1358. [Google Scholar] [CrossRef] [PubMed]
- Olson, D.E.; Udeshi, N.D.; Wolfson, N.A.; Pitcairn, C.A.; Sullivan, E.D.; Jaffe, J.D.; Svinkina, T.; Natoli, T.; Lu, X.; Paulk, J.; et al. An unbiased approach to identify endogenous substrates of "Histone" Deacetylase 8. ACS Chem. Biol. 2014, 9, 2210–2216. [Google Scholar] [CrossRef] [PubMed]
- Lopez, G.; Bill, K.L.; Bid, H.K.; Braggio, D.; Constantino, D.; Prudner, B.; Zewdu, A.; Batte, K.; Lev, D.; Pollock, R.E. Hdac8, a potential therapeutic target for the treatment of malignant peripheral nerve sheath tumors (mpnst). PLoS ONE 2015, 10, e0133302. [Google Scholar] [CrossRef] [PubMed]
- Stambolic, V.; MacPherson, D.; Sas, D.; Lin, Y.; Snow, B.; Jang, Y.; Benchimol, S.; Mak, T.W. Regulation of PTEN transcription by p53. Mol. Cell 2001, 8, 317–325. [Google Scholar] [CrossRef]
- Tournier, J.N.; Quesnel-Hellmann, A.; Cleret, A.; Vidal, D.R. Contribution of toxins to the pathogenesis of inhalational anthrax. Cell Microbiol. 2007, 9, 555–565. [Google Scholar] [CrossRef] [PubMed]
- Cote, C.K.; Welkos, S.L. Anthrax toxins in context of Bacillus anthracis spores and spore germination. Toxins (Basel) 2015, 7, 3167–3178. [Google Scholar] [CrossRef] [PubMed]
- Banks, D.J.; Barnajian, M.; Maldonado-Arocho, F.J.; Sanchez, A.M.; Bradley, K.A. Anthrax toxin receptor 2 mediates Bacillus anthracis killing of macrophages following spore challenge. Cell Microbiol. 2005, 7, 1173–1185. [Google Scholar] [CrossRef] [PubMed]
- Guidi-Rontani, C.; Levy, M.; Ohayon, H.; Mock, M. Fate of germinated Bacillus anthracis spores in primary murine macrophages. Mol. Microbiol. 2001, 42, 931–938. [Google Scholar] [CrossRef] [PubMed]
- Cote, C.K.; Van Rooijen, N.; Welkos, S.L. Roles of macrophages and neutrophils in the early host response to Bacillus anthracis spores in a mouse model of infection. Infect. Immun. 2006, 74, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Bachran, C.; Leppla, S.H. Tumor targeting and drug delivery by anthrax toxin. Toxins (Basel) 2016, 8, 197. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
HDAC8 regulates susceptibility to LeTx cytotoxicity in human monocytic THP-1 cells. (A) THP-1 cells were treated with LeTx (500 ng/mL LF and 500 ng/mL PA) for 3 h and further cultured in fresh media in the presence or absence of TM-2-51 (TM; 25 µM) or PCI-34051 (PCI; 5 µM). Trypan blue staining negative live cells were counted using a hemocytometer at the time indicated. (B) THP-1 cells were treated with different doses of LeTx for 3 h and cultured in fresh media with or without TM (25 µM) and PCI (5 µM) for 48 h. Cell viability was measured using MTT assay. (A,B) Results are expressed as the mean ± SD (n = 3). (C,D) Similarly, cells were treated with LeTx in the presence or absence of various doses of: TM for 48 h (C); and PCI for 72 h (D). Cell viability was measured by MTT assay. Data are expressed as means ± SD (n = 3; N.S., not significant; *, P < 0.05, Student’s t test).
Figure 1.
HDAC8 regulates susceptibility to LeTx cytotoxicity in human monocytic THP-1 cells. (A) THP-1 cells were treated with LeTx (500 ng/mL LF and 500 ng/mL PA) for 3 h and further cultured in fresh media in the presence or absence of TM-2-51 (TM; 25 µM) or PCI-34051 (PCI; 5 µM). Trypan blue staining negative live cells were counted using a hemocytometer at the time indicated. (B) THP-1 cells were treated with different doses of LeTx for 3 h and cultured in fresh media with or without TM (25 µM) and PCI (5 µM) for 48 h. Cell viability was measured using MTT assay. (A,B) Results are expressed as the mean ± SD (n = 3). (C,D) Similarly, cells were treated with LeTx in the presence or absence of various doses of: TM for 48 h (C); and PCI for 72 h (D). Cell viability was measured by MTT assay. Data are expressed as means ± SD (n = 3; N.S., not significant; *, P < 0.05, Student’s t test).

Figure 2.
HDAC8 activity regulates susceptibility to LeTx-induced cell cycle arrest. THP-1 cells were treated with LeTx as described in the legend to Figure 1A. Cells were then harvested at 48 h or 72 h after LeTx treatments and fixed with 70% ethanol. Cell cycle phase was measured by the FACS Calibur flow cytometry/CellQuest program using propidium iodide DNA staining, followed by data analysis using ModFit software. Data shown are representative results of two independent experiments.
Figure 2.
HDAC8 activity regulates susceptibility to LeTx-induced cell cycle arrest. THP-1 cells were treated with LeTx as described in the legend to Figure 1A. Cells were then harvested at 48 h or 72 h after LeTx treatments and fixed with 70% ethanol. Cell cycle phase was measured by the FACS Calibur flow cytometry/CellQuest program using propidium iodide DNA staining, followed by data analysis using ModFit software. Data shown are representative results of two independent experiments.

Figure 3.
HDAC8 regulates AKT phosphorylation in THP-1 cells. (A) THP-1 cells were treated with LeTx as described in the legend to Figure 1A in the presence or absence of TM (25 µM) and PCI (5 µM). AKT phosphorylation at Ser-473 and MEK3 cleavage was analyzed using Western blots (upper panel). Western blotting against β-actin was used as the loading control. Results are representative blots from three independent experiments. Immunoreactivities against phospho-AKT (Ser-473) were analyzed using the NIH Image program (lower panel) and relative phosphor-AKT immunoreactivity was normalized to those of β-actin. Data are means and SD (n = 3; *, P < 0.05, Student’s t test). (B) Similarly, cells were treated with LeTx in the presence or absence of LY294002 (LY, 10 µM) or wortmannin (Wort, 1 µM) for 48–52 h. AKT phosphorylation and MEK1 N-terminal cleavage were measured by Western blotting (upper panel). Immunoblots shown are representative images of three independent experiments. For cytotoxicity assay, cells were similarly treated with LeTx in the presence or absence of LY (10 µM). Cytotoxicity was measured using MTT assay at the time points indicated (lower panel). Data are expressed as means and SD (n = 3).
Figure 3.
HDAC8 regulates AKT phosphorylation in THP-1 cells. (A) THP-1 cells were treated with LeTx as described in the legend to Figure 1A in the presence or absence of TM (25 µM) and PCI (5 µM). AKT phosphorylation at Ser-473 and MEK3 cleavage was analyzed using Western blots (upper panel). Western blotting against β-actin was used as the loading control. Results are representative blots from three independent experiments. Immunoreactivities against phospho-AKT (Ser-473) were analyzed using the NIH Image program (lower panel) and relative phosphor-AKT immunoreactivity was normalized to those of β-actin. Data are means and SD (n = 3; *, P < 0.05, Student’s t test). (B) Similarly, cells were treated with LeTx in the presence or absence of LY294002 (LY, 10 µM) or wortmannin (Wort, 1 µM) for 48–52 h. AKT phosphorylation and MEK1 N-terminal cleavage were measured by Western blotting (upper panel). Immunoblots shown are representative images of three independent experiments. For cytotoxicity assay, cells were similarly treated with LeTx in the presence or absence of LY (10 µM). Cytotoxicity was measured using MTT assay at the time points indicated (lower panel). Data are expressed as means and SD (n = 3).

Figure 4.
HDAC8 is required for suppression of PTEN expression in LeTx-exposed THP-1 cells. (A–C) Cells were treated with LeTx as described in the legend to Figure 1 in the presence or absence of PCI (5 µM) or VO-OHpic (10 µM) as indicated. (A) PTEN mRNA expression and protein levels were measured by qPCR (upper panel) and Western blotting (lower panel), respectively. (B) At 48 h after treatments, PTEN mRNA levels were measured by qPCR (upper panel), and PTEN protein and AKT phosphorylation levels were analyzed by Western blotting. (C) Similarly, AKT phosphorylation was measured by Western blotting (upper panel) and cell survival was measured by MTT assay (lower panel). Immunoblot images are representative of three independent experiments. Data shown are expressed as means ± SD (n = 3; *, P < 0.05, Student’s t test).
Figure 4.
HDAC8 is required for suppression of PTEN expression in LeTx-exposed THP-1 cells. (A–C) Cells were treated with LeTx as described in the legend to Figure 1 in the presence or absence of PCI (5 µM) or VO-OHpic (10 µM) as indicated. (A) PTEN mRNA expression and protein levels were measured by qPCR (upper panel) and Western blotting (lower panel), respectively. (B) At 48 h after treatments, PTEN mRNA levels were measured by qPCR (upper panel), and PTEN protein and AKT phosphorylation levels were analyzed by Western blotting. (C) Similarly, AKT phosphorylation was measured by Western blotting (upper panel) and cell survival was measured by MTT assay (lower panel). Immunoblot images are representative of three independent experiments. Data shown are expressed as means ± SD (n = 3; *, P < 0.05, Student’s t test).

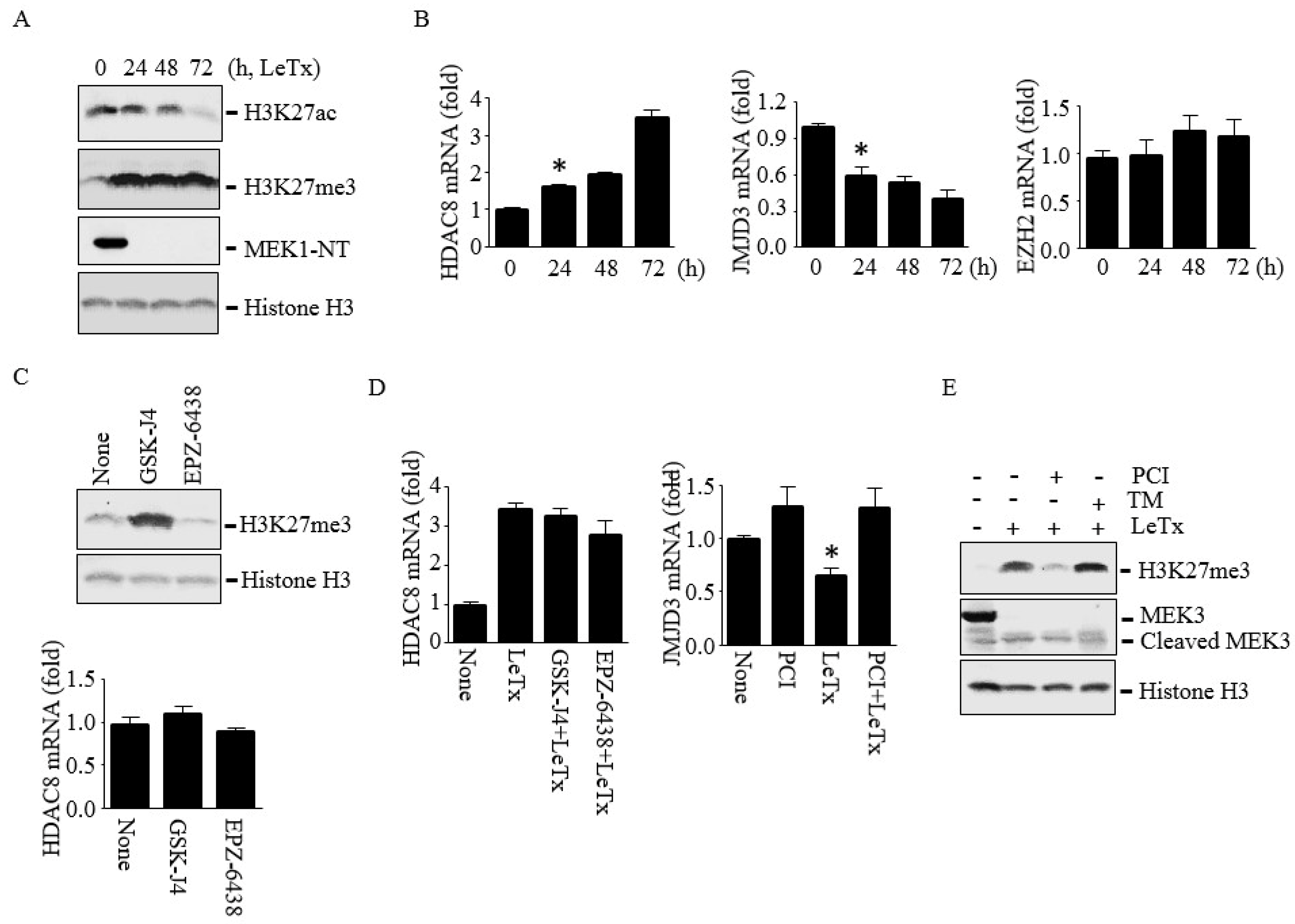
Figure 5.
HDAC8 induces H3K27me3 through down-regulating JMJD3 in THP-1 cells. Cells were treated with LeTx as described in the legend to Figure 1 in the presence or absence of PCI (5 µM), GSK-J4 (1 µM) or EPZ-6438 (500 nM) for 24–72 h. (A) Levels of H3K27Ac and H3K27me3 were analyzed by Western blotting. Histone H3 immunoblot was used as the loading control. (B) HDAC8, JMJD3 and EZH2 mRNA levels were analyzed by qPCR. Results are expressed as means ± SD (n = 3). (C) Cells were treated with LeTx in the presence or absence of GSK-J4 or EPZ-6438 for 48 h. H3K27me3 (upper panel) and HDAC8 mRNA (lower panel) levels were analyzed by Western blotting and qPCR, respectively. (D) Similarly, cells were treated with LeTx, together with PCI or GSK-J4 or EPZ-6438 for 48h, and mRNA expression of HDAC8 (left panel) and JMJD3 (right panel) were measured by qPCR. Data are expressed as means ±SD (n = 3; *, P < 0.05, Student’s t test). (E) H3K27me3 levels in LeTx treated cell with or without PCI or TM were analyzed by Western blotting.
Figure 5.
HDAC8 induces H3K27me3 through down-regulating JMJD3 in THP-1 cells. Cells were treated with LeTx as described in the legend to Figure 1 in the presence or absence of PCI (5 µM), GSK-J4 (1 µM) or EPZ-6438 (500 nM) for 24–72 h. (A) Levels of H3K27Ac and H3K27me3 were analyzed by Western blotting. Histone H3 immunoblot was used as the loading control. (B) HDAC8, JMJD3 and EZH2 mRNA levels were analyzed by qPCR. Results are expressed as means ± SD (n = 3). (C) Cells were treated with LeTx in the presence or absence of GSK-J4 or EPZ-6438 for 48 h. H3K27me3 (upper panel) and HDAC8 mRNA (lower panel) levels were analyzed by Western blotting and qPCR, respectively. (D) Similarly, cells were treated with LeTx, together with PCI or GSK-J4 or EPZ-6438 for 48h, and mRNA expression of HDAC8 (left panel) and JMJD3 (right panel) were measured by qPCR. Data are expressed as means ±SD (n = 3; *, P < 0.05, Student’s t test). (E) H3K27me3 levels in LeTx treated cell with or without PCI or TM were analyzed by Western blotting.

Figure 6.
JMJD3 inhibition suppresses PTEN expression, enhances AKT phosphorylation and protects cell cycle arrest in LeTx-treated cells even in the absence of HDAC8 activity. THP-1 cells were treated with LeTx as described in the legend to Figure 1 in the presence or absence of PCI (5 µM), GSK-J4 (1 µM) or EPZ-6438 (500 nM) for 48 h. (A) Cell cycle analysis was performed as described in the legend to Figure 2. Results shown are representative data from three independent experiments. (B,C) H3K27me3, AKT phosphorylation at Ser-473 and PTEN protein levels were analyzed by Western blotting. Data shown are representative images of three independent experiments. (D) Cell viability was analyzed using MTT assay. Results are expressed as the mean ± SD (n = 3; *, P < 0.01, Student`s t test).
Figure 6.
JMJD3 inhibition suppresses PTEN expression, enhances AKT phosphorylation and protects cell cycle arrest in LeTx-treated cells even in the absence of HDAC8 activity. THP-1 cells were treated with LeTx as described in the legend to Figure 1 in the presence or absence of PCI (5 µM), GSK-J4 (1 µM) or EPZ-6438 (500 nM) for 48 h. (A) Cell cycle analysis was performed as described in the legend to Figure 2. Results shown are representative data from three independent experiments. (B,C) H3K27me3, AKT phosphorylation at Ser-473 and PTEN protein levels were analyzed by Western blotting. Data shown are representative images of three independent experiments. (D) Cell viability was analyzed using MTT assay. Results are expressed as the mean ± SD (n = 3; *, P < 0.01, Student`s t test).

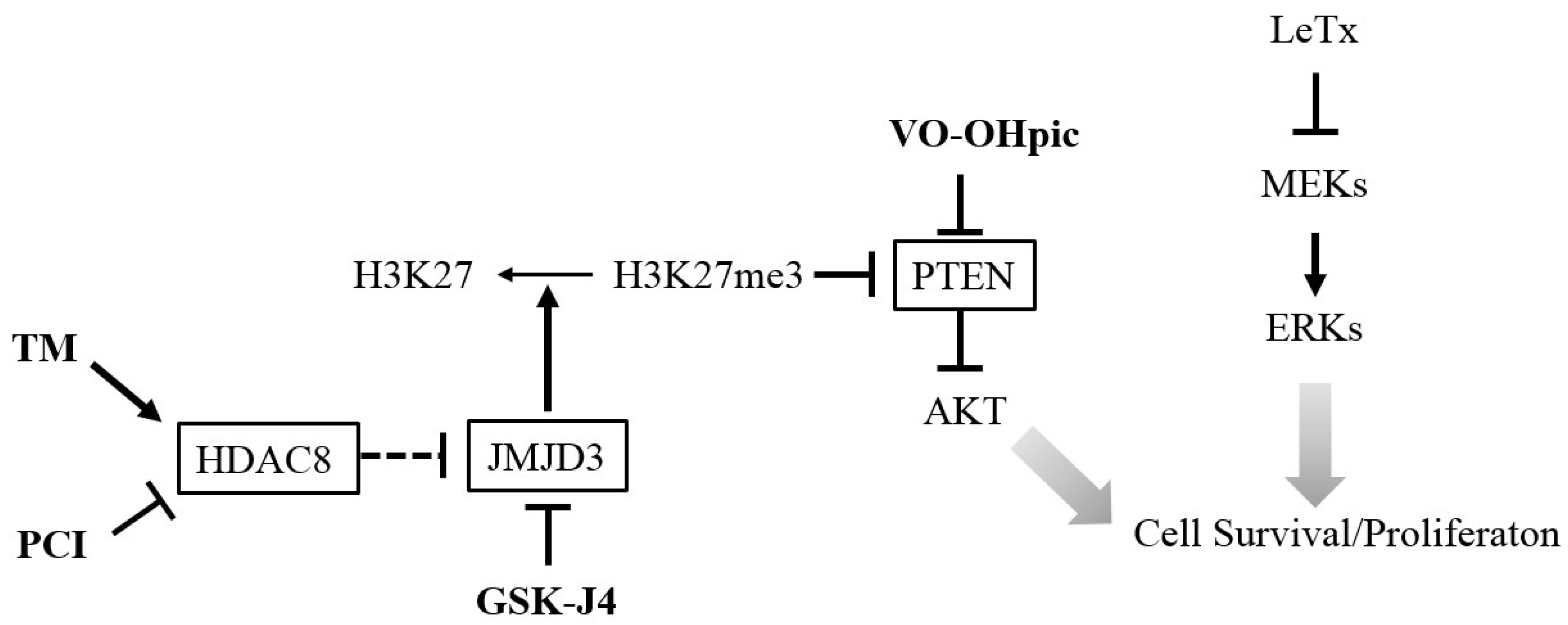
Figure 7.
A diagram illustrating the targets, and proposed effects on AKT activation and cell survival/proliferation of the inhibitors and activator. The HDAC8 activator TM and JMJD3 inhibitor GSK-J4 lead to an increase of H3K27me3 that suppress PTEN expression and enhances AKT activation. Direct inhibition of PTEN by VO-OHpic also induces AKT activation. In contrast, inhibition of HDAC8 by PCI leads to expression of PTEN and inhibits AKT activation. Activation of AKT promotes cell survival/proliferation in LeTx-exposed monocytes/macrophages. Arrows indicate a positive effect or activation and blocked arrows indicate a negative effect or inhibition.
Figure 7.
A diagram illustrating the targets, and proposed effects on AKT activation and cell survival/proliferation of the inhibitors and activator. The HDAC8 activator TM and JMJD3 inhibitor GSK-J4 lead to an increase of H3K27me3 that suppress PTEN expression and enhances AKT activation. Direct inhibition of PTEN by VO-OHpic also induces AKT activation. In contrast, inhibition of HDAC8 by PCI leads to expression of PTEN and inhibits AKT activation. Activation of AKT promotes cell survival/proliferation in LeTx-exposed monocytes/macrophages. Arrows indicate a positive effect or activation and blocked arrows indicate a negative effect or inhibition.

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ha, S.-D.; Cho, W.; Kim, S.O. HDAC8 Prevents Anthrax Lethal Toxin-induced Cell Cycle Arrest through Silencing PTEN in Human Monocytic THP-1 Cells. Toxins 2017, 9, 162. https://doi.org/10.3390/toxins9050162
AMA Style
Ha S-D, Cho W, Kim SO. HDAC8 Prevents Anthrax Lethal Toxin-induced Cell Cycle Arrest through Silencing PTEN in Human Monocytic THP-1 Cells. Toxins. 2017; 9(5):162. https://doi.org/10.3390/toxins9050162
Chicago/Turabian StyleHa, Soon-Duck, Woohyun Cho, and Sung Ouk Kim. 2017. "HDAC8 Prevents Anthrax Lethal Toxin-induced Cell Cycle Arrest through Silencing PTEN in Human Monocytic THP-1 Cells" Toxins 9, no. 5: 162. https://doi.org/10.3390/toxins9050162
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.