
Ribosomal Protein S6: A Potential Therapeutic Target against Cancer?



1
Department of Biochemistry, College of Medicine, Dankook University, Cheonan 31116, Chungcheongnam-do, Korea
2
Department of Nanobiomedical Science, Dankook University, Cheonan 31116, Chungcheongnam-do, Korea
3
Graduate School of Convergence Medical Science, Dankook University, Cheonan 31116, Chungcheongnam-do, Korea
4
Graduate School, Kyung Hee University, Seoul 02447, Korea
*
Authors to whom correspondence should be addressed.
Int. J. Mol. Sci. 2022, 23(1), 48; https://doi.org/10.3390/ijms23010048
Submission received: 26 November 2021
/
Revised: 19 December 2021
/
Accepted: 20 December 2021
/
Published: 21 December 2021
(This article belongs to the Special Issue Molecular Target and Action Mechanism of Anti-Cancer Agents)
Abstract
:Ribosomal protein S6 (RPS6) is a component of the 40S small ribosomal subunit and participates in the control of mRNA translation. Additionally, phospho (p)-RPS6 has been recognized as a surrogate marker for the activated PI3K/AKT/mTORC1 pathway, which occurs in many cancer types. However, downstream mechanisms regulated by RPS6 or p-RPS remains elusive, and the therapeutic implication of RPS6 is underappreciated despite an approximately half a century history of research on this protein. In addition, substantial evidence from RPS6 knockdown experiments suggests the potential role of RPS6 in maintaining cancer cell proliferation. This motivates us to investigate the current knowledge of RPS6 functions in cancer. In this review article, we reviewed the current information about the transcriptional regulation, upstream regulators, and extra-ribosomal roles of RPS6, with a focus on its involvement in cancer. We also discussed the therapeutic potential of RPS6 in cancer.
1. Background
Eukaryotic ribosome (80S) consists of the 60S large and 40S small ribosomal subunits, which contain four ribosomal RNAs (rRNAs; 5S, 5.8S, and 28S rRNAs in the large subunit and 18S rRNA in the small subunit) and 79 ribosomal proteins (RPs) [1,2]. RPs are RNA-binding proteins and are primarily involved in the regulation of protein translation [3]. RPs also function in ribosome biogenesis in the nucleolus. They act as RNA chaperones to ensure the stabilization and correct folding of rRNAs to assemble ribosomal subunits. Dysregulation of ribosome biogenesis may result in tumorigenesis [4]. RPs also have multiple extra-ribosomal functions, including regulation of cell-cycle arrest, cell proliferation, cell migration and invasion, apoptosis, DNA damage repair, malignant transformation, and tumorigenesis via both p53/mouse double-minute 2 homolog (MDM2)-dependent and p53-independent mechanisms [3]. For example, ribosomal protein L5 (RPL5) [5], RPL11 [6,7], and RPL23 [8,9] exert anticancer effects through the activation of the tumor suppressor p53 by specifically binding to its inhibitor MDM2.
Ribosomal protein S6 (RPS6), also known as phosphoprotein NP33 [10], is a component of the 40S small ribosomal subunit as a ribosomal RNA-binding protein [11,12]. RPS6 is evolutionarily conserved across eukaryotes from yeast to vertebrates [12] and plays important roles in ribosome biogenesis, protein translation, cell proliferation (increase in cell number), cell growth (increase in cell mass/volume), DNA repair, apoptosis, cell differentiation, and glucose metabolism in both normal and cancer cells [13,14,15]. RPS6 is the first ribosomal protein identified to be phosphorylated by protein kinases [16] and is one of the only two RPs known to be phosphorylated [17].
RPS6 is used as a readout of the mechanistic/mammalian target of rapamycin complex 1 (mTORC1) activity in many diseases, including cancers, since alleviated activity of mTORC1, an upstream regulator of RPS6, is commonly found in many types of cancer cells [2,18,19,20]. However, considering the almost five-decade-long history of research on RPS6, the functions and therapeutic implications of this protein and its downstream regulatory pathways have not been well appreciated yet. Recently, we found that dual inhibition of epidermal growth factor receptor (EGFR) and MET kinase activities resulted in the synergistic anti-proliferation effect through downregulation of RPS6 in triple-negative breast cancer (TNBC) cells [21]. In addition, knockdown of RPS6 was enough to suppress the proliferation of TNBC cells in vitro. These results motivate us to investigate previous studies on the therapeutic potential of RPS6 targeting. In this review article, we analyzed the roles of RPS6 in tumorigenesis and evaluated the potential of using it as an anticancer therapeutic target.
2. Regulation of RPS6
2.1. Transcriptional Regulation of RPS6
The human RPS6 gene is located on chromosome 9p21, comprises six exons [22], and encodes a protein of 249 amino acids [23,24]. Contrary to rRNAs, which are transcribed by RNA polymerase I or III, RP genes are transcribed by RNA polymerase II [1]. Following transcription, the produced RP mRNAs are transported from the nucleoplasm to the cytoplasm for translation, after which the de novo formed RPs are translocated into the nucleus and concentrated in the nucleolus. Interestingly, the promoter regions of most RP genes are phylogenetically well conserved [1].
The promoters of the human RPS6 and mouse Rps6 genes share 80% sequence similarity [25], lack a consensus TATA box, and have high GpC content [22,25]. Consensus binding sites for specificity protein 1 (SP1) have been found in the upstream regions and within the first introns of both human RPS6 and mouse Rps6 genes [22,25]. The SP1 sites in the human RPS6 gene have been reported to minimally contribute to the transcription of the gene in COS-1 cells [26]. Additionally, the human RPS6 promoter contains four putative binding sites for GA-binding protein (GABP), a member of the ETS DNA-binding protein family. Potential binding sequences for CCAAT/enhancer binding protein (C/EBP) and the DNA-binding protein Ikaros have also been identified. In addition, the recombinant Yin-Yang 1 (YY1) protein has been shown to bind to the consensus binding site in the first intron of the human RPS6 gene. In fact, upstream GABP and SP1 sites and downstream YY1 sites are common features of several RP gene promoters [27]. However, the functional implications of GABP and YY1 have not been determined yet [26,28,29].
Chromatin immunoprecipitation experiments have shown that the MYC protein binds to the E-box motif in the TSC2 and RPS6 genes [30]. Interestingly, MYC differentially regulates the transcription of these genes. For example, the TSC2 protein level in the MYC-deficient rat fibroblast HO15 cells is increased, whereas that of RPS6 is reduced. MYC has been shown to repress the transcription of TSC2 and thereby activates the mTORC1/S6K pathway, but the involvement of MYC in the transcriptional regulation of the RPS6 gene has not been determined yet [31].
Zinc finger BED domain-containing protein 1 (ZBED1), an E3 SUMO-protein ligase, has been reported to bind to the upstream regions of RP genes, including RPS6 [32]. Depletion of ZBED1 reduces the transcription of the RPS6, RPSL10A, and RPL12 genes. The expression of the ZBED1 and RP genes is coordinately regulated via a cell-cycle-dependent manner. ZBED1 SUMOylates promoter-bound chromodomain-helicase-DNA-binding protein 3 (CHD3) to release it from the DNA and thereby increases the recruitment of RNA polymerase II to RP gene promoters [33].
Dual-specific tyrosine-phosphorylation-regulated kinase 1A (DYRK1A) has been reported to be recruited to the promoters of genes transcribed by RNA polymerase II [34]. In fact, DYRK1A is constitutively recruited to the promoters of cell-cycle-dependent genes, such as CDK12, RPS6, RPS11, and RPS12, and depletion of DYRK1A downregulates expression of these genes.
The RPS6 gene is transcribed in the mid-to-late G1 phase, whereas rRNA genes are transcribed in the early G1 phase concurrently with the nucleolar assembly [35]. In contrast to the RPS27a gene promoter, the human RPS6 gene promoter is not occupied by SP1 or the cAMP-responsive element-binding protein (CREB) [35]. Interestingly, E2F transcription factor 1 (E2F1) has been detected to bind to the RPS6 gene promoter [35]. Since E2F1 is a well-established transcription factor that regulates the cell-cycle-dependent transcription [36], it is highly plausible that E2F1 is a key transcriptional regulator of RPS6 during cell cycle progression. Additionally, the retinoblastoma-associated protein (RB) might contribute to the transcriptional upregulation of RPS6 by binding to E2F1 on the RPS6 promoter. RPS6-knockdown (KD) is known to downregulate phospho (p)-RB in various cancer cells [37,38,39,40]; however, it is elusive whether such a positive feedback loop between RPS6 and the p-RB level is involved in cancer progression. Furthermore, the precise roles and effects of E2F1 and RB on the transcription of RPS6 remain to be determined.
2.2. Post-Translational Modifications of RPS6
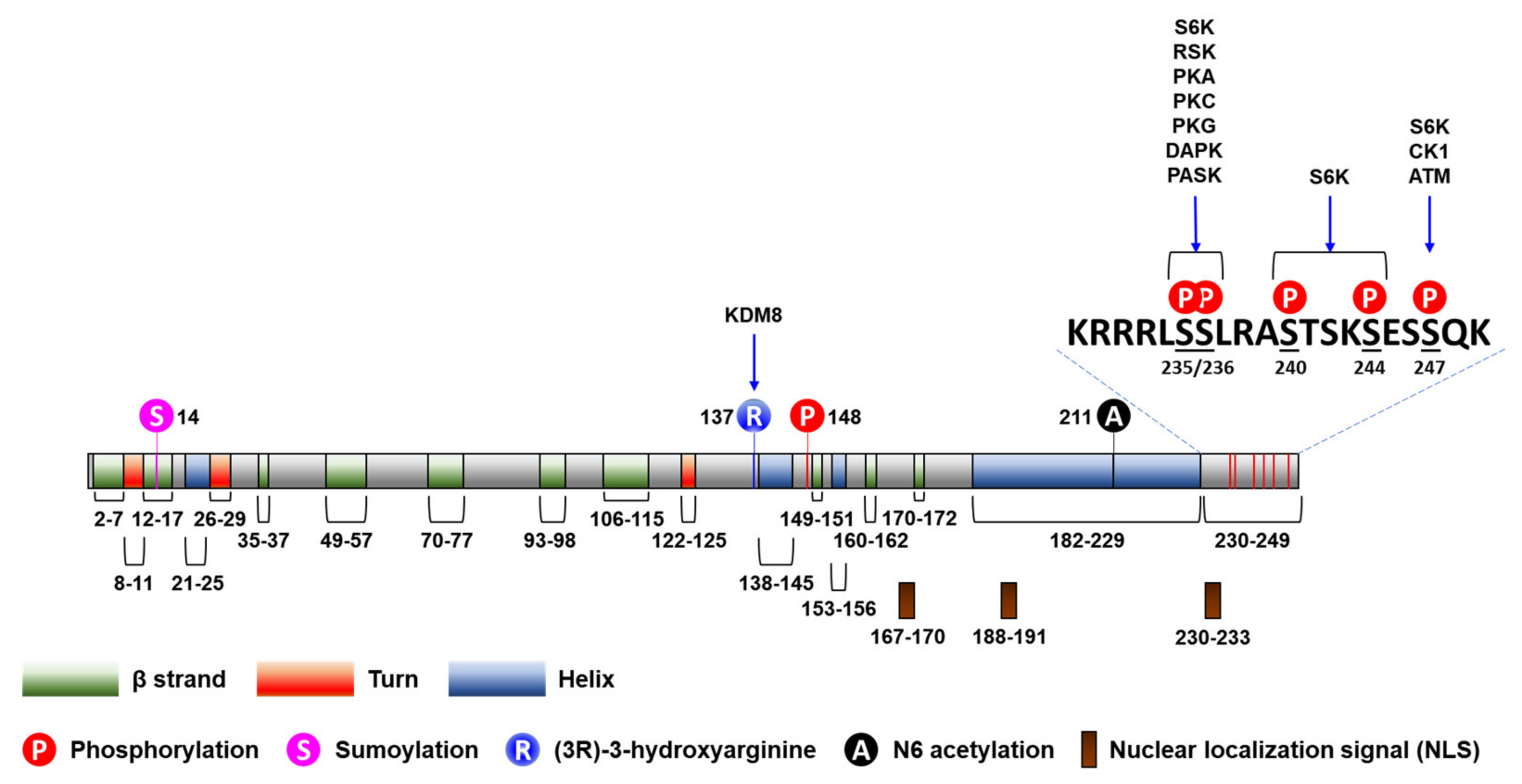
Post-translational modifications, including phosphorylation, acetylation, methylation, O-linked β-N-acetylglucosaminylation, and ubiquitinylation, have been described to participate in the regulation of the activity of RPs [41] (Figure 1). For example, ischemic preconditioning (IPC) induces S-nitrosylation of RPs, including RPS6, in perfused mouse hearts [42]. In addition, the R137 residue of RPS6 has been identified to be hydroxylated by lysine demethylase 8 (KDM8), an L-arginine (3R)-hydroxylase [43]. However, little is known about the regulation of the post-translational modifications of RPS6, except for phosphorylations, which we will discuss next.
2.2.1. Phosphorylation of RPS6
Translation is strictly regulated by signaling pathways sensing environmental stresses (e.g., heat shock or ultraviolet [UV] irradiation), extracellular stimuli (e.g., hormones or growth factors), and intracellular cues (e.g., nutrients, metabolites, energy status, or oxygen availability) [44]. The phosphoinositide 3-kinase (PI3K)/AKT and the mitogen-activated protein kinase (MAPK) pathways are two major pathways involved in the regulation of translation.
RPS6 has five evolutionarily conserved C-terminal serine residues (S235, S236, S240, S244, and S247) that are phosphorylated by various protein kinases (Figure 1) [12,45]. S6 kinase 1 (S6K1) sequentially phosphorylates all these residues, starting with S236 followed by S235, S240, S244, and S247 [46,47]. S235/236 can be also phosphorylated by ribosomal protein S6 kinases (RSKs) [48,49], protein kinase A (PKA; aka cAMP-dependent protein kinase) [50,51,52,53], protein kinase C (PKC) [54], protein kinase G (PKG) [52], and death-associated protein kinase 1 (DAPK1) [55]. S247 can be also phosphorylated by casein kinase 1 (CK1) [56] and ataxia telangiectasia mutated (ATM) [57]. RPS6 is also phosphorylated at S235/236 by PAS domain-containing serine/threonine-protein kinase (PASK), which regulates translation and glycogen synthesis in mammalian cells [58]. Accordingly, p-RPS6 (S240/244) is a more specific readout for S6K activation than the other RPS6 phosphorylation. Interestingly, all these phosphorylated serine residues are dephophorylated by a single phosphatase, protein phosphatase 1 (PP1) [56,59].
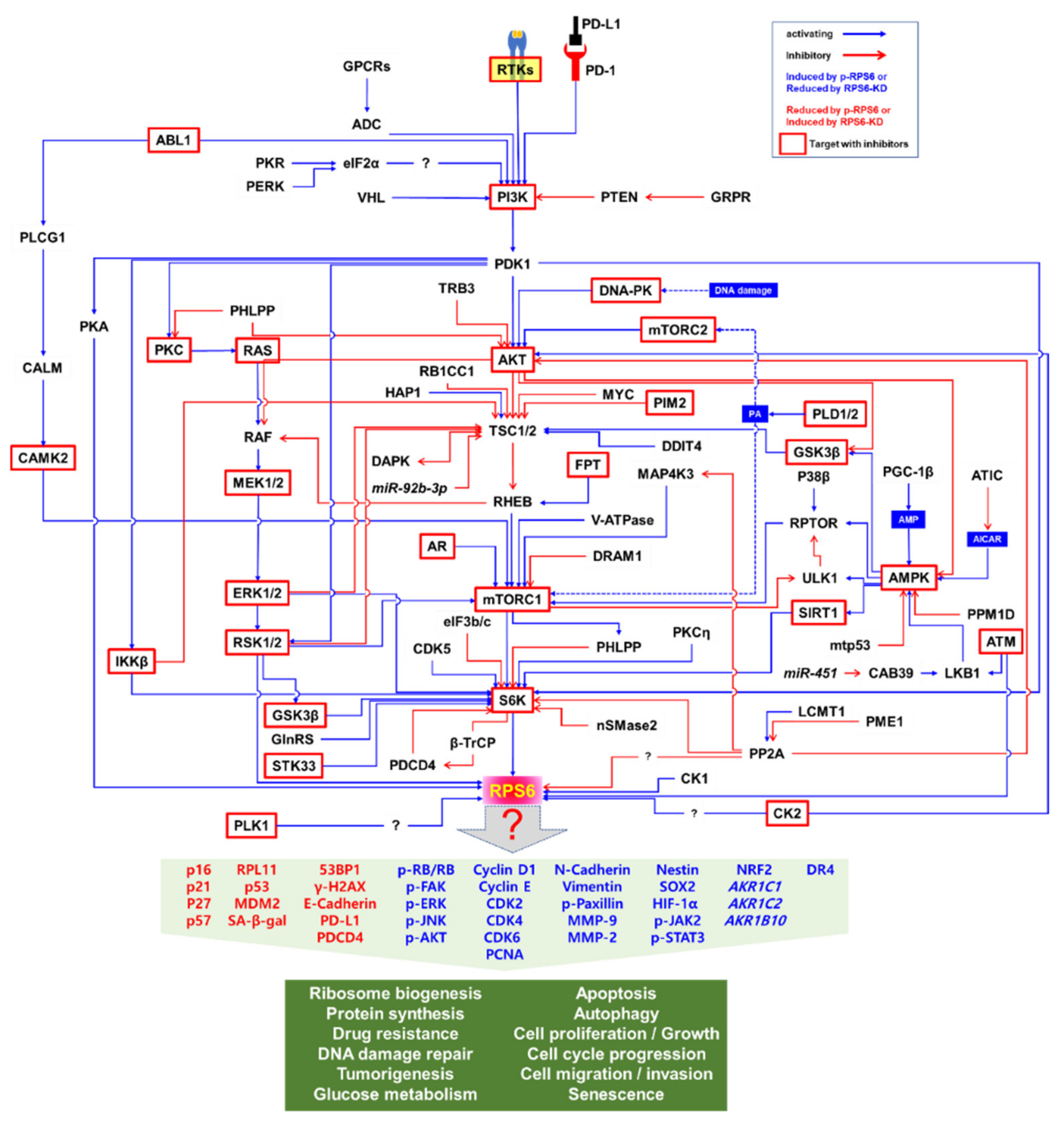
The PI3K/AKT/mTORC1/S6K pathway is the major signaling pathway that upregulates the phosphorylation of RPS6 in mammalian cells (Figure 2) [2]. RPS6 is the first identified substrate of S6K1 (p70S6Kα) [60], and phosphorylation of RPS6 has been used as a readout of mTORC1 activity [2,18,19,20]. However, a close homolog of S6K1, S6K2 (p70S6Kβ), is the major kinase of RPS6 [61,62,63,64,65]. The two S6K homologs S6K1 (S6Kα) and S6K2 (S6Kβ) in mammal share extensive but dispersed homology (43%, 84%, and 59% in N-terminus, catalytic domain, and C-terminus, respectively) [63]. S6K1 has cytosolic (p70S6K1) and nuclear (p85S6K1) isoforms [66], whereas the two isoforms of S6K2 (p54S6K2 and p56S6K2) are nuclear because of a C-terminal nuclear localization sequence (NLS) they have [61,63]. The activity of S6K is tightly controlled via rapid dephosphorylation by pleckstrin homology domain leucine-rich repeat protein phosphates (PHLPP), which is upregulated by mTORC1 [67]. However, the details of the fine regulation of RPS6 phosphorylation by these S6K isoforms are elusive.
The MAPK pathway consists of rat sarcoma (RAS)/v-raf-1 murine leukemia viral oncogene homolog (RAF)/MAPK-ERK kinase (MEK)/extracellular signal-regulated kinase (ERK) cascade. RSKs are downstream effectors of the MAPK pathway, which also regulates RPS6 activity through RPS6 phosphorylation [48]. Four RSK genes, namely RSK1—4, have been identified in mammals, and these four genes display high homology (78–90%), especially in two kinase domains [68]. In cells lacking S6K, phosphorylation of RPS6 at S235/236 is mediated by RSKs [69]. In resting cells, RSKs are mainly localized in the cytoplasm [70]. Upon stimulation of cells with a growth factor, RSK1 shows maximal kinase activity and transiently translocates to the plasma membrane and then to the nucleus [71]. Subcellular localization of RSK2 is differentially regulated. For example, upon mitogen stimulation of cells, RSK2 localizes to the nucleus; whereas oxidative stress induces RSK2 localization to the cytoplasmic stress granules to promote the survival of the cells [72].
Since S6K and RSK are the downstream effectors of the PI3K/AKT/mTORC1 and MAPK pathways, respectively, RPS6 constitutes a convergence node of these two signaling pathways (Figure 2). In addition, synergistic crosstalk between the mTORC1 and MAPK signaling in the regulation of RPS6 phosphorylation has been reported (see below). ERK enhances S6K activity by phosphorylating it at T421/424 [73]. Additionally, ERK phosphorylates and thereby inhibits tuberous sclerosis 2 (TSC2), consequently leading to mTORC1 activation [74]. ERK also indirectly activates mTORC1 through RSK-mediated phosphorylation of the regulatory-associated protein of mTOR (RPTOR) [75]. RPS6 is also phosphorylated in hippocampal neurons by S6K, which is activated by cyclin-dependent kinase 5 (CDK5) in these cells [76]. Knocking-in the gene of a non-phosphorylatable RPS6 (rpS6P−/−) mutant protein resulted in small cell size, hypoinsulinemia, decreased β cell size, and muscle weakness in mice [77,78]. However, the exact roles of RPS6 phosphorylations still remain enigmatic.
Upstream Effectors of RPS6 Phosphorylation
Various factors regulating the phosphorylation of RPS6 have been identified, including extracellular proteins, kinases and phosphatases, membrane receptors, transcription factors, and adaptor proteins. For example, a genome-scale siRNA screening against 21,121 genes revealed multiple regulators for p-RPS6 (S235/236) in the pancreatic cancer cell line MIA PaCa-2, which contains a constitutively high p-RPS6 level, followed by confirmation in TSC1-null mouse embryonic fibroblasts (MEFs) [79]. Of the 1046 identified candidate regulators of p-RPS6 (S235/236), 632 were validated via individual RNAi. However, most of these p-RPS6 regulators should be further experimentally validated.
- (1)
- Extracellular proteins that regulate RPS6 phosphorylation
Various extracellular proteins such as cytokines and growth factors have been identified as regulators of RPS6 phosphorylation (Table 1). Notably, mitogens, including epidermal growth factor (EGF) and insulin, are well-established activators of RPS6 phosphorylation. However, spatiotemporal fine regulation of RPS6 phosphorylation by multiple extracellular proteins still remains to be elucidated.
- (2)
- Kinases and phosphatases that regulate RPS6 phosphorylation
The human genome is known to encode 656 kinases and approximately 184 phosphatases [97]. Multiple kinases and phosphatases contribute to fine regulation of RPS6 phosphorylation (Table 2). The upstream regulators of RPS6 are controlled in a context- and/or cell-type-dependent manner. In addition, the spatiotemporal regulation is more complex than the simple snapshot illustrated in Figure 2. For example, the activity of AKT is generally regulated by PI3K in response to growth factors, leading to activation of the mTORC1/S6K/RPS6 signaling by inhibiting TSC2 [98,99]. However, DNA damage induces AKT activation via the DNA-dependent protein kinase (DNA-PK) and activated AKT suppresses the RAF/MEK/ERK/S6K1 signaling, thereby reducing the p-RPS6 (S235/236) level, independently of TSC2, mTORC1, or p53 [100]. Under these conditions, the DNA damage-inducible transcript 4 protein (DDIT4)/regulated in development and DNA damage responses 1 (REDD1) inhibits the AKT signal to mTORC1 by sequestering the 14-3-3 protein to activate TSC. In addition, the interaction of Sestrin-2 with the 5′-AMP-activated protein kinase (AMPK) inhibits the AKT-mediated inhibition of AMPK upon DNA damage. Therefore, DNA damage downregulates p-RPS6 (S235/236) by inhibiting the RAF/MEK/ERK/S6K1 signaling through DNA-PK-activated AKT even in the presence of growth factors. These results suggest that changes in p-RPS6 level that are induced by drugs or other stimuli do not always reflect changes in the mTORC1 signaling [100]. The downstream effectors of mTORC1, S6K1, and eukaryotic translation initiation factor 4E-binding protein 1 (4E-BP1) have been also reported to be differentially regulated by exogenous signals. For example, DNA damage suppresses S6K1-mediated RPS6 phosphorylation in a DNA-PK/AKT-dependent manner, whereas p-4E-BP1 does in a p53 and p63 activity-dependent manner [100].
AMPK both negatively and positively regulates p-RPS6. Activated AMPK has been reported to phosphorylate TSC2, which inhibits mTORC1 activity to downregulate p-RPS6 (S240//244) [96]. Conversely, in the TNBC cell line MDA-MB-231, it has been demonstrated that overexpression of peroxisome proliferator-activated receptor-γ (PPAR- γ) coactivator-1β (PGC-1β) increases the concentration of AMP to activate AMPK, which then induces p-RPTOR (S792) and p-RPS6 (S240/244) [101].
Regulation of p-RPS6 by glycogen synthase kinase-3β (GSK3β) is also complex. AMPK-priming phosphorylated GSK3β activates TSC2, leading to downregulation of p-RPS6 (S240/244) [96]. However, GSK3β also induces p-RPS6 through the activation of S6K1 by directly phosphorylating S6K1 at S371 residue in HEK293 cells in response to insulin [83].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 2.
Selected examples of the kinases and phosphatases regulating RPS6 phosphorylation.
| Protein | Effects |
|---|---|
| ABL1 (Abelson murine leukemia viral oncogene homolog 1) |
|
| AKT |
|
| AMPK (AMP-activated protein kinase) |
|
| CK2 (casein kinase 2) |
|
| DAPK1 (death-associated protein kinase 1) |
|
| DNA-PK (DNA-dependent protein kinase) |
|
| ERK1/2 (extracellular signal-regulated kinase 1/2) | |
| FGFR4 (fibroblast growth factor receptor 4) |
|
| GSK3β (glycogen synthase kinase-3β) |
|
| IKKβ (inhibitor of NF-κB kinase β) |
|
| MAP4K3 (mitogen-activated protein kinase kinase kinase kinase 3) |
|
| mTOR |
|
| P38β | |
| PDK1 (3-phosphoinositide-dependent protein kinase 1) |
|
| PERK (PRKR-like endoplasmic reticulum kinase) |
|
| PI3K p110α (phosphoinositide 3-kinase 110kDa catalytic subunit α) |
|
| PIM2 (serine/threonine-protein kinase PIM2) |
|
| PKA (protein kinase A; also known as cyclic AMP-dependent protein kinase) |
|
| PKR (protein kinase RNA-activated) |
|
| PHLPP1/2 (PH domain leucine-rich repeat-containing protein phosphatase 1/2) |
|
| PLK1 (Polo-like kinase 1) |
|
| PP2A (serine/threonine-protein phosphatase-2A) |
|
| PPM1D/WIP1 (protein phosphatase magnesium-dependent 1 delta/wild-type p53-induced phosphatase) |
|
| PTEN (phosphatase and tensin homolog) |
|
| RACK1 (receptor of activated protein kinase C kinase 1) |
|
| RSK1/2 (ribosomal protein S6 kinase 1/2) |
|
| S6K1 (ribosomal protein S6 kinase beta-1) | |
| STK33 (serine/threonine kinase 33) |
|
| ULK1 (Unc-51-like kinase 1) |
|
- (3)
- Membrane proteins that regulate RPS6 phosphorylation
Beyond receptor tyrosine kinases, membrane proteins that regulate phosphorylation of RPS6 have also been studied (Table 3). Interestingly, the programmed cell death protein 1 (PD-1)/programmed death ligand 1 (PD-L1) pathway has been found associated with p-RPS6. PD-1 binds to both RPS6 and eIF-4E to promote their phosphorylation in a hepatocellular carcinoma (HCC) cell line [148]. In addition, a recombinant PD-L1 fragment crystallizable (Fc) fusion protein induces p-RPS6 in a murine melanoma cell line in an mTOR-dependent, but not PI3K-dependent manner [149]. In contrast, PD-L1 expression has been reported to be negatively correlated with the p-RPS6 (S235/236) level in non-small cell lung cancer (NSCLC) clinical samples [150]. Since RPS6-KD induced PD-L1 expression and immune-resistance in breast and prostate cancer cells [151], further studies will clarify the potential role of p-RPS6 in immuno-oncological regulation.
- (4)
- Transcription factors that regulate RPS6 phosphorylation
Transcription factors have also been reported to regulate RPS6 phosphorylation via various mechanisms (Table 4). The Forkhead box protein O17 (FOXO17), MYC, and mutant (mt) p53P151S have been reported to induce p-RPS6 indirectly [30,153,154]. MYC transcriptionally suppressed TSC2 gene expression to activate S6K kinase activity, leading to an increase in p-RPS6 [30]. In addition, mtp53 blocked the AMPK activity through direct binding to the AMPKα subunit, leading to upregulation of p-RPS6 (S240/244) in head and neck squamous cell carcinoma (HNSCC) cells [154]. In contrast, androgen receptor (AR), basic transcription factor 3 (BTF3), and FOXO3 negatively regulated p-RPS6. The mechanism of AR- and BTF3-mediated suppression remains to be determined [155,156], whereas FOXO3 has been reported to activate TSC1 transcription [157].
- (5)
- Other proteins that regulate RPS6 phosphorylation
In addition to the above-mentioned factors, various proteins have also been reported to regulate RPS6 phosphorylation (Table 5). Notably, DNA damage-related proteins, such as DNA damage-binding protein 1 (DDB1) and DDIT4, contribute to the regulation of RPS6 phosphorylation. Mechanistically, DDIT4 negatively regulates p-RPS6 by activating TSC function [100,158].
It has also been reported that tumor suppressor proteins negatively regulate p-RPS6. Programmed cell death 4 (PDCD4) was reported to downregulate p70S6K1 phosphorylation and translation, but not p70S6K2, leading to chemosensitivity of colorectal cancer (CRC) cells to the insulin-like growth factor 1 receptor (IGF1R) inhibitor linsitinib (OSI-906) [103]. Interestingly, the expression of PDCD4 is negatively regulated by p70S6K1-mediated phosphorylation and the subsequent proteasomal degradation mediated by beta-transducin repeat containing protein (β-TrCP) [159]. In contrast, von Hippel–Lindau disease tumor suppressor (VHL) activates the PI3K/AKT/mTOR/S6K pathway by direct binding to PI3K p110, thereby upregulating p-RPS6 (S235/236) in HEK293 cells [160].
Table 5.
Selected examples of the other proteins regulating RPS6 phosphorylation.
| Protein | Effects |
|---|---|
| ADAR1 (adenosine deaminase acting on RNA 1) |
|
| AMOG (adhesion molecule in glia) |
|
| ATIC (AICAR transformylase/inosine monophosphate cyclohydrolase) |
|
| DDB1 (DNA damage-binding protein 1) |
|
| DDIT4/REDD1 (DNA damage-inducible transcript 4 protein/regulated in development and DNA damage responses 1) | |
| DRAM1 (DNA damage-regulated autophagy modulator protein 1) |
|
| eIF3 (eukaryotic translation initiation factor 3) |
|
| ETV6/RUNX1 (E/R) fusion protein |
|
| FASN (fatty acid synthase) |
|
| Gal-13/PLAC8 (galectin-13/placenta-specific 8) |
|
| GlnRS/QARS (glutaminyl-tRNA synthase) | |
| HAP1 (huntingtin-associated protein 1) |
|
| IKAP (IκB kinase complex-associated protein) | |
| JAK2 (Janus kinase 2) |
|
| LCMT1 (leucine carboxyl methyltransferase 1) |
|
| LPIN1 (lipin 1) |
|
| nSMase2 (neutral sphingomyelinase-2) | |
| p62/SQSTM (sequestome-1) | |
| PDCD4 (programmed cell death 4) |
|
| PGC-1β (peroxisome proliferator-activated receptor-γ (PPAR- γ) coactivator-1β) |
|
| PLCG1 (phospholipase C-γ1) |
|
| PME1 (protein phosphatase methylesterase 1) |
|
| RAS | |
| RB1CC1 /FIP200 (RB1-inducible coiled-coil protein 1/FAK family kinase-interacting protein of 200 kDa) |
|
| RHEB (RAS homolog enriched in brain) | |
| SHE1 (nucleoporin SHE1) | |
| SIRT1 (NAD-dependent protein deacetylase sirtuin-1) | |
| TRB3 (Tribbles homolog 3) | |
| TSC1 (Tuberous sclerosis 1 protein) |
|
| TSC2 (Tuberous sclerosis 2 protein) | |
| URM1 (ubiquitin-related modifier 1) |
|
| V-ATPase (vacuolar proton-ATPase) | |
| VHL (von Hippel-Lindau disease tumor suppressor) |
MicroRNAs That Regulate RPS6 Level
Only a few microRNAs (miRNAs) have been identified as regulators of RPS6 expression (Table 6). MiR-92b-3p and miR-451 have been reported to target TSC1 [203] and calcium-binding protein 39 (CAB39) [204], respectively, to activate mTORC1. In addition, miR-451 further activated the AMPK/mTOR pathway to induce p-RPS6 (S235/235) in CRC cells via an unknown mechanism [105]. Further studies will be needed to identify additional miRNAs involved in the regulation of RPS6.
Natural Ligands or Stimuli That Regulate RPS6 Phosphorylation
Endogenous natural ligands and stimuli also regulate RPS6 phosphorylation (Table 7). Mitogenic ligands or stimuli including amino acids, all-trans-retinoic acid (ATRA), 5α-dihydrotestosterone (DHT; androgen), and 17-β estradiol (E2) induce p-RPS6, whereas alpha-linoleic acid (ALA), hypoxia, mannitol, and oleic acid (OA) downregulate p-RPS6. The effect of hydrogen peroxide is controversial. Hydrogen peroxide has been shown to induce p-RPS6 (S235/236) in MCF7 and HCT116 cells in an mTOR-independent manner (Jia et al., 2013), but it has also been shown to downregulate p-RPS6 (S235/235) in the cytoplasm of MCF7 cells through a mechanism involving ATM/liver kinase B1 (LKB1)/AMPK/TSC2 [205]. The detailed mechanism of this differential regulation remains elusive.
Pharmacological Perturbation of the Upstream Effectors of RPS6
To date, various agents including antibodies and small molecules have been reported to perturbate the upstream effectors of RPS6 (Table 8). Antibodies against cytokines, growth factors, or membrane proteins have been reported to modulate RPS6 phosphorylation. For example, the anti-cathepsin L/MEP antibody [81] and the anti-PD-1 monoclonal antibody (mAb) [149] have been reported to inhibit RPS6 phosphorylation. Conversely, IFNα and IFNβ upregulate p-RPS6 (Table 1), and anti-IFNα/β receptor 2 antibody (anti-IFNAR2 Ab) induces p-RPS6 [87].
Notably, many small-molecule protein kinase inhibitors (PKIs) downregulate p-RPS6 by blocking specific protein kinases. Several of these PKIs, such as alpelisib, dabrafenib, everolimus, imatinib, nilotinib, rapamycin, selumatinib, trametinib, and vemurafenib, have been approved by the US Food and Drug Administration (FDA) and clinically applied to treat various diseases including cancers [213].
Unlike AMPK itself [see Section Upstream Effectors of RPS6 Phosphorylation. (2) Kinases and phosphatases that regulate RPS6 phosphorylation], all AMPK activators (5-aminoimidazole-4-carboxamide ribonucleotide (AICAR), metformin, and phenformin) downregulate p-RPS6 [105,214]. Conversely, the AMPK inhibitor dorsomorphin abolishes GSK28030371-mediated downregulation of p-RPS6 in the presence of PDGF-BB [92].
Results derived from experiments involving small-molecule PKIs have suggested new protein kinases as regulators of RPS6 phosphorylation. For example, pan-PIM kinase inhibitors such as AZD1208 [215], LGB321 [132], and SGI-1776 [133], have been reported to downregulate p-RPS6. BI2536, an inhibitor of Polo-like kinases (PLKs), also inhibits RPS6 phosphorylation [136]. These results suggest that PIM and PLKs regulate p-RPS6 either directly or indirectly. Other potential upstream regulators of RPS6 phosphorylation, such as STK33 [143] and IKK [115], may also be identified by the applications of kinase-specific PKIs.
Various protein kinases crosstalk with each other, forming both positive and negative feedback and feedforward loops (Figure 2). For example, pharmacological inhibition of mTORC1/S6K suppresses the negative feedback loops leading to the activation of compensatory upstream signaling molecules including AKT [216,217,218]. Notably, targeting mTORC1 with rapalogs as monotherapies to treat tumors has had limited success in hundreds of clinical trials [219]. Therefore, targeting RPS6 and its downstream rather than upstream effectors might be an alternative approach for cancer treatment.
Table 8.
Selected examples of the agents regulating the upstream of the RPS6 signaling.
| Agent | Effects |
|---|---|
| Antibodies | |
| Anti-cathepsin L/MEP Ab |
|
| Anti-IFNAR2 Ab (Anti-IFNα/β receptor 2 antibody) |
|
| Anti-PD-1 mAb |
|
| Small molecule inhibitors or activators | |
| AICAR (5-aminoimidazole-4-carboxamide ribonucleoside; Acadesine) | |
| Alpelisib (BYL719) | |
| AR-A014418 | |
| Aspirin |
|
| AZD1208 | |
| AZD8055 |
|
| BI2536 |
|
| Bicalutamide (Casodex) |
|
| BMS-754807 |
|
| BPTES | |
| Buparlisib (BKM120, NVP-BKM120) | |
| Butyrate |
|
| 1-butanol (1-BtOH) |
|
| C75 trans (trans-C75, (±)-C75) | |
| Caffeine | |
| CB-839 (Telaglenastat) |
|
| Cisplatin | |
| Cocaine |
|
| Compound 44 | |
| CX-5011 |
|
| Cycloheximide |
|
| Dabrafenib (GSK2118436) | |
| Dactolisib (BEZ235) | |
| Deferasirox | |
| 2-deoxyglucose (2-DG) |
|
| Dexamethasone | |
| DG2 | |
| Dorsomorphin (Compound C) |
|
| Doxorubicin |
|
| Etoposide | |
| Everolimus (RAD001) | |
| FRI00705 | |
| FTI 277 | |
| FMK | |
| Forskolin (colforsin) | |
| GNE-140 | |
| GSK28030371 |
|
| Imatinib (STI571, CGP057148B, Gleevec) |
|
| IR (ionizing radiation) |
|
| KN93 | |
| KU-0063794 | |
| LiCl (lithium chloride) |
|
| Linsitinib (OSI-906) | |
| LGB321 | |
| Lonafarnib (SCH66336) | |
| luteolin |
|
| LY294002 |
|
| Mesalamine (5-aminosalicylic acid, 5-ASA) | |
| Metformin | |
| MK-2206 | |
| ML281 | |
| Nilotinib (AMN-107) | |
| Okadaic acid | |
| Omipalisib (GSK2126458) | |
| Oxamate |
|
| L-norvaline | |
| PD184352 (CI-1040) | |
| Phenformin | |
| PI-103 |
|
| PIK-75 |
|
| PF-4708671 |
|
| PKC412 | |
| PMA (phorbol 12-myristate 13-acetate) | |
| Propranolol | |
| RAME (rosmarinic acid methyl ester) | |
| Rapamycin |
|
| Resveratrol |
|
| Sapanisertib (MLN0128, INK 128, TAK-228) | |
| SB415286 | |
| Selumetinib (AZD6244) | |
| SGI-1776 |
|
| Silmitasertib (CX-4945) | |
| SRT2183 |
|
| Topotecan | |
| Torin 1 |
|
| Torkinib (PP242) | |
| Trametinib (GSK1120212) | |
| U0126 | |
| Vemurafenib (PLX4032) |
|
| Wedelolactone | |
| WYE-354 | |
| YM-024 | |
2.3. Subcellular Localization of RPS6
As a component of the 40S ribosome, RPS6 is transported from the cytoplasm, where its translation occurs, to the nucleolus, where it is assembled into the 40S ribosome [321]. Then, the 40S ribosome is released to the nucleoplasm before its transport into the cytoplasm through the nuclear pores. RPS6 has three putative NLSs (Figure 1), and removal of all these NLSs results in the failure of its nuclear import [322]. The functions of RPS6 in all these cellular locations remain largely unknown.
In addition, p-RPS6 has been found in both the cytoplasm and nucleus [69], but differential nuclear/cytoplasmic distribution of p-RPS6, which depends on the phosphorylated sites, has also been demonstrated [323] with still unknown physiological significance. The nucleocytoplasmic localization of total (t)-RPS6 and p-RPS6 (S240/244) has been investigated. In TSC1 wildtype MEFs, the level of p-RPS6 (S240/244) was regulated in a cell cycle-dependent manner; the lowest levels were observed in the G1 phase, a strong increase and a moderate decline in S phase, and the second peak in the G2/M phase [324]. The levels of p-RPS6 and the accompanying t-RPS6 were increased in the nucleus during the G1 phase. The nuclear level of p-RPS6 reached the maximum in the early S phase, whereas the level of t-RPS6 rapidly declined. Concomitantly, the cytoplasmic RPS6 was strongly upregulated and peaked in the mid (t-RPS6)-to-late S (p-RPS6) phase.
2.4. Proteasome-Dependent Degradation of RPS6
RPs have been found excessively expressed beyond the amount required for efficient ribosome biogenesis, and their levels are controlled through continuous proteasomal degradation in the nucleoplasm [325]. However, the corresponding E3 ligase responsible for RPS6 stability has not been identified yet. Although the Drosophila Pallbearer (PALL) has been identified as an F-box protein that regulates proteasome-dependent RPS6 turnover [326], the role of the human homolog has not been investigated yet (see Section 3.2.11. Functions of RPS6 in Other Higher Eukaryotes).
Cyclic accumulation of RPS6 in the nucleolus, in accordance with the cell cycle, has been reported; it starts in the S phase, culminates in the G2 phase, and diminishes in the M phase with the disintegration of the nucleoli [327]. In addition, mammalian RPS6 is dephosphorylated upon heat shock [328,329]. Heat shock protein 90 (HSP90) has been reported to interact with RPS3 and RPS6 and protect them from proteasomal degradation [330]. Consistently, the HSP90 inhibitor geldanamycin blocked the HSP90-RPS6 interaction and induced the degradation of RPS6. However, the mechanism for the regulation of RPS6 stability during cell cycle progress and by phosphorylation remains elusive.
Downregulation of t-RPS6 has been found in TNBC cells treated with the EGFR inhibitor (EGFRi) gefitinib and the MET inhibitor (METi) SU11274 [21] (see Section 5.2.1. RPS6-KD in Breast Cancer Cells). Although the reduction in t-RPS6 level was augmented until 16 h post-treatment, 4 h treatment of the cells with the proteasome inhibitor MG132 was not sufficient to inhibit the reduction in t-RPS6 level in the presence of gefitinib and SU11274 [21]. Further studies will be necessary to reveal the role of RPS6 stability in the combinatorial PKI therapies against cancer.
3. Functions of RPS6
3.1. Ribosome Biogenesis and Protein Synthesis
RPS6 plays a role in the maturation of pre-rRNA. The siRNA-based RPS6-KD in HeLa cells resulted in the accumulation of 30S pre-rRNAs and the decrease in mature 18S rRNAs without perturbing the formation of mature 28S and 5.8S rRNAs [331]. Similar to this observation, in a mouse model with conditional deletion of both Rps6 alleles in liver, the decease in 18S rRNAs was observed, and 34S pre-rRNAs (equivalents of human 30S pre-rRNAs) were accumulated in liver cells [332]. However, the hepatic hypertrophy but not hyperplasia in fasted animals in response to nutrients was not blocked by the absence of RPS6. A recent study has demonstrated that p-RPS6 is involved in the endonuclease processing of 30S pre-rRNAs in the nucleolus [327]. The splicing of 30S pre-rRNAs was only mediated by fully phosphorylated RPS6 in the C-terminal five serine residues. Additional genetic manipulations of RPS6, including tissue-specific knockouts of both alleles or conditional deletion of one allele, further support its indispensable role in the ribosome in the thymus, spleen, and lymph nodes [333]. More importantly, Rps6 gene haploinsufficiency leads to embryonic lethality during gastrulation, preceded by failure in entering mitosis and induced apoptosis [334]. As in heterozygous Rps6 T cells [333], the embryonic lethality of heterozygous Rps6 mice might be due to a p53-mediated checkpoint during gastrulation [334].
P-RPS6 controls translation at the level of mRNA-binding in dividing cells [335,336,337]. P-RPS6 (S235/236) is also induced by RAS/RAF/ERK/RSK signaling, and p-RPS6 is recruited to the 7-methylguanosine cap structure [48].
RPS6 has been reported to play a role in the translation of mRNAs with a polypyrimidine tract at their 5′-terminal oligopyrimidine track (5′-TOP mRNAs) [338]. High levels of RPS6 have been found in primary diffuse large B-cell lymphoma (DLBCL) cell lines and patient samples. RPS6-KD reduced the proliferation of DLBCL cell lines [338] and increased the 5′-TOP mRNA translation [338,339]. A study reports that the translation of 5′-TOP mRNAs is downregulated by rapalog treatment, suggesting a role of p-RPS6 in the regulation of 5′-TOP mRNA translation [93]. Another study has reported that MEFs with the non-phosphorylated mutant RPS6 in all five serine sites (rpS6P−/−) and 70 kDa S6K1-null (p70S6K1−/−) display efficient translation of 5′-TOP mRNAs in response to mitogens [90]. RPS6 has been found to associate with 5′-TOP mRNAs, such as RPS8, RPL11, RPL16, and RPS24, through the 5′-TOP sequences to inhibit their translation [338]. RPS6-KD in the human breast cancer cell MCF7 and the human cervical carcinoma cell HeLa increases the number of these 5′-TOP mRNAs in actively translating polysomes.
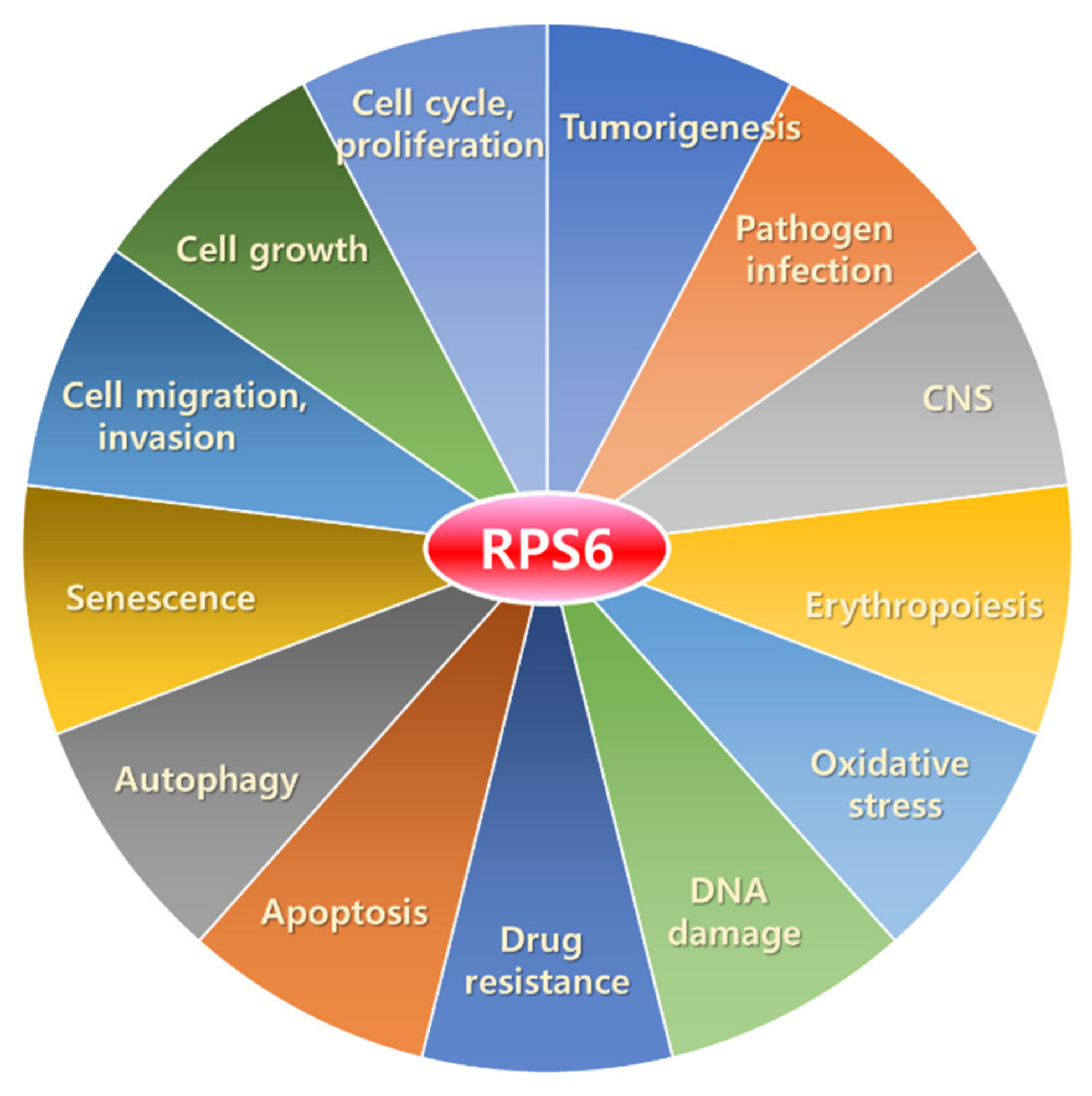
3.2. Extra-Ribosomal Functions of RPS6
In addition to the roles in translational control, RPS6 has extra-ribosomal functions (Figure 3). A study has reported that approximately 5% of endogenous RPS6 is detected in ribosome-free subcellular fractions [340]. Here, we briefly describe the functions of RPS6 in various cellular processes.
3.2.1. Regulation of Cell Cycle, Proliferation, and Growth
The relationship between p-RPS6 and cell cycle regulation was first identified in Xenopus eggs [341]. Hyperphosphorylation of RPS6 was correlated with the oncogenic RAS-induced cell-cycle arrest.
RPS6 deficiency has been demonstrated to induce the p53-dependent cell-cycle arrest that is antagonized by the depletion of p53 [339]. This induction of p53 is dependent on the upregulation of ribosomal protein L11 (RPL11). Loss of RPS6 disrupts the 40S ribosome biogenesis, leading to selective upregulation of the translation of 5′-TOP mRNAs [339]. RPL11, which is translated from one of these 5′-TOP mRNAs, binds to MDM2, the E3 ligase of p53. Under normal conditions, the activity of the tumor suppressor p53 is tightly regulated by MDM2, through a ubiquitin-dependent proteasome pathway [342,343]. RPL11-MDM2 association stabilizes and activates p53 by preventing MDM2-p53 binding [6]. Induction of p53 led to the expression of its target genes including cyclin-dependent kinase inhibitor 1a (Cdkn1a; the gene encoding p21), BCL2-associated X (Bax), and Mdm2, in the liver of Rps6-deleted mice [339]. Previous results also support the p53-dependent cell-cycle arrest and apoptosis in other tissues with the deletion of one Rps6 allele [333,334]. Notably, RPS6-KD is also known to upregulate RPL11 in the human breast cancer cell line, MCF7, and the human cervical carcinoma cell line, HeLa [338].
Failure of hepatic cell proliferation or cyclin E induction after partial hepatectomy has been reported in RPS6-deficient mice despite the presence of active cyclin D-CDK4 complexes [332]. These results suggest the induction of a checkpoint control by abrogation of 40S ribosome biogenesis, leading to the prevention of cell cycle progression. The depletion of RPS6 induced the p53-dependent cell-cycle arrest [339]. Newborn rpS6P−/− mice have been demonstrated to contain high DNA content [77]. In addition, rpS6P−/− MEFs displayed a shorter population-doubling time with a short G1 phase compared with those of the wild-type MEFs. However, the mechanism of its regulation of cell proliferation remains to be determined.
The suppressive effect of short hairpin RNA (shRNA)-based RPS6-KD on cell proliferation has been observed to increase with time in two lung cancer cell lines, A549 and H520 [37]. Inversely, the expression of senescence-associated β-galactosidase (SA-β-gal) was increased by RPS6-KD in both cell lines. Additionally, RPS6-KD increased the number of cells in the G0/G1 phase and decreased the number of cells in the G2/M phase, concurrently with reduced levels of p-RB and cyclin D1, whereas no significant changes were observed in the levels of cyclin A, cyclin E, and total RB. More interestingly, the levels of CDK inhibitors (CKIs), including p16, p21, p27, and p57, were increased. However, RPS6-KD did not induce apoptosis and altered the expression of B-cell lymphoma-extra large (BCL-xL), BAX, and caspase-3. The anticancer effect of RPS6-KD was reproduced in a xenograft model: A549 lung cancer cells with RPS6-KD resulted in reduced tumorigenicity with an increase in the number of SA-β-gal(+) cells in xenograft tissues. Similar changes were observed in the expression of cell cycle regulators and CKIs [37].
Functional implications of RPS6 in cell cycle regulation were also demonstrated by RPS6-KD in a variety of cancer cells. RPS6-KD induced G0/G1 cell cycle-arrest in human NSCLC cells [37,38], ovarian cancer cells [40], and drug-resistant melanoma cells [39] (see Section 5.2. RPS6-KD in Cancer Cells).
The levels of p-RB, cyclin D1, cyclin E, CDK2, CDK4, or CDK6 by RPS6-KD were reduced in NSCLC cells [38], ovarian cancer cells [40], and drug-resistant melanoma cells [39]. Hypophosphorylated RB inhibits cell cycle progression by binding to the transcription factor E2F and repressing the transcription of E2F target genes that are required for G1-to-S transition [36]. The phosphorylation of RB is mediated by the cyclin D/CDK4/6 complex and derepresses the transcription of E2F target genes [36]. It remains to be studied how RPS6 regulates the G0/G1 checkpoint regulators in NSCLC, ovarian cancer cells, and melanoma cells. In contrast, the levels of the CKIs p21 and p27 were increased by RPS6-KD in NSCLC cells [38]. These CKIs inhibit CDK activity either by disrupting the CDK4/6-cyclin complexes (p16) or by binding to both the cyclin and CDK in the complexes (p21, p27, and p57) [344]. Inversely, overexpression of RPS6 in the normal human bronchial epithelial (HBE) cell line promoted the cell proliferation with concurrent increases in the levels of p-RPS6, p-RB, cyclin D1, cyclin E, CDK2, and CDK4 and decreases in CKIs, p21 and p27, and the number of cells at the G0/G1 phase [38].
It has been demonstrated that cell growth (increase in size/volume) is regulated mainly by the mTORC1 pathway. RPS6 phosphorylation by S6K1 has been demonstrated to be directly involved in the positive control of cell size. Various cells derived from rpS6P−/− mice, including MEFs, fetal liver cells, pancreatic β-cells [345], and muscle myotubes [78], have been reported to be significantly smaller than the wild-type controls. However, pancreatic acinar cells displayed a similar size regardless of S6K1 deficiency [346] or RPS6 phosphorylation defect [77].
3.2.2. Regulation of Cell Migration
The migration of NSCLC cells is also reduced by RPS6-KD, which has been shown to downregulate the level of proteins involved in cell migration, including N-cadherin, vimentin, matric metallopeptidase-9 (MMP-9), MMP-2, and p-paxillin. Conversely, E-cadherin is increased by RPS6-KD [38]. In contrast, the migration of HBE cells is enhanced by RPS6 overexpression. Additionally, RPS6 overexpression upregulates N-cadherin, vimentin, MMP-2, and p-paxillin in HBE cells [38]. Induction of the epithelial–mesenchymal transition (EMT) markers suggests that overexpression of RPS6 contributes to metastasis of cancers [347].
Cell migration is also linked to angiogenesis and the tumor microenvironement. In fact, activation of the mTOR/RPS6 pathway has been associated with cancer cell survival, inflammation, and neoangiogenesis through various upstream regulators [84,95,310,348]. Since tumor growth is a result of the constant crosstalk between a tumor and its surrounding microenvironment, leading to neoangiogenesis and immune escape, further studies on the role of RPS6 in this crosstalk contribute to developing additional strategies combining anti-angiogenic therapy and immunotherapy against cancer [349,350].
3.2.3. Regulation of Apoptosis
Unphosphorylated RPS6 has been demonstrated to induce apoptosis via a mechanism involving the tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) by inducing the expression of death receptor 4 (DR4) [340] (see Section 5.2.2. RPS6-KD in Cervical Carcinoma Cells and Section 5.2.7. RPS6-KD in Hematopoietic Cancer Cells). MEFs that express unphosphorylated RPS6 have been demonstrated to be more sensitive to TRAIL-induced apoptosis than the control MEFs. Similarly, the human HCC cell line, SK-HEP-1, expresses a low level of p-RPS6 and is more vulnerable to TRAIL-induced apoptosis than other tumor cells that express a high level of p-RPS6. Consistently, ectopic expression of the phospho-defective RPS6 mutant (RPS6SS235/236AA) in HeLa cells resulted in an increase in TRAIL-induced apoptosis compared to that of the phospho-mimic RPS6 mutant (RPS6SS235/236DD) [340]. Conversely, the overexpression of RPS6SS235/236DD had no effect on the TRAIL-induced apoptosis. Since S6K1, the upstream kinase of RPS6, has anti-apoptotic activity, other apoptotic regulators, rather than RPS6, may contribute to the S6K1-dependent apoptosis induced by TRAIL [340]. More importantly, the N-terminus of RPS6 (aa 1–70) without the phosphorylation residues has been reported to carry pro-apoptotic activity to sensitize HeLa cells to TRAIL. These results imply a phosphorylation-dependent negative regulatory effect of the C-terminus of RPS6 on the pro-apoptotic activity of RPS6 [340].
DNA damage-regulated autophagy modulator protein 1 (DRAM1) is a direct target of p53 during DNA damage. It induces autophagy and is essential for the p53-dependent apoptosis [351]. Recently, negative regulation of p-RPS6 by DRAM1 has been reported [166]. Overexpression of DRAM1 downregulated the levels of p-RPS6 (S235/236) and p-RPS6 (S240/244) in an mTORC1-dependent manner in HEK293T cells. DRAM1 was also found to be localized at the plasma membrane to regulate IGF1R phosphorylation. Additionally, the overexpression of DRAM1 reduced the viability and inhibited the colony formation of the human colon cancer cell line SW480 [166]. The role of the DRAM1/RPS6 axis in apoptotic regulation remains to be determined.
3.2.4. Regulation of Drug Resistance
The overexpression of RPS6 confers intrinsic or acquired drug resistance to cancer cells [39,352,353,354,355,356,357,358,359,360]. RPS6 has been reported to confer drug resistance through nuclear factor erythroid 2-related factor 2 (NRF2) [354]. NRF2 is a master transcription factor that activates genes during oxidative stress response, detoxification, and drug resistance [361,362,363,364,365,366,367,368]. In human epidermal growth factor receptor 2 (HER2)-amplified gastric cancer (GC) cell lines with resistance to the HER2 inhibitors (HER2is) lapatinib and trastuzumab, RPS6-KD reduced the cell viability and the cellular resistance to HER2is [354]. More importantly, RPS6-KD led to downregulation in the levels of mRNAs of NRF2 target genes such as aldo-keto reductase (AKR) family 1 member B10 (AKR1B10), C1 (AKR1C1), and C2 (AKR1C2). The RPS6-NRF link was further confirmed by pharmacological inhibition of p-RPS6 in a xenograft model; treatment of the xenografted animals with the PI3K/mTOR inhibitor GSK458 alongside lapatinib reduced the growth of the HER2i-resistant tumor concurrently with downregulation of p-RPS6 and NRF2 proteins and the AKR1C1, AKR1C2, and AKR1B10 mRNAs in the xenograft tumor [354]. Human AKRs are NAD(P)H-dependent oxidoreductases, and their overexpression leads to drug resistance [369]. Previously, RPS6 has been reported to induce the translation of NRF2 by binding to the NRF2 mRNA through interaction with Sjögren syndrome antigen B (SSB) [370]. Notably, HER2 induced drug resistance in human breast cancer cells by direct physical binding to and activation of NRF2 [361].
3.2.5. Roles in the DNA Damage Response
The expression of rpS6P−/− in mice with the oncogenic Kristen rat sarcoma (KRAS) gene background has shown an increase in p53 expression along with the increased staining of phosphorylated H2A histone family member X (γ-H2AX) and p53-binding protein 1 (53BP1) in areas of acinar ductal metaplasia [17]. First, the administration of rapamycin, the selective inhibitor of mTORC1, once every other day for 1 month to mice implanted with a 7,12-dimethylbenz[a]anthracene (DMBA)-soaked cotton pledget in the pancreas significantly decreased the score of pancreatic intraepithelial neoplasia (PanIN) lesions compared with the score in the untreated mice. Histological examination has revealed that no RPS6 phosphorylation is observed in the pancreas of DMBA-treated mice when treated with rapamycin, whereas strong p-RPS6 is observed without rapamycin treatment. Transgenic mice expressing the oncogenic KRASG12D in the pancreatic epithelium develop PanIN lesions that infrequently progress to pancreatic ductal adenocarcinoma (PDAC) [371]. These mice have a strong and uniform expression of p-RPS6 in the acinar cells [17]. Additionally, rpS6P−/− mice treated with DMBA showed attenuated development of PanIN lesions. Furthermore, mice with both a KRASG12D mutation and rpS6P−/− had a significantly lower score of PanIN lesions compared with the score in the mice with a KRASG12D mutation and one or two wild-type RPS6 alleles. Further analysis revealed that frequent p53-positive cells were detected in mice with both KRASG12D mutation and rpS6P−/−. Finally, the DNA damage markers, γ-H2AX and 53BP1, were highly expressed in the mice with both KRASG12D mutation and rpS6P−/−. γ-H2AX is recruited to the chromatin domains near DNA double-strand breaks (DSBs) [372] and 53BP1 is colocalized with γ-H2AX and required for p53 accumulation in response to DNA damage [373]. Taken together, these results suggest the potential role of p-RPS6 in the attenuation of DNA damage in a mutant KRAS background, leading to the reduction of p53-dependent tumor suppression [17]. In relation to this observation, BEZ235, a dual inhibitor of PI3K and mTOR, has previously been reported to downregulate p-53BP1 (S25) and 53BP1 foci induced by the poly (ADP-ribose) polymerase inhibitor (PARPi) olaparib in breast cancer type 1 susceptibility protein (BRCA1)-mutant breast cancer cells [374].
ATM, the master regulator of the DNA damage response, is a serine/threonine protein kinase [375]. Upon DSB formation, ATM is phosphorylated and activated to induce cell-cycle arrest, DNA repair, or apoptosis. The S247 of RPS6 is a substrate of ATM in response to UV irradiation or the treatment with the genotoxic drug doxorubicin and is inhibited by the ATM inhibitor KU-55933 [57]. ATM-mediated RPS6 phosphorylation has been reported to be S6K-independent.
Phosphorylation of RPS6 has been reported to attenuate DSBs in BRCA1-deficient breast cancer cells both in vitro and in vivo [376]. In olaparib-resistant BRCA1-deficient breast cancer cells, ionizing radiation (IR) decreased the number of γ-H2AX foci and increased the number of DNA repair protein RAD51 homolog 1 (RAD51) foci compared with the numbers of them in the parental cells. A remarkable increase in p-RPS6 is the distinct feature of olaparib-resistant BRCA1-deficient cells, and abrogation of RPS6 phosphorylation completely reverses the formation of foci for γ-H2AX or RAD51. These results suggest that p-RPS6 is crucial to load the RAD51 recombinase onto DNA damage sites in BRCA1-deficient cells. In fact, BRCA1 is indispensable for RAD51 loading onto DNA damage sites, leading to homologous recombination repair (HRR) [377]. More importantly, data support that p-RPS6 confers olaparib-resistance to BRCA1-deficient breast cancer cells [see Section 4.3.4. RPS6 in Resistance to PARP Inhibitors (PARPis)].
3.2.6. Response to Oxidative Stress
Insulin has been demonstrated to induce the interaction of mTORC2 with the ribosome, leading to enhancement of mTORC2 activity in a translation-independent manner [378]. It has been reported that RPS6 plays a cardioprotective role in response to oxidative stress [52]. IPC, and insulin or opioid treatment, induced phosphorylation of RPS6 at S235/236 through the AKT/mTORC1/S6K pathway in perfused mouse hearts or neonatal rat ventricular myocytes. The p-RPS6 interacted with the rapamycin-insensitive companion of mTOR (RICTOR) to enhance mTORC2 kinase activity. RPS6-KD reduced the insulin-induced phosphorylation of mTORC2 and AKT (S473). Inversely, RPS6 overexpression upregulated p-AKT (S473). Additionally, RPS6-KD abrogated insulin-induced cardioprotection against the H2O2-induced oxidative stress [52]. Interestingly, S6K1 negatively regulates mTORC2 via the phosphorylation of RICTOR in the mouse adipocyte cell line 3T3-L1 [379]. Taken together, mTORC2 activity may be tightly controlled by RPS6-mediated positive feedback and S6K-induced negative feedback.
3.2.7. RPS6 in Cellular Senescence
Cellular senescence is one of the nine hallmarks for aging, which is characterized by constant G1 cell-cycle arrest and an inflammatory response, the senescence-associated secretory phenotype (SASP) [380,381,382]. The mTORC1/S6K pathway has been recognized as the key signaling pathway responsible for aging and cellular senescence [383,384]. Since RPS6 is the downstream effector of this mTORC1/S6K pathway, p-RPS6 has been used as a marker for aging and premature senescence [385].
Contrary to this notion, a study found that there are no significant differences in the levels of mTOR or p-mTOR (S2448) between young and old human dermal fibroblasts (HDFs), whereas downregulation of S6K1, p-S6K1 (T389), and p-RPS6 was observed in replicative senescent HDFs [386]. In addition, p-RPS6, S6K1, and p-S6K1 were also downregulated in HDFs with autophagy impairment-induced premature senescence (AIPS) that was induced by siRNA-based knockdown of the autophagy related 7 (ATG7) or lysosome-associated membrane glycoprotein 2 (LAMP2). Interestingly, reactive oxygen species (ROS) scavenging (through treatment of the antioxidant N-acetyl cysteine (NAC)) or p53 inhibition (either through treatment of a p53 inhibitor, pifithrin-α, or knockdown) restored the levels of p-RPS6, S6K1, and p-S6K1 in AIPS HDFs. However, the exact roles of RPS6 and its phosphorylation in cellular senescence have not been elucidated yet.
3.2.8. Roles of RPS6 in Erythropoiesis
Congenital (Diamond-Blackfan anemia; DBA) and acquired (5q-syndrome) hypoproliferative macrocytic anemia share a common erythroid phenotype of RP haploinsufficiency [387]. Although mutations of the RPS6 gene have not been reported in this anemia, mice lacking one Rps6 allele postnatally display features of the 5q-syndrome, such as macrocytic anemia, erythroid hypoplasia, and megakaryocytic dysplasia with thrombocytosis [388]. Additionally, mice with heterozygously deleted Rps6 have also been demonstrated to phenocopy the 5q-syndrome [389]. However, the clinical significance of these findings remains elusive.
3.2.9. Roles of RPS6 in the Central Nervous System
Increasing evidence indicates p-RPS6 as a marker of neuronal activation during synaptic plasticity [390,391]. Various pharmacological stimuli also induced the p-RPS6 in neurons [392]. For example, massive phosphorylation of RPS6 at S235/236 and S240/244 by a proconvulsant drug, such as kainite, pilocarpine, pentylenetetrazol, or dopamine D1 receptor (DRD1) agonist SKF81297, has been demonstrated. In addition, drugs of abuse and antipsychotics also regulate RPS6 phosphorylation [392]. Interestingly, the p-RPS6 level in schizophrenia is reduced [393]. Elevated expression or phosphorylation of RPS6 has also been found in multiple sclerosis [394]. The roles of RPS6 in diseases of the central nervous system remain to be determined.
3.2.10. Roles of RPS6 in Response to Infection
Mounting evidence suggests the roles of RPS6 during pathogen infection [395]. The induction of p-RPS6 in HeLa cells by vaccinia virus infection was reported as early as 1976 [396].
RPS6 has been demonstrated to be associated with latency-associated nuclear antigen (LANA). LANA is a multifunctional protein of Kaposi’s sarcoma-associated herpesvirus (KSHV) that is tightly associated with primary effusion lymphoma (PEL), Kaposi’s sarcoma I, and multicentric Castleman’s disease (MCD) [397]. RPS6 binds to the N-terminal domain of LANA in a nucleic acid-independent manner in a PEL cell line, BC-3 [398]. Interestingly, RPS6 increased the transcriptional activity of the LANA protein on the LANA promoter. Moreover, shRNA-based RPS6-KD reduced the stability of the LANA protein, while increasing the stability of p53 in BC-3 cells. The half-life of the LANA protein was markedly reduced from several days [399] to 0.6 h by RPS6-KD [398]. Consistent with the increased p53 stability, the upregulation of p21 protein levels was also induced by RPS6-KD. These effects led to anti-proliferative effects in BC-3 cells [399]. The underlying mechanism of the RPS6-dependent regulation of LANA stability is subject to further study.
The open reading frame 45 (ORF45) of KSHV has been reported to induce p-RPS6 (S235/236) by directly activating RSKs through the mTORC1/S6k-dependent signaling [400]. Phosphorylation of RPS6 and eukaryotic translation initiation factor 4B (eIF4B) through the ORF45/RSK axis has been suggested to contribute to the translational control during KSHV lytic replication.
An indispensable role of RPS6 has been found in hepatitis C virus (HCV) propagation. HCV is a single-stranded RNA virus whose translation is initiated in a cap-independent manner by its internal ribosome entry site (IRES) [401]. Reduction of the 40S ribosome abundance by RPS6-KD was reported to selectively suppress HCV IRES-mediated translation without affecting the translation of the host cells [402]. Only knockdown of the 40S RPS genes, but not the 60S RPL genes, of the ribosomal subunit inhibited HCV translation.
Protein phosphatase 2A (PP2A) is a heterotrimeric serine/threonine phosphatase, which is composed of a structural A subunit, a catalytic C subunit, and >20 regulatory B-type subunits resulting in >80 different PP2A holoenzymes [403]. The composition and function of the PP2A holoenzyme are regulated through methylation and demethylation by leucine carboxyl methyltransferase 1 (LCMT1) and protein phosphatase methylesterase 1 (PME1), respectively [404]. Cellular transformation either by polyomavirus or by simian virus 40 (SV40) occurs through activation of p-RPS6 (S235/236) via inhibiting PP2A by replacing the regulatory B-type subunits from the PP2A heterotrimer by viral oncoproteins such as the polyomavirus middle (PyMT) and small (PyST) tumor antigen and the SV40 small tumor antigen (SVST) [137]. PyST and SVST preferentially bind to PP2A instead of the methylated PP2A catalytic subunit, which is mediated by LCMT1, whereas the binding of PyMT is not. The methylated PP2A downregulates p-RPS6 by inactivating AKT and S6K. The methylated PP2A also reduced p-RPS6 in an S6K-independent manner. Similarly, PyMT and PyST enhance RPS6 phosphorylation [405,406].
RPS6 phosphorylation is also increased in cells infected by pathogens such as Rift Valley Fever (RVF) Virus, Herpesvirus, Plasmodium, and Toxoplasma [407,408,409,410]. Pharmacological inhibitions of the mTORC1/S6K/RPS6 pathway inhibits the infection or pathogenesis of these pathogens [407,408]. Additionally, p-RPS6 is activated by an unknown regulator of the non-canonical pathway in Plasmodium-infected hepatocytes [407]. These data suggest that parasites override the RPS6 pathway to support their growth in infected cells.
3.2.11. Functions of RPS6 in Other Higher Eukaryotes
In the plant Arabidopsis, RPS6 negatively regulates rDNA transcription by associating with the histone deacetylase, AtHD2B [411], which might be antagonized by the interaction of RPS6 with the histone chaperone, AtNAP1 [412].
RPS6 has been identified as a negative regulator of efferocytosis in Drosophila. Efferocytosis is a process of clearing apoptotic cells via phagocytosis by professional or amateur phagocytes [413]. Since approximately 3 × 1011–5 × 1011 cells in our body die daily via apoptosis, clearing dead cells is crucial to maintain tissue homeostasis [414,415]. PALL is an F-box protein in Drosophila, and a component of the Skp-Cullin-F-box (SCF) E3 ubiquitin ligase complex [416]. PALL has been found to bind to p-RPS6 to promote RPS6 ubiquitylation and proteasomal degradation [326]. Degradation of RPS6 promoted efferocytosis through Rac family small GTPase 2 (RAC2) activation and F-actin remodeling in Drosophila cells. Consistently, RPS6-KD enhanced the efferocytosis and RPS6 overexpression reduced the clearance of apoptotic cells. Although F-box only protein 28 (FBXO28) has been identified as a human homolog of PALL [417], whether FBXO28 binds to and degrades p-RPS6 in mammalian cells remains to be determined.
4. RPS6 in Cancer
Although it still needs further investigation whether p-RPS6 is just a byproduct of tumorigenic pathway activation or a prerequisite for tumorigenesis in various types of cancers, we provide evidence here supporting the potential roles of RPS6 in tumorigenesis in humans and suggesting this protein as a potential therapeutic target against cancer.
4.1. Roles of RPS6 in Tumorigenesis
A knock-in mouse model of a non-phosphoylatable RPS6 mutant (rpS6P−/−) has demonstrated that p-RPS6 is indispensable to develop PDAC in KRASG12D mutation background [17]. The importance of p-RPS6 in tumorigenesis was further demonstrated in mice expressing constitutively active AKT (MyrAKT1) in pancreatic β-cells in the background of rpS6P−/−. Defects in the phosphorylation of RPS6 rescued the MyrAKT1-induced reduction in the nuclear level of p27 [418], a CKI [419,420]. In addition, p-RPS6 deficiency reduced the development of the MyrAKT1-induced hyperplasia and tumor formation in β cells (insulinoma). RPS6 phosphorylation deficiency also increased the overall protein synthesis with concomitant reduction in translation fidelity, leading to unexpected resistance to proteotoxic (MG132) or genotoxic (etoposide) stress-induced apoptosis in MyrAKT1-expressing cells. However, p-RPS6 deficiency failed to inhibit, and instead enhanced, the MyrAKT1-triggered aneuploidy, and increased the number of β cells and amount of insulin secretion [418]. Although several nuclear proteins, including PC4 and SFRS1-interacting protein 1 (PSIP1), serine/arginine-rich splicing factor 1 (SRSF1), and DNA topoisomerase IIβ (TOP2B), have been identified as binding partners of unphosphorylated RPS6, the functional significance of these interactions has not been elucidated yet [418]. Since rpS6P−/− could not prevent thymic lymphomatogenesis in mice with a constitutively active AKT2 in immature T cells [421], the anti-tumorigenic effect of rpS6P−/− may be tissue-specific.
P-RPS6 has been reported to promote the translation, not transcription, of hypoxia-inducible factor 1-alpha (HIF1α) upon the activation of an environmental carcinogen, arsenite, leading to carcinogen-induced transformation of the normal mouse epidermal Cl41 cells [422]. Arsenite treatment induced the increase in p-RPS6 (S235/236). P-RPS6-dependent HIF1α expression was negatively regulated by the CKI, p27, via the RAS/RAF/MEK/ERK/RSK pathway, but not via the AKT/S6K pathway. P27 is a cell cycle regulator that binds and inhibits cyclin D/CDK4 [423,424], cyclin D/CDK6 [425], and cyclin E/CDK2 complexes [426]. CDKN1B (the gene encoding p27)-KD induced p-RPS6 even in the absence of arsenite. Arsenite-induced p-RPS6 was augmented by CDKN1B-KD and in an arsenite dose-dependent manner. Mutational analyses further demonstrated that the induction of HIF1α translation in p27-deficient cells was p-RSP6 (S235/236)-dependent. Interestingly, CDKN1B depletion abrogated the expression of PHPLL, a negative regulator of RAS [427], RAF1 [428], and AKT [429,430], resulting in the activation of the RAS/RAF/MEK/ERK/RSK pathway [422]. Notably, SRC, the upstream regulator of the RAS/RAF/MEK/ERK pathway, phosphorylates p27 and induced its proteolysis [431]. In addition, hypoxia is an important inducer of neoangiogenesis, and activated RPS6 may play an important role in the crosstalk between endothelial cells and tumor cells in the tumor microenvironment [84,95,310,348]. For example, it has been reported that RPS6 is activated by tumor microenvironment conditions and hypoxia in endothelial cells [432].
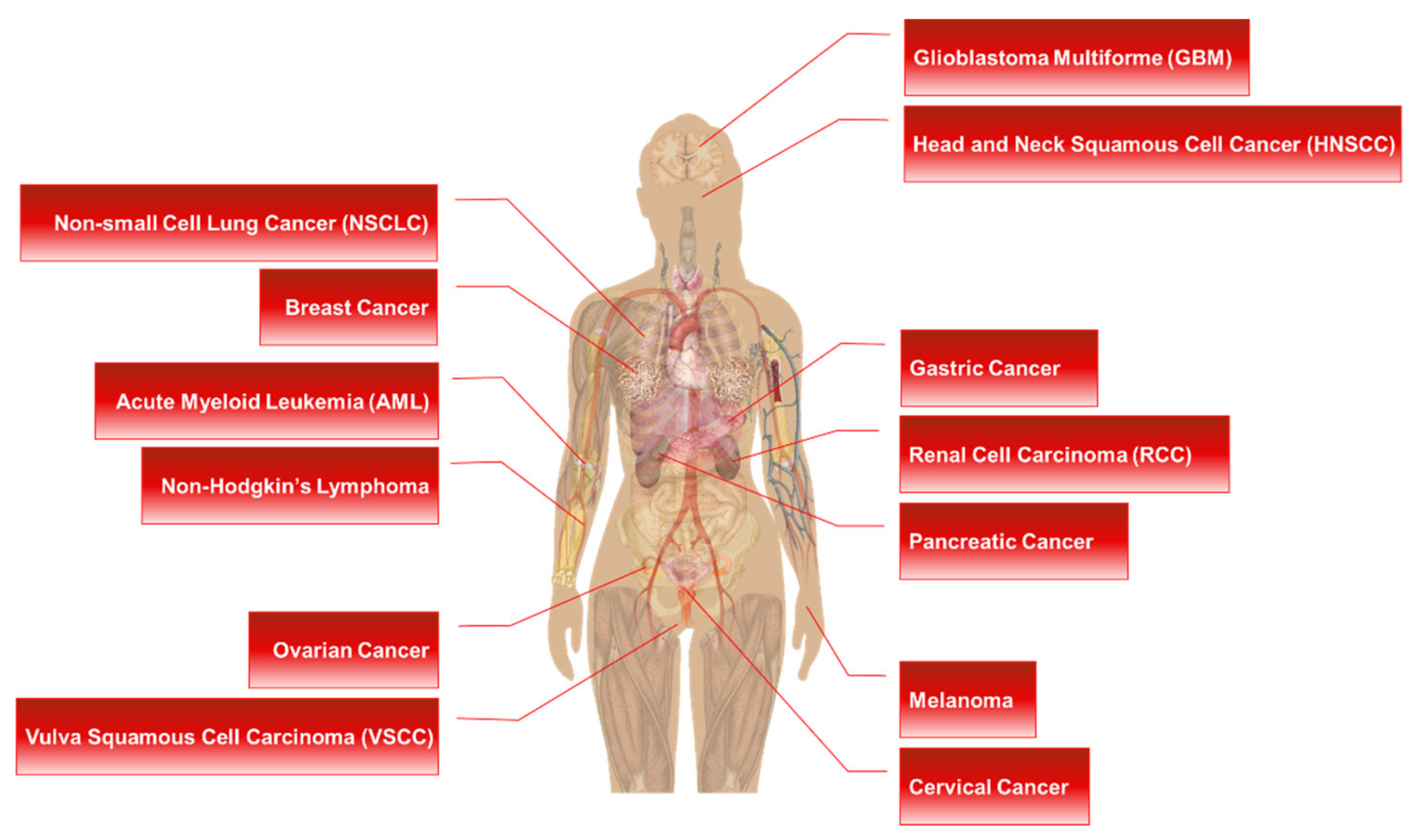
4.2. RPS6 as a Predictive Marker in Cancers
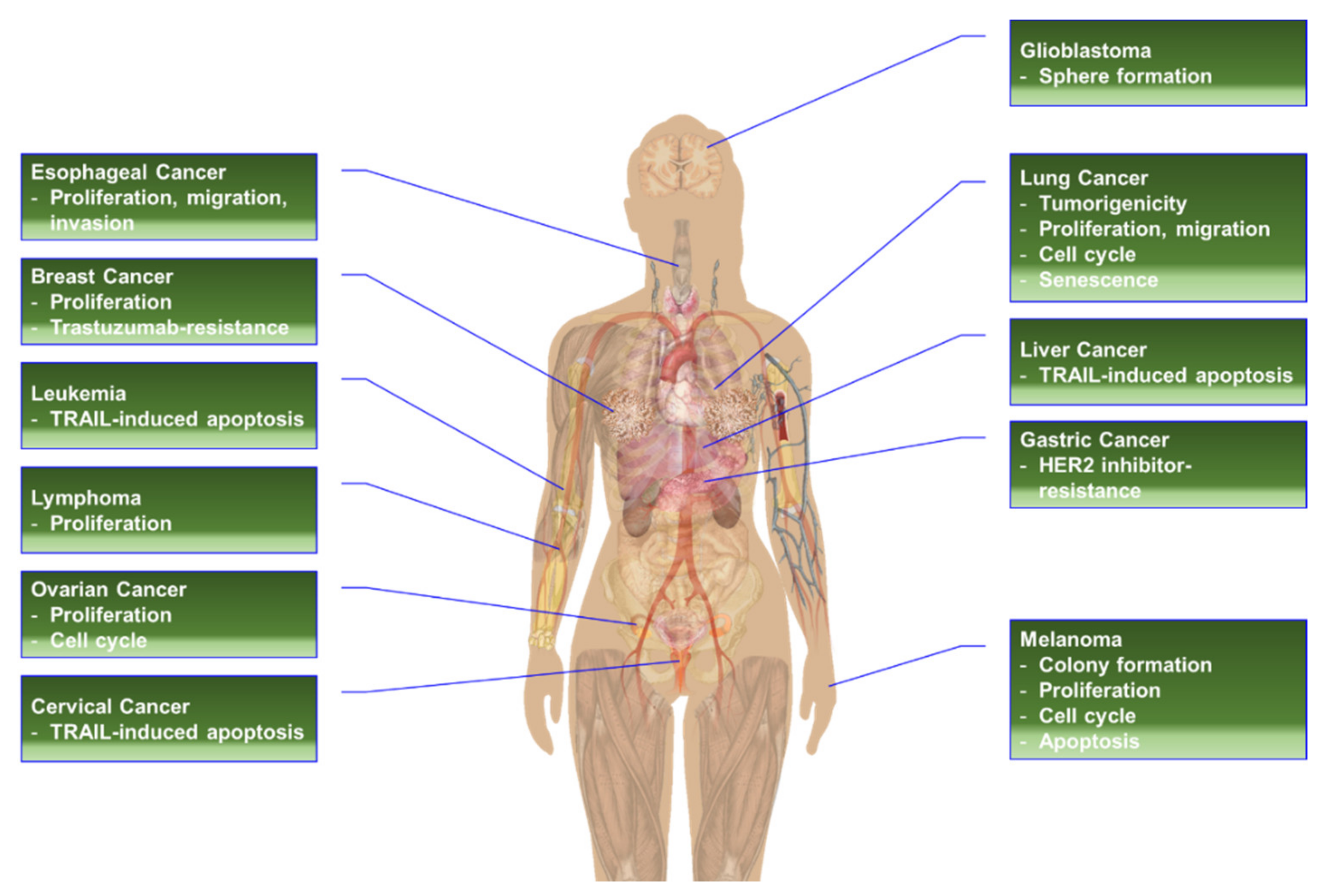
High-level phosphorylation and/or overexpression of RPS6 has been reported in various types of cancers, including acute myeloid leukemia (AML) [433], breast cancer [434], cervical cancer [435], esophageal squamous cell carcinoma (ESCC) [436], GC [437], glioblastoma multiforme (GBM) [438], HNSCC [439], melanoma [440], non-Hodgkin’s lymphoma [338], NSCLC [38], oral squamous cell carcinoma (OSCC) [441,442], ovarian epithelial cancer (OEC) [40,443], pancreatic cancers [13,17,444], renal cell carcinoma (RCC) [445], sarcoma [446], and vulva squamous cell carcinoma (VSCC) [447]. More interestingly, mTOR-independent phosphorylation of RPS6 was frequently identified in primary central nervous system lymphoma (PCNSL) and DLBCL [338,355]. PASK, but not RSK, was found as a potential kinase of RPS6 in these lymphomas [355]. Additionally, RPS6 has been proposed as a predictive biomarker in cancers. The level of RPS6 or p-RPS6 was also correlated with the pathological grade and/or disease progression in distinct human cancers (Figure 4). The change in the expression level and the status of p-RPS6 and/or t-RPS6 could be monitored by various methods to predict drug response and resistance, and disease progression after drug treatment [440].
4.2.1. RPS6 in Acute Myeloid Leukemia (AML)
The shRNA-based knockdown of CSNK1 (the gene encoding CK1) downregulated p-RPS6 (S244/247) concurrently with increased p53 activity in primary mouse MLL-AF9 leukemia cells [448]. CSNK1-KD had anti-leukemia efficacy both in vitro and in vivo. The anti-leukemic effects of CSNK1-KD were further supported by the rescue of CK1 functions by an shRNA-resistant CSNK1 cDNA, which inhibits the anti-leukemic effects of CSNK1-KD, whereas kinase-dead CK1D136N [449] cDNA did not [448]. The regulation of RPS6 activity by CK1-mediated phosphorylation has been reported previously [56]. Interestingly, CSNK1-KD also induced the levels of p53 and p21 with concurrent induction of apoptosis and G1 arrest in MLL-AF9 leukemia cells [448]. Previously, it has been demonstrated that dephosphorylation of RPS6 causes p53 activation in pancreatic cancer cells in response to DNA damage [17]. In addition, RPS6 depletion has been reported to induce p53-dependent cell-cycle arrest [339]. The p53 induction was mediated by the upregulation of RPL11, which binds to and inhibits MDM2 in RPS6 depleted cells [6,339] (see Section 3.2.1. Regulation of Cell Cycle, Proliferation, and Growth). Overexpression of a phosphomimetic mutant RPS6S5D [56] partially rescued the CSNK1-KD-induced proliferation defect in leukemia cells [448]. Taken together, the relationship between p-RPS6 and p53 in AML or other cancer cells will be an interesting topic in the future.
4.2.2. RPS6 in Breast Cancer
High levels of p-RPS6 (S235/236), but not p-ERK (T202/Y204) or p53, have been found to strongly correlate with high Ki-67 expression in ER(+)/HER2(−) breast cancer clinical samples [434]. However, there was no significant difference in relapse-free survival (RFS) between cancer patients with high p-RPS6 levels and those with low p-RPS6 levels. In addition, high-level expression of p-RPS6 (S240) correlated with high Ki67 expression and short overall survival (OS) of breast cancer patients [450].
The t-RPS6 protein has been reported to be downregulated in TNBC cells treated by the EGFRi gefitinib and the AKT inhibitor MK2206 [451]. TNBC cells, especially mesenchymal stem-like (MSL) subtype cells, have intrinsic resistance to EGFRi [21,451,452,453,454,455,456]. Treatment of gefitinib alone did not reduce the viability or colony-forming ability of the MSL subtype cells [21,451,455]. The combination of gefitinib and MK2206 induced synergistic anti-proliferative and anti-colony forming effects in HS578T and MDA-MB-231 cells. No significant suppression of the PI3K/AKT and MAPK pathways was observed in TNBC cells treated with gefitinib alone. The treatment of MK2206 alone reduced the levels of p-AKT (S473), p-mTOR (S2448), p-PRAS40 (T246), p-4E-BP1 (T37/46), and p-RPS6 (S235/236). Interestingly, MK2206 alone induced downregulation of t-RPS6 [451]. The reduction of t-RPS6 was induced by the gefitinib+MK2206 treatment in a dose-dependent manner. Since no significant change in the level of p-ERK1/2 (T202/Y204) was observed, RSK may not contribute to the t-RPS6 downregulation by the gefitinib+MK2206 treatment. Moreover, siRNA-based RPTOR-KD, but not RICTOR (rapamycin-insensitive companion of mTOR), downregulated the t-RPS6 protein and this reduction was potentiated by gefitinib addition, supporting the idea that mTORC1 controls the level of t-RPS6 in TNBC cells. RPTOR-dependent downregulation of t-RPS6 inhibited the proliferation of TNBC cells in the presence of gefitinib [451]. Although these results suggest RPS6 as an attractive therapeutic target to treat TNBC, the underlying mechanism maintaining the RPS6 level and its contribution to EGFRi resistance remain to be determined.
RPS6 has been reported to contribute to the regulation of intraductal colonization of basal-like breast cancer cells [457]. In an immortalized basal-like breast epithelial cell line, RPS6 dephosphorylation initiated the keratinization process. In addition, dephosphorylated RPS6 triggered detachment-induced keratin 5 (KRT5) upregulation post-transcriptionally. P-RPS6 was not detected in suspension cells in 3D cultures, whereas reconstitution of p-RPS6 by a constitutively active S6K mutant attenuated keratinization. Consistently, pharmacological inhibition of RPS6 phosphorylation caused sporadic keratinization in attached cells. Reduced RPS6 phosphorylation was also identified in a prolonged detached TNBC cell line, MDA-MB-468 cells, and pharmacological inhibition of p-RPS6 also caused keratinization in attached MDA-MB-468 cells [457].
4.2.3. RPS6 in Gastric Cancer
AMPK negatively regulates mTORC1 activity through direct phosphorylation of TSC2 and RPTOR (Figure 2): (1) It phosphorylates and activates TSC2, which inhibits the mTORC1 kinase activity [96,458]; and (2) it phosphorylates RPTOR, leading to its binding to 14-3-3, which suppresses the mTORC1 activity [459]. High levels of p-RPS6 and low levels of p-AMPKα have been reported to be associated with gastric tumor progression and to be independent predictors of patient survival after resection of primary cancer [437]. The median OS values of 28 vs. 73 months and 28 vs. 78 months in patients with positive vs. negative expression of p-RPS6 and with negative vs. positive expression of p-AMPKα, respectively.
4.2.4. RPS6 in Glioblastoma
High-level expression of RPS6 is strongly correlated with GBM stem cell (GSC) markers, Nestin and SRY-box transcription factor 2 (SOX2), and an oligodendrocyte progenitor cell marker, oligodendrocyte transcription factor 2 (OLIGO2), in GBM tissues [438]. High levels of RPS6 and SOX2 are detected in high-grade GMB samples compared with the levels in the low-grade samples. RPS6 may contribute to the stemness of GSC through activation of the JAK/STAT3 pathway to induce these stemness-related proteins (see Section 5.2.5. RPS6-KD in Glioblastoma Cells). RPS6-KD reduced p-JAK2 and p-STAT3 and suppressed the tumor sphere formation, a characteristic of GSCs, of GBM cells in vitro. Inversely, RPS6 overexpression facilitated the tumor sphere formation, and the STAT3 inhibitor, AG490, antagonized the RPS6-enhanced sphere formation.
4.2.5. RPS6 in Head and Neck Cancer
In patients with HNSCC, double-positive p21 and p-RPS6 present better disease-specific survival [439]. The p21-p-RPS6 double-positiveness was determined to be mTORC1-dependent but not p53-dependent. The inhibitory phosphorylation of 4E-BP1 by mTORC1 stabilizes p21 by inhibiting its degradation that is induced by its interaction with 4E-BP1 [460]. Consistently, the PI3K/AKT/mTORC1 pathway is frequently activated in HNSCC [439].
An earlier study suggested that RPS6 phosphorylation is associated with early events of OSCC tumor progression [441]. Compared with healthy control samples (15/30; 50%), clinical samples of epithelial dysplasia (15/15; 100%) and OSCC (47/53; 88.68%) have displayed higher frequencies of p-RPS6 (S40/244). High-level expression of p-RPS6 has been also identified in OSCC clinical samples [442]. P-RPS6 is correlated with p21 expression and inversely correlated with the tumor size and local infiltration.
High levels of p-RPS6 (240/244) are associated with shorter disease-free survival (DFS) than low levels in patients with ESCC [436]. There was no difference in the OS rate. The association was more significant in the early-stage ESCC patients than in the late-stage patients. In addition, the high ratio of p-RPS6/t-RPS6 resulted in a more significant correlation with adverse prognosis than the p-RPS6 level alone [436]. Interestingly, RPS6-KD resulted in anticancer effects in ESCC cell lines (see Section 5.2.3. RPS6-KD in Head and Neck Cancer Cells).
4.2.6. RPS6 in Lung Cancer
The positivity of RPS6 and p-RPS6 (S235/236) is significantly higher in NSCLC clinical samples than in normal tissues (82.4% vs. 55.8% and 62.6% vs. 53.3%, respectively) [37]. It has been correlated with shorter median OS and DFS in NSCLC patients with high p-RPS6 levels than in patients with low levels (10 months vs. 60 months and 21 months vs. 48 months, respectively). However, although the positiveness of t-RPS6 showed differences, they did not reach statistical significance (30 months vs. 43 months and 26 months vs. 40 months, respectively; p > 0.05) [37]. In a separate study, hyperphosphorylation of RPS6 (S235/236) or the ratio of p-RPS6 to t-RPS6 was reported to be a predictive marker for survival of patients with NSCLC [38]. From the analysis of patient samples, the 5-year survival rate and median OS of patients with high levels of p-RPS6 were found to be significantly lower than those with p-RPS6(-) (3.0% vs. 26.5%; 20 months vs. 42 months, respectively). The significance was greater in patients with a high p-RPS6/t-RPS6 vs. patients with a low p-RPS6/t-RPS6 (2.1% vs. 32.0%; 12 months vs. 48 months, respectively) [38]. The predictability of the p-RPS6/t-RPS6 ratio was more powerful in patients of stage I NSCLC (8 months vs. 61 months). In addition, hyperphosphorylation of RPS6 is correlated with unfavorable clinical survival outcomes in NSCLC, lung adenocarcinoma, and GC [38,437,461].
4.2.7. RPS6 in Melanoma
The suppression of p-RPS6 has been identified as a predictive marker for improved progression-free survival (PFS) in BRAF-mutant melanoma treated with RAF or MEK inhibitors. A study conducted sensitivity assays of growth inhibition and apoptosis induction for the RAF inhibitor (RAFi), vemurafenib, and the MEK inhibitor (MEKi), selumetinib, against a panel of 16 BRAF-mutant melanoma cell lines. The results suggested that the inhibition of p-ERK is necessary but not sufficient to predict the sensitivity of melanoma cell lines to the RAFi or MEKi [440]. Further analysis revealed that the sensitivity of melanoma cell lines to these drugs correlates well with the decrease in the levels of p-RPS6 (S235/236). Resistant cell lines maintain the p-RPS6 levels after RAFi or MEKi treatment. The degree of suppression of RPS6 phosphorylation was a predictable indication of the sensitivity to these drugs not only in xenograft models but also in melanoma patients in a prospective evaluation. Although the number of patients was small (reduction vs. no reduction, n = 6 vs. n = 3, respectively), the reduction of p-RPS6 levels after treatment of the RAFi vemurafenib was correlated with an approximately 5-fold improvement in PFS [440].
4.2.8. RPS6 in Ovarian Cancer
High levels of RPS6 are associated with poor OS and PFS in OEC [40]. The RPS6 protein level was evaluated in clinical tissue samples, and poor clinical outcomes were found with median OS values of 30 vs. 42 months and median PFS values of 15 vs. 21 months in patients with high vs. low RPS6 levels, respectively [40]. High levels of p-RPS6 and NOTCH3 are associated with poor prognosis and a shorter OS (12.3 months) than the OS (81.9 months) of patients with low p-RPS6 and NOTCH3 levels in OEC [443]. Although NOTCH is known to activate the PI3K/mTORC1 pathway, the details are still largely unknown [462]. In addition, the roles of RPS6 in conjunction with NOTCH-related cancers remains to be determined.
4.2.9. RPS6 in Renal Cell Carcinoma (RCC)
In RCCs, RPS6 has been identified as the key mediator of the antitumor effects of everolimus (RAD001), a rapalog [445]. Everolimus binds to the 12 kDa FK506-binding protein (FKBP12) to form a complex, which binds to mTOR to destabilize and inactivate the mTORC1 complex [348,463]. Everolimus reduces the clonogenicity and proliferation of RCC cells by blocking protein biosynthesis but does not induce apoptosis. Knockdown of RPS6, but not EIF4EBP1 (the gene encoding 4E-BP1) or CDKN1B, abolishes the everolimus-induced anti-proliferative effect and blocks translation. More importantly, high levels of RPS6 and p-RPS6 have been found inversely correlated with the survival of patients with RCC [445].
4.2.10. RPS6 in Vulvar Squamous Cell Carcinoma (VSCC)
The non-viral-associated VSCC results from genetic and epigenetic changes in the absence of papillomavirus infection. Genetic alterations in the differentiated vulvar intraepithelial neoplasia (VIN) include allelic imbalance, microsatellite instability, and TP53 (the gene encoding p53) mutations [464,465]. P-RPS6 (S235/236)-positiveness has been identified in VSCC and differentiated VINs but not in basal and parabasal cells of the healthy vulvar epithelium. Additionally, laminin γ2 expression has solely been identified within p-RPS6 positive regions [447]. The expression of laminin γ2 is not detectable via immunohistochemical analysis in the normal epithelial basal cells, whereas its expression is a well-established marker of carcinoma [466,467,468].
4.3. RPS6 in Anticancer Drug Resistance
As mentioned earlier, the overexpression of RPS6 has been implicated in intrinsic or acquired drug resistance of cancer cells (see Section 3.2.4. Regulation of Drug Resistance) [39,352,353,354,355,356,357,358,359,360]. More importantly, constitutive phosphorylation of RPS6 has been found to confer drug resistance in breast cancer cells, GC cells, and melanoma cells [39,359,360].
4.3.1. RPS6 in Resistance to HER2 Inhibitors (HER2is)
The level of p-RPS6 in trastuzumab-resistant HER2-positive (+) breast cancer cells inversely correlated with their susceptibility to HER2-targeting drugs [360]. Trastuzumab (Herceptin®), a US FDA-approved drug, is a humanized monoclonal antibody against HER2 to treat HER2+ breast cancer [469]. However, the benefit of trastuzumab is limited by intrinsic or acquired resistance [470,471]. The phospho-proteins in the PI3K/mTORC1 pathway, including p-mTOR (S2448), p-AKT (S473), and p-RPS6 (S235/236), have been tested in response to trastuzumab in both sensitive and resistant HER2+ breast cancer cells [360]. Among these proteins, the p-RPS6 level and the degree of its response to the treated drugs could predict the susceptibility of breast cancer cells to not only trastuzumab but also other drugs, including rapamycin, AZD2014, BEZ235, erlotinib, lapatinib, MK226, and OSI-906. Drugs that could not downregulate p-RPS6, such as trastuzumab, erlotinib, MK2206, and OSI-906, failed to inhibit the growth of trastuzumab-resistant breast cancer cells [360]. Moreover, the combination of trastuzumab and lapatinib further reduced the p-RPS6 level, alongside concurrent synergistic inhibition of cell growth. These results suggested the p-RPS6 level as a predictive marker for the susceptibility of HER2+ breast cancers to anti-HER2 drugs and a feasibility marker of drugs for a combination strategy for the trastuzumab-resistant HER2+ breast cancers [360].
4.3.2. RPS6 in Resistance to MEK Inhibitors (MEKis)
Selumetinib (KoselugoTM) is a very recently FDA-approved MEK1/2 inhibitor for patients with neurofibromatosis [472]. Anticancer activity of selumetinib has been tested in a panel of 12 CRC cell lines [353]. The study found that the levels of p-S6K1 (T3889) and p-RPS6 (S235/236) remained unaffected or increased by selumetinib treatment in cell lines with intrinsic resistance. Resistant primary tumors from patients also demonstrated a similar response. Pharmacological inhibition of the PI3K/AKT/mTORC1/S6K1 pathway or S6K1-KD overcame the selumetinib resistance in these CRC cells [353].
The phosphorylation level of mTORC1 downstream effectors, such as S6K1, 4E-BP1, and RPS6, has been found to be associated with resistance to the MEKi PD0325901 in a panel of 48 GC cell lines [359]. Interestingly, MEKi sensitivity was independent of the level of p-ERK suppression but dependent on the degree of suppression in the levels of p-S6K1 (T389), p-4E-BP1 (S65), and p-RPS6 (S235). On the contrary, no significant suppression of phosphorylation in these proteins was observed in resistant cells after MEKi treatment. Among them, the change in p-RPS6 level by MEKi showed the strongest correlation with MEKi sensitivity in vitro. This resistance of GC cells to MEKi was similarly recapitulated in xenograft models [359]. In addition, since the p-AKT level in sensitive GC cells was not affected by MEKi, the MEKi-dependent suppression of p-RPS6 was suggested to be mediated by mTORC1 in these cells. Interestingly, ERK regulates the level of p-RPS6 not only through RSK but also the TSC1/2 complex, the upstream negative regulator of mTORC1: (1) ERK1 induces inhibitory phosphorylation of TSC2 [74]; (2) ERK also phosphorylates and activates S6K [73]; and (3) ERK activates mTORC1 through RSK-mediated RPTOR phosphorylation (Figure 2) [75]. Previously, p-RPS6 has been demonstrated to be a potential predictive marker for the responsiveness of RCC cells to everolimus [445]. Taken together, further evaluation is warranted for RPS6 and/or p-RPS6 as a companion diagnostic for the therapeutic use of various inhibitors for mTOR and MEK.
4.3.3. RPS6 in Resistance to RAF Inhibitors (RAFis)
Currently, BRAFV600E-selective inhibitors (BRAFis), such as dabrafenib, encorafenib, and vemurafenib, and MEK inhibitors (MEKis) including binimetinib, cobimetinib, and trametinib, have been approved by the US FDA for the treatment of melanoma [213]. However, similar to other clinical PKIs, acquired resistance to these inhibitors is rapidly developed after a transient clinical benefit [473]. The constitutive phosphorylation of RPS6 has been found in BRAFV600E-mutant melanoma cell clones with acquired resistance to PKIs [39]. In two human melanoma clones, A375-DR and A375-TR, with resistance to the BRAFi, dabrafenib, and the MEKi trametinib, respectively, p-RPS6 could not be downregulated by other MAPK pathway inhibitors. In contrast, an S6K inhibitor LY2584702 downregulated p-RPS6 in these cells. Additionally, the PI3K/mTOR inhibitor BEZ235 and the mTOR inhibitor AZD2014 could also inhibit the RPS6 phosphorylation in the resistant cells. Therefore, the switched regulation of p-RPS6, from the RAS/RAF/MEK/ERK pathway to the PI3K/AKT/mTORC1/S6K pathway, led to the resistance of these cells to BRAFis and MEKis by inducing the expression of G0/G1 cell cycle checkpoint proteins, including RB, cyclin D1, and CDK6 [39]. The mechanism underlying this mechanistic switched regulation of p-RPS6 remains to be evaluated.
4.3.4. RPS6 in Resistance to PARP Inhibitors (PARPis)
The poly(ADP-ribose) polymerase (PARP) is a well-established target to treat cancers with HRDs, including mutations in BRCA1/2 (breast cancer susceptibility gene 1/2) [474]. Currently, four small-molecule PARPis, olaparib (Lynparza®), talazoparib (Talzenna®), rucaparib (Rubraca®), and niraparib (Zejula®), have been approved by the US FDA to treat breast, ovarian, pancreatic, and prostate cancer [475]. P-RPS6 has been reported to confer the BRCA1-deficient breast cancer HCC1937 cells with resistance to the PARPi olaparib [376]. Long-term treatment of HCC1937 cells with olaparib upregulated p-RPS6, whereas restoration of BRCA1 abrogated the olaparib-induced RPS6 phosphorylation. In addition, RPS6P−/− breast cancer cells with a BRCA1-defective background (HCC1937) demonstrated olaparib sensitivity. More interestingly, in olaparib-resistant HCC1937 cells, IR decreased and increased γ-H2AX foci and RAD51 foci, respectively, whereas IR elicited exactly the opposite effects in RPS6P−/− HCC1937 cells. Since γ-H2AX is a surrogate marker of DNA DSBs, and RAD51 is a marker of HR, these results imply that p-RPS6 plays a critical role in inducing HRR to eliminate DSBs in BRCA1-deficient cells. More importantly, blocking phosphorylation of RPS6 by rapamycin or BEZ235 in combination with olaparib results in synergistic anticancer activity both in vitro and in vivo [374,376].
5. RPS6 as a Therapeutic Target in Cancer
Substantial evidence suggests that RPS6 is a potential therapeutic target against cancers. Knockdown experiments have demonstrated that RPS6 itself, not only p-RPS6, is indispensable for the proliferation or survival of various cancer cells (Table 9).
5.1. MicroRNAs Targeting RPS6
MiR-129-5p sensitized HER2(+) breast cancer to trastuzumab (Herceptin) by targeting RPS6 [476]. The level of miR-129-5p is reduced in the trastuzumab-resistant human breast cancer cells, JIMT-1, and patient serum samples [476,481]. MiR-129-5p reduced RPS6 expression by targeting the 3′-untranslated region (3′-UTR) of RPS6 mRNA, and transient transfection of the miR-129-5p mimic increased the trastuzumab sensitivity of JIMT-1 cells via downregulating RPS6 expression [476]. Consistent with this observation, the inhibition of miR-129-5p in the trastuzumab-susceptible breast cancer cell line, SK-BR-3, induced trastuzumab resistance, whereas concurrent treatment of RPS6-siRNA abolished this resistance. Similarly, the miR-129-5p overexpression-induced trastuzumab-sensitivity was revered by RPS6 overexpression in JIMT-1 cells [476].
5.2. RPS6-KD in Cancer Cells
RPS6 or its phosphorylation has been proposed as an alternative target to treat cancer types with high levels of RPS6 [21,482] (Figure 5). The siRNA- or shRNA-based RPS6-KD has demonstrated anticancer effects in various cancer cells (Table 9).
5.2.1. RPS6-KD in Breast Cancer Cells
Studies on the TNBC cells HS578T and MDA-MB-231 have suggested RPS6 as a therapeutic target against TNBC [21]. Synergistic downregulation of both p-RPS6 (S235236) and t-RPS6 has been identified as a unique feature in TNBC cells co-treated with the EGFRi gefitinib and the METi SU11274, whereas no significant or consistent changes have been observed in the levels of p-EGFR (Y1068), t-EGFR, t-MET, p-AKT (S473), t-AKT, p-ERK1/2 (T202/Y204), and t-ERK1/2. Additionally, 4 h treatment with the proteasome inhibitor MG132 was not sufficient to inhibit the reduction of t-RPS6 in the TNBC cells in the presence of gefitinib and SU11274. The siRNA-based RPS6-KD reproduced the reduction of cell proliferation observed in TNBC cells co-treated with the gefitinib alongside SU11274. Unfortunately, the functional consequence of RPS6-KD in TNBC cells has not been further studied. However, it is noteworthy that the downregulation of t-RPS6 was augmented over time, whereas the initial reduction in the levels of p-EGFR and p-AKT was recovered with time in the TNBC cells co-treated with gefitinib and SU11274 [21]. Further research is needed to address the mechanism of regulating RPS6 stability in TNBC cells and its implication for TNBC treatment. RPS6-KD or rapamycin treatment has been reported to upregulate p-4E-BP1 (T37/46) and acetylated histone H3 (Ac-H3) in TNBC cells [477]. Co-treatment of the cells with valproic acid (VA) and tamoxifen suppresses RPS6-KD-mediated induction of p-4E-BP1 and Ac-H3 (K56) levels, and the triple combination of rapamycin, VA, and tamoxifen suppresses p-RPS6 (S235/236), p-4E-BP1 (T37/46), and Ac-H3 (K56) levels concurrently with a decrease in the viability of TNBC cells. Interestingly, this triple combination restored the expression of estrogen receptor alpha (ERα). In addition, both the triple combination and RPS6-KD with VA and tamoxifen reduced the cancer stem cell (CSC) population in TNBC cells. Furthermore, the triple combination reduced tumor growth, CSC subpopulation, and tumorigenesis in vivo [477].
5.2.2. RPS6-KD in Cervical Carcinoma Cells
Downregulation of RPS6 in the cervical carcinoma cell line HeLa resulted in the inhibition of TRAIL-dependent apoptosis in a DR4-dependent manner [340]. RPS6-KD downregulated DR4 and desensitized these cancer cells to TRAIL-dependent apoptosis (see Section 5.2.7. RPS6-KD in Hematopoietic Cancer Cells). Conversely, the overexpression of DR4 sensitized TRAIL-induced apoptosis in RPS6-KD cells. The detailed mechanism underlying the regulation of DR4 expression by RPS6 remains to be described. Similar to Jurkat cells expressing antisense RPS6, HeLa cells stably expressing antisense RPS6 or RPS6-shRNA were not resistant to apoptosis induced by doxorubicin or tunicamycin. In contrast, RPS6 overexpression in HeLa cells resulted in sensitization of the cells to TRAIL-dependent apoptosis, but not to apoptosis induced by doxorubicin, etoposide, staurosporine, or tunicamycin. Overexpression of other RPSs, such as RPS2, RPS3, and RPS20, did not affect TRAIL-induced apoptosis in HeLa cells. As mentioned earlier (see Section 3.2.3. Regulation of Apoptosis), the sensitization of TRAIL-induced apoptosis is mediated by unphosphorylated RPS6 not by p-RPS6 in HeLa cells.
5.2.3. RPS6-KD in Head and Neck Cancer Cells
The level of p-RPS6 (S240/244) and the ratio of p-RPS6/t-RPS6 are correlated with unfavorable prognosis in ESCC patients [436]. The important role of RPS6 in ESCC cells was further demonstrated by a knockdown experiment. RPS6-KD in the ESCC cell lines TE8 and TE10 reduced the numbers of these cells over time compared with the numbers of the control KD cells. Western blot analyses showed downregulation of cyclin D and CDK2 proteins in the RPS6-KD ESCC cells. In contrast, the levels of p21 and p27 were induced by RPS6-KD. Furthermore, RPS6-KD reduced the migration, invasion, and focal adhesion formation of ESCC cells, concurrently with a reduction of p-FAK (Y397), p-paxillin (Y118), p-ERK, and p-JNK (T183/Y185). S6K1-KD, an upstream kinase of RPS6, also resulted in similar effects in ESCC cells [436].
5.2.4. RPS6-KD in Gastric Cancer Cells
As mentioned earlier, RPS6-KD, in HER2i-resistant GC cell lines, resulted in a reduction in viability and induced sensitivity to HER2is through the downregulation of NRF2 and its target genes such as AKR1C1, AKR1C2, and AKR1B10 [354] (see Section 3.2.4. Regulation of Drug Resistance). The mechanisms of NRF2 downregulation by RPS6-KD remain largely unknown. Notably, RPS6 has been reported to bind to the La autoantigen (also known as Sjögren syndrome type B; SSB) upon oxidative stress, leading to increased translation of NRF2 [370]. La/SSB binds to the 5′-UTR of NRF2 mRNA in response to oxidative stress, and RPS6 consequently associates with the ribosomes.
5.2.5. RPS6-KD in Glioblastoma Cells
RPS6-KD suppressed the tumor-sphere formation of GBM cells through the inhibition of p-JAK2 and p-STAT3 and concomitantly downregulated the stemness-related proteins Nestin and SOX2 [438]. The size of the tumor spheres was also reduced by RPS6-KD. Inversely, transient overexpression of RPS6 enhanced the tumor-sphere formation of these cells [438]. Furthermore, the interleukin 6 (IL6)-induced tumor sphere formation was reduced by RPS6-KD. Taken together, RPS6 may regulate the IL6/IL6 receptor (IL6R) pathway. Since the tumor sphere formation and the expression of Nestin and SOX2 are the characteristics of GSCs, these results suggest that RPS6 plays critical roles in the development and maintenance of GSCs [438]. Interestingly, the stemness-related proteins Nestin and SOX2 have been established as transcriptional targets of STAT3 [483,484].
5.2.6. RPS6-KD in HCC Cells
Similar to HeLa cells (see Section 5.2.2. RPS6-KD in Cervical Carcinoma Cells), RPS6-KD in the human HCC cell line SK-HEP-1 suppressed TRAIL-induced apoptosis and downregulated DR4 [340]. The reconstitution of DR4 expression rescued the TRAIL-resistance in this cell line.
5.2.7. RPS6-KD in Hematopoietic Cancer Cells
A functional genetic screening for the death-rescue modifier has identified TRAIL-resistant T cell leukemia Jurkat clones that contain antisense RPS6 cDNA in their genome [340]. The reduced RPS6 expression in these cells resulted in the attenuation of TRAIL-induced apoptosis. This resistance was specific against TRAIL. No attenuation was observed in cell death caused by other signals, such as tumor necrosis factor-α (TNFα), FAS ligand (FASL), etoposide, and staurosporine.
As previously mentioned, RPS6-KD reduces the proliferation of the PEL cell line BC-3 (see Section 3.2.10. Roles of RPS6 in Response to Infection) [399]. Since BC-3 cells were infected by KSHV, the cells contain the viral genome and express the viral proteins as well [485]. RPS6 may contribute to the extraordinary stability (several days) of LANA. RPS6-KD remarkably reduced the LANA stability to 0.6 hr, leading to p53 stabilization and a p53-target gene, CDKN1A (the gene encoding p21) expression [399]. However, the molecular mechanism of RPS6 in the LANA stabilization needs further study.
RPS6-KD has decreased the levels of the proliferating cell nuclear antigen (PCNA) in p53 wild-type DLBCL cell lines [338]. However, RPS6-KD did not induce apoptosis in these cells.
5.2.8. RPS6-KD in Lung Cancer Cells
The suppression of proliferation by shRNA-based RPS6-KD was exaggerated by time in two lung cancer cell lines, A549 and H520 [37]. Inversely, the expression of senescence-associated β-galactosidase (SA-β-gal) was increased by RPS6-KD in both cell lines. Additionally, RPS6-KD increased the G0/G1 phase and decreased the G2/M phase with reduced protein levels of p-RB and cyclin D1, whereas no significant changes were observed in the levels of cyclin A, cyclin E, and total RB. More interestingly, the levels of CKIs, including p16, p21, p27, and p57, were increased. However, RPS6-KD did not induce apoptosis or altered the expression of BCL-xL, BAX, or caspase-3. The anticancer effect of RPS6-KD was reproduced in a xenograft model in which A549 lung cancer cells with RPS6-KD resulted in reduced tumorigenicity with an increase in the number of SA-β-gal(+) cells in the xenograft tissues. Similar changes were observed in the expression of cell cycle regulators and CKIs [37].
RPS6-KD induced the G0/G1 cell-cycle arrest, decreasing cell viability in the NSCLC cell lines SK-MEK-1 and H1650 [38]. The levels of p-RB, cyclin D1, cyclin E, CDK2, and CDK4 were also reduced, whereas the levels of CKIs, p21, and p27 were increased by RPS6-KD in these cells. The migration of NSCLC cells was also reduced by RPS6-KD, which also downregulated the proteins involved in cell migration, including N-cadherin, vimentin, MMP-9, MMP-2, and p-paxillin. In contrast, E-cadherin was upregulated by RPS6-KD. Inversely, the overexpression of RPS6 in a normal HBE cell line promoted cell proliferation concurrently with the upregulation of p-RPS6, p-RB, cyclin D1, cyclin E, CDK2, and CDK4, and decreases in CKIs and the number of cells in the G0/G1 phase. The migration of HBE cells was also enhanced by RPS6 overexpression. Additionally, RPS6 overexpression upregulated N-cadherin, vimentin, MMP-2, and p-paxillin in these cells [38].
As mentioned earlier (see Section 3.2.1. Regulation of Cell Cycle, Proliferation, and Growth), RPS6-KD by siRNA in A549 cells induced the p53-mediated checkpoint in an RPL11-depenent manner [339]. Although RPS6-siRNA transfection was not enough to completely deplete the RPS6 protein due to the long half-life of ribosomes, it induced the expression of p53 and p21, leading to a partial decrease in DNA synthesis and an increase in the number of cells arrested at the G1 phase. The concurrent depletion of TP53 by siRNA abrogated the effects of RPS6-siRNA transfection. RPS6 depletion increased the translation of RPL11 mRNA and the amount of ribosome-free RPL11, which can bind to MDM2, leading to the inhibition of MDM2 function to degrade p53. Consequently, the mRNAs and protein levels of p53 targets, such as CDKN1A (the gene encoding p21) and MDM2, were increased.
5.2.9. RPS6-KD in Melanoma Cells
RPS6-KD alone reduced the proliferation of two human melanoma cells A375-DR and A375-TR, which are resistant to the BRAFV600E-selective inhibitor dabrafenib and the MEK1/2 inhibitor trametinib [39]. In addition, RPS6-KD induced the G0/G1 cell-cycle arrest in these cells. More interestingly, RPS6-KD was sufficient to reduce the levels of G0/G1 checkpoint regulators such as RB, cyclin D1, and CDK6. Additionally, the enhanced downregulation of p-RPS6 by drug combinations exerted a synergistic antitumor activity [39].
In a separate study, RPS6-KD significantly reduced the colony formation and induced the apoptosis of cutaneous malignant melanoma (CMM) cells in vitro [478]. Additionally, the same authors found that S6K1 was upregulated in clinical samples with advanced stages of CMM. Furthermore, the mTORC1 signaling pathway was identified to be downregulated by the combination of the EGFRi afatinib and the ALK/ROS1 dual inhibitor crizotinib in malignant melanoma. More interestingly, the level of t-RPS6 itself, in addition to p-RPS6 level, was also downregulated by the afatinib+crizotinib treatment in melanoma cell lines in vitro [478].
5.2.10. RPS6-KD in Ovarian Cancer Cells
It has been reported that the transfection of ovarian cancer cells with RPS6-siRNA reduces the activities of glutaminase (GLS) and glutamate dehydrogenase (GDH), leading to a reduction in glutamine-induced proliferation of the cells [479]. Since glutamine is the main nutrient used by cancer cells, targeting the glutamine metabolism has been recognized as a promising therapeutic alternative [486]. Glutamine depletion inhibits cell proliferation concurrently with the induction of G1 cell-cycle arrest and apoptosis in ovarian cancer cells [479]. Glutamate increases cell proliferation in the presence of glucose. Interestingly, rapamycin reduced the glutamine-induced p-RPS6 (S235/236) and GLS expression in a dose-dependent manner in ovarian cancer cells. In the ovarian cancer cell line HEY, transfection of RPS6-siRNA was sufficient to deplete p-RSP6 (S235/236) although not t-RPS6 [479]. Under these conditions, RPS6-siRNA transfection reduced the glutamine-induced GDH activity and cell proliferation in HEY cells, whereas no significant effect of RPS6-siRNA transfection was observed in the absence of glutamine. Notably, rapamycin treatment only partially mimicked the effects of RPS6-siRNA transfection. The significance of this discrepancy remains to be elucidated.
RPS6-KD also inhibits the proliferation and long-term colony formation of the ovarian cancer cell lines SKOV-3 and HO-8910 [40]. The migration and invasion of these ovarian cancer cells are also reduced by RPS6-KD. Additionally, RPS6-KD induces the G0/G1 cell-cycle arrest. Consistently, the G0/G1 checkpoint regulators, such as cyclin D1, cyclin E, CDK2, CDK4, CDK6, and p-RB, are diminished by RPS6-KD in ovarian cancer cells [40].
5.2.11. RPS6-KD in Sarcoma Cells
An siRNA-based screening identified RPS6 as an effector of IGF1R inhibition in sarcoma cells [480]. RPS6-KD by siRNA reduced the survival of Ewing sarcoma cell lines over time. Interestingly, MTOR-KD recapitulated RPS6-KD, whereas TSC1-KD had little or no effect. In addition, RPS6-KD also induced cell death in rhabdomyosarcoma (RMS) cell lines [480]. The significance of p-RPS6 in these sarcoma cell lines is underlined by their sensitivity to the IGF1R inhibitor BMS-536924. RMS cell lines were resistant to BMS-536924 compared to Ewing sarcoma cell lines. BMS-536924 failed to downregulate p-RPS6 (S235/236) and p-4EBP1 in RMS cell line, but dose-dependently downregulated p-RPS6 (S235/236) and p-4EBP1 in sensitive Ewing sarcoma cell lines. Additional siRNA screening has found that the knockdown of macrophage stimulating 1 receptor (MST1R) sensitizes RMS cell lines to BMS-536924 with concordant reduction of p-RPS6 (S235/236). These data suggest that MST1R may be an alternative activator of RPS6 in IGF1R inhibitor-resistance sarcoma cells.
6. Conclusions
In addition to its role in protein synthesis, RPS6 may activate various pathways to induce the oxidative stress response, drug resistance, cancer stem cell stemness, and cell-cycle arrest (Figure 6). Evidence from knockdown experiments supports the therapeutic potential of RPS6 targeting in various cancer cells including breast cancer, cervical carcinoma, head and neck cancer, gastric cancer, glioblastoma, HCC, hematopoietic cancer, lung cancer, melanoma, ovarian cancer, and sarcoma (Figure 5). However, the signaling pathways downstream of RPS6 are currently largely unknown. In addition, the roles of activated RPS6 in tumorigenesis still remain elusive. The positive and negative feedback and/or feedforward mechanisms and novel regulatory pathways controlled by RPS6 may provide alternative therapeutic interventions to overcome the current limitations in cancer treatment.
Author Contributions
Concept and writing of the manuscript: Y.W.Y., S.-G.L. and Y.-S.S.; investigation: Y.W.Y., K.S.Y., S.-G.L. and Y.-S.S.; critical revision and edition of the manuscript: Y.W.Y., K.S.Y., J.-S.P., S.-G.L. and Y.-S.S. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by grants from the National Research Foundation of Korea (NRF), funded by the Korea Ministry of Science and ICT (MSIT), Republic of Korea: NRF-2021R1A4A1028268 (Y.W.Y. and Y.-S.S.); NRF-2021R1I1A2054560 (S.G.L.); NRF-R-2021-01167 (K.S.Y.); and NRF-2018R1D1A1B07048931 (Y.-S.S.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
2-DG, 2-deoxyglucose; 4E-BP1, eukaryotic translation initiation factor 4E-binding protein 1; 5-ASA, 5-aminosalicylic acid; 5′-TOP, 5′-terminal oligopyrimidine track; 53BP1, p53-binding protein 1; ABL1, Abelson murine leukemia viral oncogene homolog 1; ADAR1, adenosine deaminase acting on RNA 1; AICAR, 5-aminoimidazole-4-carboxamide ribonucleotide; AIPS, autophagy impairment-induced premature senescence; AKR, aldo-keto reductase; AKR1B10, AKR family 1 member B10; AKR1C, AKR family 1 member C; ALA, alpha-linoleic acid; AML, acute myeloid leukemia; AMOG, adhesion molecule in glia; AMPK, 5′-AMP-activated protein kinase; AMPKK, AMPK kinase; AR, androgen receptor; ATG7, autophagy related 7; ATIC, AICAR transformylase/inosine monophosphate cyclohydrolase; ATM, ataxia telangiectasia mutated; ATRA, all-trans-retinoic acid; BAX, BCL2 associated X; BCL-xL, B-cell lymphoma-extra large; BDNF, brain-derived neurotrophic factor; β-TrCP, beta-transducin repeat containing protein; BRCA1, breast cancer type 1 susceptibility protein; BRD4, bromodomain 4; BTF3, basic transcription factor 3; CAB39, calcium-binding protein 39; CDK, cyclin-dependent kinase; CDKN1A, CDK inhibitor 1A; CDKN1B, CDK inhibitor 1B; C/EBP, CCAAT/enhancer binding protein; CG, chorionic gonadotropin; CHD3, chromodomain-helicase-DNA-binding protein 3; CK1, casein kinase 1; CKI, CDK inhibitor; CMM, cutaneous malignant melanoma; CRC, colorectal cancer; CREB, cAMP-responsive element-binding protein; CSC, cancer stem cell; DAPK1, death-associated protein kinase 1; DBA, Diamond-Blackfan anemia; DDB1, DNA damage-binding protein 1; DDIT4, DNA damage-inducible transcript 4 protein; DFS, disease-free survival; DHT, 5α-dihydrotestosterone; DLBCL, diffuse large B-cell lymphoma; DMBA, 7,12-dimethylbenz[a]anthracene; DNA-PK, DNA-dependent protein kinase; DR4, death receptor 4; DRAM1, DNA damage-regulated autophagy modulator protein 1; DRD1, dopamine D1 receptor; DSB, double-strand break; DYRK1A, dual-specific tyrosine-phosphorylation-regulated kinase 1A; E2, 17β estradiol; E2F1, E2F transcription factor 1; EGF, epidermal growth factor; EGFRi, EGFR inhibitor; eIF (3 and 4B), eukaryotic translation initiation factor (3 and 4B); ERα, estrogen receptor alpha; ERK, extracellular signal-regulated kinase; ESCC, esophageal squamous cell carcinoma; FASL, FAS ligand; FASN, fatty acid synthase; FBXO28, F-box-only protein 28; Fc, fragment crystallizable; FDA, Food and Drug Administration; FGF, fibroblast growth factor; FGFR, FGF receptor; FIP200, FAK family kinase-interacting protein of 200 kDa; FKBP12, 12 kDa FK506-binding protein; FOXO, Forkhead box protein O; FPT, farnesyl protein transferase; Gal-13, galetin-13; γ-H2AX, phosphorylated H2A histone family member X; GABP, GA-binding protein; GBM, glioblastoma multiforme; GDH, glutamate dehydrogenase; GlnRs, glutaminyl-tRNA synthase; GLS, glutaminase; GRPR, gastrin-releasing peptide receptor; GSK3β, glycogen synthase kinase-3β; HAP1, huntingtin-associated protein 1; HBE, human bronchial epithelial; HCC, hepatocellular carcinoma; HCV, hepatitis C virus; HDF, human dermal fibroblast; HER2, human EGFR 2; HER2i, HER2 inhibitor; HIF1α, hypoxia-inducible factor 1-alpha; HNSCC, head and neck squamous cell carcinoma; HRAS, Harvey rat sarcoma; HRR, homologous recombination repair; HRD, HRR deficiency; HSP90, heat shock protein 90; IGF1R, insulin-like growth factor 1 receptor; IFN, interferon; IFNAR2, interferon α/β receptor 2; IKAP, IκB kinase complex-associated protein; IKK, inhibitor of NF-κB kinase; IL (1 and 6), interleukin (1 and 6); IL6R, IL6 receptor; IPC, ischemic preconditioning; IR, ionizing radiation; IRES, internal ribosome entry site; KD, knockdown; KRAS, Kristen rat sarcoma; KRT5, keratin 5; KSHV, Kaposi’s sarcoma-associated herpesvirus; LAMP2, lysosome-associated membrane glycoprotein 2; LANA, latency-associated nuclear antigen; LCMT1, leucine carboxyl methyltransferase 1; LDHA, L-lactate dehydrogenase A chain; LKB1, liver kinase B1; LPIN1, lipin 1; mAb, monoclonal antibody; MAPK, mitogen-activated protein kinase; MAP4K3, mitogen-activated protein kinase kinase kinase kinase 3; MCD, multicentric Castleman’s disease; MDM2, mouse double minute 2; MEF, mouse embryonic fibroblast; MEK, MAPK-ERK kinase; MEKi, MEK inhibitor; METi, mesenchymal epithelial transition inhibitor; miRNA, microRNA; MK, midkine; MMP, matrix metallopeptidase; MST1R, macrophage-stimulating 1 receptor; mt, mutant; mTORC1, mechanistic/mammalian target of rapamycin complex 1; NAC, N-acetyl cysteine; NGF, nerve growth factor; NHDF, normal human dermal fibroblast; NLS, nuclear localization signal; NRF2, nuclear factor erythroid 2-related factor 2; NSCLC, non-small cell lung cancer; nSMase 2, neutral sphingomyelinase-2; OA, oleic acid; ORF45, open reading frame 45; OS, overall survival; OSCC, oral squamous cell carcinoma; OSM, oncostatin M; OEC, ovarian epithelial cancer; PA, phosphatidic acid; PALL, Pallbearer; PanIN, pancreatic intraepithelial neoplasia; PARP, poly (ADP-ribose) polymerase; PARPi, PARP inhibitor; PASK, PAS domain-containing serine/threonine-protein kinase; PCNA, proliferating cell nuclear antigen; PCNSL, primary central nervous system lymphoma; PD-1, programmed cell death protein 1; PDAC, pancreatic ductal adenocarcinoma; PDCD4, programmed cell death 4; PDGF, platelet-derived growth factor; PDK1, 3-phosphoinositide-dependent protein kinase 1; PD-L1, programmed death ligand 1; PEL, primary effusion lymphoma; PERK, protein kinase RNA-activated (PRKR)-like endoplasmic reticulum kinase; PFS, progression-free survival; PGC-1β, peroxisome proliferator-activated receptor-γ (PPAR-γ) coactivator-1β; PHLPP, pleckstrin homology domain leucine-rich repeat protein phosphatases; PI3K, phosphoinositide 3-kinase; PIM2, serine/threonine-protein kinase PIM2; PK (A, C, and G), protein kinase (A, C, and G); PKI, protein kinase inhibitor; PRKR, protein kinase RNA-activated; PLAC, placenta-specific 8; PLCG1, phospholipase C-γ1; PLD, phospholipase D; PLK, polo-like kinase; PMA, phorbol 12-myristate 13-acetate; PME1, protein phosphatase methylesterase 1; PP1, protein phosphatase 1; PP2A, serine/threonine-protein phosphatase-2A; PPAR-γ, peroxisome proliferator-activated receptor-γ; PPM1D, protein phosphatase magnesium-dependent 1 delta; PSIP1, PC4 and SFRS1-interacting protein 1; PTEN, phosphatase and tensin homolog; PyMT, polyomavirus middle tumor antigen; PyST, polyomavirus small tumor antigen; QARS, glutaminyl-tRNA synthase; RAC2, Rac family small GTPase 2; RACK1, receptor of activated PKC kinase 1; RAD51, DNA repair protein RAD51 homolog 1; RAF, v-raf-1 murine leukemia viral oncogene homology; RAFi, RAF inhibitor; RAME, rosmarinic acid methyl ester; RAS, rat sarcoma; RB, retinoblastoma-associated protein; RB1CC1, RB1-inducible coiled-coil protein 1; RCC, renal cell carcinoma; REDD1, regulated in development and DNA damage responses 1; RHEB, RAS homolog enriched in brain; RICTOR, rapamycin-insensitive companion of mTOR; RMS, rhabdomyosarcoma; ROS, reactive oxygen species; RP, ribosomal protein; RPL (5, 11, and 16), ribosomal protein L (5, 11, and 16); RPS (6, 8, and 24), ribosomal protein S (6, 8, and 24); rRNA, ribosomal RNA; RPTOR, regulatory-associated protein of mTOR; RSK, ribosomal protein S6 kinase; RVF, Rift Valley Fever; S6K, S6 kinase; SA-β-gal, senescence associated β-galactosidase; SASP, senescence-associated secretory phenotype; SCF, Skp-Cullin-F-box; SGK, serum and glucocorticoid-inducible kinases; SHE1, nucleoprotein sensitive to high expression protein 1; shRNA, short hairpin RNA; SIRT1, NDA-dependent protein deacetylase sirtuin-1; SOX2, SRY-box transcription factor 2; SSB, Sjögren syndrome antigen B; STK33, serine/threonine kinase 33; SP1, specificity protein 1; SQSTM, sequestome-1; SRSF1, serine/arginine-rich splicing factor 1; SV40, simian virus 40; SVST, SV40 small tumor antigen; TRAIL, tumor necrosis factor-related apoptosis-inducing ligand; TRB3, tribbles homolog 3; TNBC, triple-negative breast cancer; TNFα, tumor necrosis factor-α; TOP2B, DNA topoisomerase IIβ; t-RPS6, total RPS6; TSC, tuberous sclerosis; ULK1, Unc-51-like kinase 1; URM1, ubiquitin-related modifier 1; UTR, untranslated region; UV, ultraviolet; V-ATPase, vacuolar proton-ATPase; VIN, vulvar intraepithelial neoplasia; VHL, von Hippel-Lindau disease tumor suppressor; VSCC, vulva squamous cell carcinoma; WIP1, wild-type p53-induced phosphatase 1; WNT3A, wingless-related integration site family member 3A; YY1, Yin-Yang 1; ZBED1, zinc finger BED domain-containing protein 1.
References
- Zhou, X.; Liao, W.-J.; Liao, J.-M.; Liao, P.; Lu, H. Ribosomal proteins: Functions beyond the ribosome. J. Mol. Cell Biol. 2015, 7, 92–104. [Google Scholar] [CrossRef] [Green Version]
- Meyuhas, O. Ribosomal Protein S6 Phosphorylation: Four Decades of Research. Int. Rev. Cel Mol. Biol. 2015, 320, 41–73. [Google Scholar] [CrossRef]
- Xu, X.; Xiong, X.; Sun, Y. The role of ribosomal proteins in the regulation of cell proliferation, tumorigenesis, and genomic integrity. Sci. China Life Sci. 2016, 59, 656–672. [Google Scholar] [CrossRef] [PubMed]
- Barna, M.; Pusic, A.; Zollo, O.; Costa, M.; Kondrashov, N.; Rego, E.; Rao, P.H.; Ruggero, D. Suppression of Myc oncogenic activity by ribosomal protein haploinsufficiency. Nature 2008, 456, 971–975. [Google Scholar] [CrossRef] [PubMed]
- Dai, M.-S.; Lu, H. Inhibition of MDM2-mediated p53 Ubiquitination and Degradation by Ribosomal Protein L5. J. Biol. Chem. 2004, 279, 44475–44482. [Google Scholar] [CrossRef] [Green Version]
- Lohrum, M.A.; Ludwig, R.L.; Kubbutat, M.H.; Hanlon, M.; Vousden, K.H. Regulation of HDM2 activity by the ribosomal protein L11. Cancer Cell 2003, 3, 577–587. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Wolf, G.W.; Bhat, K.; Jin, A.; Allio, T.; Burkhart, W.A.; Xiong, Y. Ribosomal Protein L11 Negatively Regulates Oncoprotein MDM2 and Mediates a p53-Dependent Ribosomal-Stress Checkpoint Pathway. Mol. Cell. Biol. 2003, 23, 8902–8912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, A.; Itahana, K.; O’Keefe, K.; Zhang, Y. Inhibition of HDM2 and Activation of p53 by Ribosomal Protein L23. Mol. Cell. Biol. 2004, 24, 7669–7680. [Google Scholar] [CrossRef] [Green Version]
- Dai, M.-S.; Zeng, S.X.; Jin, Y.; Sun, X.-X.; David, L.; Lu, H. Ribosomal Protein L23 Activates p53 by Inhibiting MDM2 Function in Response to Ribosomal Perturbation but Not to Translation Inhibition. Mol. Cell. Biol. 2004, 24, 7654–7668. [Google Scholar] [CrossRef] [Green Version]
- Macfarlane, D.; Gailani, D. Identification of phosphoprotein NP33 as a nucleus-associated ribosomal S6 protein and its phosphorylation in hematopoietic cells. Cancer Res. 1990, 50, 2895–2900. [Google Scholar] [PubMed]
- Vladimirov, S.N.; Ivanov, A.; Karpova, G.G.; Musolyamov, A.K.; Egorov, T.A.; Thiede, B.; Wittmann-Liebold, B.; Otto, A. Characterization of the Human Small-Ribosomal-Subunit Proteins by N-Terminal and Internal Sequencing, and Mass Spectrometry. JBIC J. Biol. Inorg. Chem. 1996, 239, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Meyuhas, O. Chapter 1 Physiological Roles of Ribosomal Protein S6: One of Its Kind. Int. Rev. Cell Mol. Biol. 2008, 268, 1–37. [Google Scholar] [CrossRef]
- Hirashita, T.; Hirashita, Y.; Iwashita, Y.; Endo, Y.; Kiyonaga, M.; Matsumoto, S.; Hijiya, N.; Moriyama, M.; Murakami, K.; Inomata, M. S6 ribosomal protein phosphorylation is associated with malignancy of intraductal papillary mucinous neoplasm of the pancreas. Ann. Gastroenterol. Surg. 2020, 4, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Ruvinsky, I.; Meyuhas, O. Ribosomal protein S6 phosphorylation: From protein synthesis to cell size. Trends Biochem. Sci. 2006, 31, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Warner, J.R.; McIntosh, K.B. How Common Are Extraribosomal Functions of Ribosomal Proteins? Mol. Cell 2009, 34, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Gressner, A.M.; Wool, I.G. The phosphorylation of liver ribosomal proteins in vivo. Evidence that only a single small subunit protein (S6) is phosphorylated. J. Biol. Chem. 1974, 249, 6917–6925. [Google Scholar] [CrossRef]
- Khalaileh, A.; Dreazen, A.; Khatib, A.; Apel, R.; Swisa, A.; Kidess-Bassir, N.; Maitra, A.; Meyuhas, O.; Dor, Y.; Zamir, G. Phosphorylation of Ribosomal Protein S6 Attenuates DNA Damage and Tumor Suppression during Development of Pancreatic Cancer. Cancer Res. 2013, 73, 1811–1820. [Google Scholar] [CrossRef] [Green Version]
- Chow, S.; Minden, M.D.; Hedley, D.W. Constitutive phosphorylation of the S6 ribosomal protein via mTOR and ERK signaling in the peripheral blasts of acute leukemia patients. Exp. Hematol. 2006, 34, 1182–1190. [Google Scholar] [CrossRef]
- Dieterlen, M.-T.; Bittner, H.B.; Klein, S.; Von Salisch, S.; Mittag, A.; Tárnok, A.; Dhein, S.; Mohr, F.W.; Barten, M.J. Assay validation of phosphorylated S6 ribosomal protein for a pharmacodynamic monitoring of mTOR-inhibitors in peripheral human blood. Cytom. Part B Clin. Cytom. 2011, 82, 151–157. [Google Scholar] [CrossRef]
- Perl, A.E.; Kasner, M.T.; Shank, D.; Luger, S.M.; Carroll, M. Single-Cell Pharmacodynamic Monitoring of S6 Ribosomal Protein Phosphorylation in AML Blasts during a Clinical Trial Combining the mTOR Inhibitor Sirolimus and Intensive Chemotherapy. Clin. Cancer Res. 2011, 18, 1716–1725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, Y.W.; You, K.; Bae, E.J.; Kwak, S.-J.; Seong, Y.-S.; Bae, I. Dual inhibition of EGFR and MET induces synthetic lethality in triple-negative breast cancer cells through downregulation of ribosomal protein S6. Int. J. Oncol. 2015, 47, 122–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antoine, M.; Fried, M. The organization of the intron-containing human S6 ribosomal protein (rpS6) gene and determination of its location at chromosome 9p21. Hum. Mol. Genet. 1992, 1, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Heinze, H.; Arnold, H.H.; Fischer, D.; Kruppa, J. The primary structure of the human ribosomal protein S6 derived from a cloned cDNA. J. Biol. Chem. 1988, 263, 4139–4144. [Google Scholar] [CrossRef]
- Lott, J.B.; Mackie, G.A. Isolation and characterization of cloned cDNAs that code for human ribosomal protein S6. Gene 1988, 65, 31–39. [Google Scholar] [CrossRef]
- Pata, I.; Metspalu, A. Structural characterization of the mouse ribosomal protein S6-encoding gene. Gene 1996, 175, 241–245. [Google Scholar] [CrossRef]
- Antoine, M.; Kiefer, P. Functional characterization of transcriptional regulatory elements in the upstream region and intron 1 of the human S6 ribosomal protein gene. Biochem. J. 1998, 336, 327–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, R.P. The architecture of mammalian ribosomal protein promoters. BMC Evol. Biol. 2005, 5, 15. [Google Scholar] [CrossRef] [Green Version]
- Ishii, K.; Washio, T.; Uechi, T.; Yoshihama, M.; Kenmochi, N.; Tomita, M. Characteristics and clustering of human ribosomal protein genes. BMC Genom. 2006, 7, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roepcke, S.; Zhi, D.; Vingron, M.; Arndt, P.F. Identification of highly specific localized sequence motifs in human ribosomal protein gene promoters. Gene 2006, 365, 48–56. [Google Scholar] [CrossRef]
- Ravitz, M.J.; Chen, L.; Lynch, M.; Schmidt, E.V. c-myc Repression of TSC2 Contributes to Control of Translation Initiation and Myc-Induced Transformation. Cancer Res. 2007, 67, 11209–11217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, E.V.; Ravitz, M.J.; Chen, L.; Lynch, M. Growth controls connect: Interactions between c-myc and the tuberous sclerosis complex-mTOR pathway. Cell Cycle 2009, 8, 1344–1351. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, D.; Sano, Y.; Adachi, Y.; Okamoto, Y.; Osada, H.; Takahashi, T.; Yamaguchi, T.; Osumi, T.; Hirose, F. hDREF Regulates Cell Proliferation and Expression of Ribosomal Protein Genes. Mol. Cell. Biol. 2007, 27, 2003–2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashita, D.; Moriuchi, T.; Osumi, T.; Hirose, F. Transcription Factor hDREF Is a Novel SUMO E3 Ligase of Mi2α. J. Biol. Chem. 2016, 291, 11619–11634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Vona, C.; Bezdan, D.; Islam, A.B.; Salichs, E.; López-Bigas, N.; Ossowski, S.; de la Luna, S. Chromatin-wide Profiling of DYRK1A Reveals a Role as a Gene-Specific RNA Polymerase II CTD Kinase. Mol. Cell 2015, 57, 506–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nosrati, N.; Kapoor, N.R.; Kumar, V. Combinatorial action of transcription factors orchestrates cell cycle-dependent expression of the ribosomal protein genes and ribosome biogenesis. FEBS J. 2014, 281, 2339–2352. [Google Scholar] [CrossRef]
- Giacinti, C.; Giordano, A. RB and cell cycle progression. Oncogene 2006, 25, 5220–5227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.; Zhang, W.; Gao, J.; Chen, H.; Jiang, L.; Liu, D.; Cao, Y.; Zhao, S.; Qiu, Z.; Zeng, J.; et al. Downregulation of ribosomal protein S6 inhibits the growth of non-small cell lung cancer by inducing cell cycle arrest, rather than apoptosis. Cancer Lett. 2014, 354, 378–389. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Tan, Z.; Gao, J.; Wu, W.; Liu, L.; Jin, W.; Cao, Y.; Zhao, S.; Zhang, W.; Qiu, Z.; et al. Hyperphosphorylation of ribosomal protein S6 predicts unfavorable clinical survival in non-small cell lung cancer. J. Exp. Clin. Cancer Res. 2015, 34, 126. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.-Z.; Wang, H.-B.; Chen, X.-L.; Cao, W.-T.; Fu, L.; Li, Y.; Quan, H.-T.; Xie, C.-Y.; Lou, L.-G. Aberrant modulation of ribosomal protein S6 phosphorylation confers acquired resistance to MAPK pathway inhibitors in BRAF-mutant melanoma. Acta Pharmacol. Sin. 2018, 40, 268–278. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Xu, L.; Yang, Y.-E.; Xiong, C.; Yu, J.; Wang, Y.; Lin, Y. Knockdown of ribosomal protein S6 suppresses proliferation, migration, and invasion in epithelial ovarian cancer. J. Ovarian Res. 2020, 13, 100. [Google Scholar] [CrossRef]
- Xue, S.; Barna, M. Specialized ribosomes: A new frontier in gene regulation and organismal biology. Nat. Rev. Mol. Cell Biol. 2012, 13, 355–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohr, M.J.; Aponte, A.M.; Sun, J.; Wang, G.; Murphy, E.; Gucek, M.; Steenbergen, C. Characterization of potentialS-nitrosylation sites in the myocardium. Am. J. Physiol. Circ. Physiol. 2011, 300, H1327–H1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkins, S.E.; Islam, S.; Gannon, J.M.; Markolovic, S.; Hopkinson, R.J.; Ge, W.; Schofield, C.J.; Chowdhury, R. JMJD5 is a human arginyl C-3 hydroxylase. Nat. Commun. 2018, 9, 1180. [Google Scholar] [CrossRef] [PubMed]
- Roux, P.P.; Topisirovic, I. Signaling Pathways Involved in the Regulation of mRNA Translation. Mol. Cell. Biol. 2018, 38, e00070-18. [Google Scholar] [CrossRef] [Green Version]
- Kozma, S.C.; Ferrari, S.; Thomas, G. Unmasking a growth factor/oncogene-activated S6 phosphorylation cascade. Cell. Signal. 1989, 1, 219–225. [Google Scholar] [CrossRef]
- Wettenhall, R.; Erikson, E.; Maller, J. Ordered multisite phosphorylation of Xenopus ribosomal protein S6 by S6 kinase II. J. Biol. Chem. 1992, 267, 9021–9027. [Google Scholar] [CrossRef]
- Flotow, H.; Thomas, G. Substrate recognition determinants of the mitogen-activated 70K S6 kinase from rat liver. J. Biol. Chem. 1992, 267, 3074–3078. [Google Scholar] [CrossRef]
- Roux, P.P.; Shahbazian, D.; Vu, H.; Holz, M.K.; Cohen, M.S.; Taunton, J.; Sonenberg, N.; Blenis, J. RAS/ERK Signaling Promotes Site-specific Ribosomal Protein S6 Phosphorylation via RSK and Stimulates Cap-dependent Translation. J. Biol. Chem. 2007, 282, 14056–14064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, B.; Roux, P.P. Translational control by oncogenic signaling pathways. Biochim. Biophys. Acta (BBA) Bioenerg. 2015, 1849, 753–765. [Google Scholar] [CrossRef]
- Moore, C.E.; Xie, J.; Gomez, E.; Herbert, T.P. Identification of cAMP-Dependent Kinase as a Third in Vivo Ribosomal Protein S6 Kinase in Pancreatic β-Cells. J. Mol. Biol. 2009, 389, 480–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valjent, E.; Bertran-Gonzalez, J.; Bowling, H.; Lopez, S.; Santini, E.; Matamales, M.; Bonito-Oliva, A.; Hervé, D.; Hoeffer, C.; Klann, E.; et al. Haloperidol Regulates the State of Phosphorylation of Ribosomal Protein S6 via Activation of PKA and Phosphorylation of DARPP-32. Neuropsychopharmacology 2011, 36, 2561–2570. [Google Scholar] [CrossRef] [Green Version]
- Yano, T.; Ferlito, M.; Aponte, A.; Kuno, A.; Miura, T.; Murphy, E.; Steenbergen, C. Pivotal Role of mTORC2 and Involvement of Ribosomal Protein S6 in Cardioprotective Signaling. Circ. Res. 2014, 114, 1268–1280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biever, A.; Puighermanal, E.; Nishi, A.; David, A.; Panciatici, C.; Longueville, S.; Xirodimas, D.; Gangarossa, G.; Meyuhas, O.; Herve, D.; et al. PKA-Dependent Phosphorylation of Ribosomal Protein S6 Does Not Correlate with Translation Efficiency in Striatonigral and Striatopallidal Medium-Sized Spiny Neurons. J. Neurosci. 2015, 35, 4113–4130. [Google Scholar] [CrossRef] [Green Version]
- House, C.; Wettenhall, R.; Kemp, B. The influence of basic residues on the substrate specificity of protein kinase C. J. Biol. Chem. 1987, 262, 772–777. [Google Scholar] [CrossRef]
- Schumacher, A.M.; Velentza, A.V.; Watterson, D.; Dresios, J. Death-Associated Protein Kinase Phosphorylates Mammalian Ribosomal Protein S6 and Reduces Protein Synthesis. Biochemistry 2006, 45, 13614–13621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutchinson, J.A.; Shanware, N.P.; Chang, H.; Tibbetts, R.S. Regulation of Ribosomal Protein S6 Phosphorylation by Casein Kinase 1 and Protein Phosphatase 1. J. Biol. Chem. 2011, 286, 8688–8696. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Mitsuhashi, S.; Ikejo, M.; Miura, N.; Kawamura, T.; Hamakubo, T.; Ubukata, M. Relationship between ATM and Ribosomal Protein S6 Revealed by the Chemical Inhibition of Ser/Thr Protein Phosphatase Type 1. Biosci. Biotechnol. Biochem. 2012, 76, 486–494. [Google Scholar] [CrossRef]
- Schläfli, P.; Tröger, J.; Eckhardt, K.; Borter, E.; Spielmann, P.; Wenger, R.H. Substrate preference and phosphatidylinositol monophosphate inhibition of the catalytic domain of the Per-Arnt-Sim domain kinase PASKIN. FEBS J. 2011, 278, 1757–1768. [Google Scholar] [CrossRef]
- Belandia, B.; Brautigan, D.; Martín-Pérez, J. Attenuation of ribosomal protein S6 phosphatase activity in chicken embryo fibroblasts transformed by Rous sarcoma virus. Mol. Cell. Biol. 1994, 14, 200–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeno, P.; Ballou, L.M.; Novak-Hofer, I.; Thomas, G. Identification and characterization of a mitogen-activated S6 kinase. Proc. Natl. Acad. Sci. USA 1988, 85, 406–410. [Google Scholar] [CrossRef] [Green Version]
- Koh, H.; Jee, K.; Lee, B.; Kim, J.; Kim, D.; Yun, Y.-H.; Kim, J.W.; Choi, H.-S.; Chung, J. Cloning and characterization of a nuclear S6 kinase, S6 kinase-related kinase (SRK); A novel nuclear target of Akt. Oncogene 1999, 18, 5115–5119. [Google Scholar] [CrossRef] [Green Version]
- Gout, I.; Minami, T.; Hara, K.; Tsujishita, Y.; Filonenko, V.; Waterfield, M.D.; Yonezawa, K. Molecular Cloning and Characterization of a Novel p70 S6 Kinase, p70 S6 Kinase β Containing a Proline-rich Region. J. Biol. Chem. 1998, 273, 30061–30064. [Google Scholar] [CrossRef] [Green Version]
- Lee-Fruman, K.K.; Kuo, C.J.; Lippincott, J.; Terada, N.; Blenis, J. Characterization of S6K2, a novel kinase homologous to S6K1. Oncogene 1999, 18, 5108–5114. [Google Scholar] [CrossRef] [Green Version]
- Saitohab, M.; Dijke, P.T.; Miyazonoa, K.; Ichijo, H. Cloning and Characterization of p70S6KβDefines a Novel Family of p70 S6 Kinases. Biochem. Biophys. Res. Commun. 1998, 253, 470–476. [Google Scholar] [CrossRef]
- Shima, H.; Pende, M.; Chen, Y.; Fumagalli, S.; Thomas, G.; Kozma, S.C. Disruption of the p70s6k/p85s6k gene reveals a small mouse phenotype and a new functional S6 kinase. EMBO J. 1998, 17, 6649–6659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinhard, C.; Thomas, G.; Kozma, S.C. A single gene encodes two isoforms of the p70 S6 kinase: Activation upon mitogenic stimulation. Proc. Natl. Acad. Sci. USA 1992, 89, 4052–4056. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Stevens, P.D.; Gao, T. mTOR-Dependent Regulation of PHLPP Expression Controls the Rapamycin Sensitivity in Cancer Cells. J. Biol. Chem. 2011, 286, 6510–6520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lara, R.; Seckl, M.J.; Pardo, O. The p90 RSK Family Members: Common Functions and Isoform Specificity. Cancer Res. 2013, 73, 5301–5308. [Google Scholar] [CrossRef] [Green Version]
- Pende, M.; Um, S.H.; Mieulet, V.; Sticker, M.; Goss, V.L.; Mestan, J.; Mueller, M.; Fumagalli, S.; Kozma, S.C.; Thomas, G. S6K1 −/−/S6K2 −/− Mice Exhibit Perinatal Lethality and Rapamycin-Sensitive 5′-Terminal Oligopyrimidine mRNA Translation and Reveal a Mitogen-Activated Protein Kinase-Dependent S6 Kinase Pathway. Mol. Cell. Biol. 2004, 24, 3112–3124. [Google Scholar] [CrossRef] [Green Version]
- Romeo, Y.; Zhang, X.; Roux, P.P. Regulation and function of the RSK family of protein kinases. Biochem. J. 2011, 441, 553–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, S.A.; Dreisbach, V.C.; Murphy, L.O.; Blenis, J.; Ogiso, Y.; Sugiura, R.; Kamo, T.; Yanagiya, S.; Lu, Y.; Okazaki, K.; et al. Characterization of Regulatory Events Associated with Membrane Targeting of p90 Ribosomal S6 Kinase 1. Mol. Cell. Biol. 2001, 21, 2324–2331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisinger-Mathason, T.K.; Andrade, J.; Groehler, A.L.; Clark, D.E.; Muratore-Schroeder, T.L.; Pasic, L.; Smith, J.A.; Shabanowitz, J.; Hunt, D.F.; Macara, I.G.; et al. Codependent Functions of RSK2 and the Apoptosis-Promoting Factor TIA-1 in Stress Granule Assembly and Cell Survival. Mol. Cell 2008, 31, 722–736. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, N.; Price, D.; Kyriakis, J.; Pelech, S.; Sanghera, J.; Avruch, J. An array of insulin-activated, proline-directed serine/threonine protein kinases phosphorylate the p70 S6 kinase. J. Biol. Chem. 1992, 267, 3325–3335. [Google Scholar] [CrossRef]
- Ma, L.; Chen, Z.; Erdjument-Bromage, H.; Tempst, P.; Pandolfi, P.P. Phosphorylation and Functional Inactivation of TSC2 by Erk: Implications for Tuberous Sclerosisand Cancer Pathogenesis. Cell 2005, 121, 179–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrière, A.; Cargnello, M.; Julien, L.-A.; Gao, H.; Bonneil, E.; Thibault, P.; Roux, P. Oncogenic MAPK Signaling Stimulates mTORC1 Activity by Promoting RSK-Mediated Raptor Phosphorylation. Curr. Biol. 2008, 18, 1269–1277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, K.-O.; Liang, Z.; Fei, E.; Huang, H.; Ip, N.Y. Cyclin-dependent Kinase 5 (Cdk5)-dependent Phosphorylation of p70 Ribosomal S6 Kinase 1 (S6K) Is Required for Dendritic Spine Morphogenesis. J. Biol. Chem. 2015, 290, 14637–14646. [Google Scholar] [CrossRef] [Green Version]
- Ruvinsky, I.; Sharon, N.; Lerer, T.; Cohen, H.; Stolovich-Rain, M.; Nir, T.; Dor, Y.; Zisman, P.; Meyuhas, O. Ribosomal protein S6 phosphorylation is a determinant of cell size and glucose homeostasis. Genes Dev. 2005, 19, 2199–2211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruvinsky, I.; Katz, M.; Dreazen, A.; Gielchinsky, Y.; Saada, A.; Freedman, N.; Mishani, E.; Zimmerman, G.; Kasir, J.; Meyuhas, O. Mice Deficient in Ribosomal Protein S6 Phosphorylation Suffer from Muscle Weakness that Reflects a Growth Defect and Energy Deficit. PLoS ONE 2009, 4, e5618. [Google Scholar] [CrossRef] [Green Version]
- Papageorgiou, A.; Rapley, J.; Mesirov, J.P.; Tamayo, P.; Avruch, J. A Genome-Wide siRNA Screen in Mammalian Cells for Regulators of S6 Phosphorylation. PLoS ONE 2015, 10, e0116096. [Google Scholar] [CrossRef]
- Takei, N.; Inamura, N.; Kawamura, M.; Namba, H.; Hara, K.; Yonezawa, K.; Nawa, H. Brain-Derived Neurotrophic Factor Induces Mammalian Target of Rapamycin-Dependent Local Activation of Translation Machinery and Protein Synthesis in Neuronal Dendrites. J. Neurosci. 2004, 24, 9760–9769. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Yuan, K.; Dai, W.; Sun, K.; Li, G.; Tan, M.; Song, W.; Yuan, Y. The mTORC1 signaling modulated by intracellular C3 activation in Paneth cells promotes intestinal epithelial regeneration during acute injury. Int. Immunopharmacol. 2018, 67, 54–61. [Google Scholar] [CrossRef]
- Palaniappan, M.; Menon, K. Human Chorionic Gonadotropin Stimulates Theca-Interstitial Cell Proliferation and Cell Cycle Regulatory Proteins by a cAMP-Dependent Activation of AKT/mTORC1 Signaling Pathway. Mol. Endocrinol. 2010, 24, 1782–1793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, S.; Wolgamott, L.; Yu, Y.; Blenis, J.; Yoon, S.-O. Glycogen synthase kinase (GSK)-3 promotes p70 ribosomal protein S6 kinase (p70S6K) activity and cell proliferation. Proc. Natl. Acad. Sci. USA 2011, 108, E1204–E1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gauglhofer, C.; Sagmeister, S.; Schrottmaier, W.; Fischer, C.; Rodgarkia-Dara, C.; Mohr, T.; Stättner, S.; Bichler, C.; Kandioler, D.; Wrba, F.; et al. Up-regulation of the fibroblast growth factor 8 subfamily in human hepatocellular carcinoma for cell survival and neoangiogenesis. Hepatology 2010, 53, 854–864. [Google Scholar] [CrossRef]
- Arsham, A.M.; Howell, J.J.; Simon, M.C. A Novel Hypoxia-inducible Factor-independent Hypoxic Response Regulating Mammalian Target of Rapamycin and Its Targets. J. Biol. Chem. 2003, 278, 29655–29660. [Google Scholar] [CrossRef] [Green Version]
- Parmar, S.; Smith, J.; Sassano, A.; Uddin, S.; Katsoulidis, E.; Majchrzak, B.; Kambhampati, S.; Eklund, E.A.; Tallman, M.S.; Fish, E.N.; et al. Differential regulation of the p70 S6 kinase pathway by interferon α (IFNα) and imatinib mesylate (STI571) in chronic myelogenous leukemia cells. Blood 2005, 106, 2436–2443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmeisser, H.; Fey, S.B.; Horowitz, J.; Fischer, E.R.; Balinsky, C.A.; Miyake, K.; Bekisz, J.; Snow, A.L.; Zoon, K.C. Type I interferons induce autophagy in certain human cancer cell lines. Autophagy 2013, 9, 683–696. [Google Scholar] [CrossRef] [Green Version]
- Kroczynska, B.; Kaur, S.; Katsoulidis, E.; Majchrzak-Kita, B.; Sassano, A.; Kozma, S.C.; Fish, E.N.; Platanias, L.C. Interferon-Dependent Engagement of Eukaryotic Initiation Factor 4B via S6 Kinase (S6K)- and Ribosomal Protein S6K-Mediated Signals. Mol. Cell. Biol. 2009, 29, 2865–2875. [Google Scholar] [CrossRef] [Green Version]
- Komata, Y.; Tsubota, S.; Sakamoto, K.; Ikematsu, S.; Kadomatsu, K. Screening of Novel Midkine Binding Protein by BioID2-Based Proximity Labeling. Nagoya J. Med. Sci. 2021, 83, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Stolovich, M.; Tang, H.; Hornstein, E.; Levy, G.; Cohen, R.; Bae, S.S.; Birnbaum, M.; Meyuhas, O. Transduction of Growth or Mitogenic Signals into Translational Activation of TOP mRNAs Is Fully Reliant on the Phosphatidylinositol 3-Kinase-Mediated Pathway but Requires neither S6K1 nor rpS6 Phosphorylation. Mol. Cell. Biol. 2002, 22, 8101–8113. [Google Scholar] [CrossRef] [Green Version]
- Ng, G.; Winder, D.; Muralidhar, B.; Gooding, E.; Roberts, I.; Pett, M.; Mukherjee, G.; Huang, J.; Coleman, N. Gain and overexpression of the oncostatin M receptor occur frequently in cervical squamous cell carcinoma and are associated with adverse clinical outcome. J. Pathol. 2007, 212, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Li, S.; Gan, X.; Qiu, C.; Chen, K.; Pei, H.; Wang, Q.; Li, D.; Li, X.; Yang, D.; et al. Wild-type p53-induced phosphatase 1 promotes vascular smooth muscle cell proliferation and neointima hyperplasia after vascular injury via p-adenosine 5′-monophosphate-activated protein kinase/mammalian target of rapamycin complex 1 pathway. J. Hypertens. 2019, 37, 2256–2268. [Google Scholar] [CrossRef]
- Jefferies, H.B.; Reinhard, C.; Kozma, S.C.; Thomas, G. Rapamycin selectively represses translation of the “polypyrimidine tract” mRNA family. Proc. Natl. Acad. Sci. USA 1994, 91, 4441–4445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ming, X.-F.; Rajapakse, A.G.; Carvas, J.; Ruffieux, J.; Yang, Z. Inhibition of S6K1 accounts partially for the anti-inflammatory effects of the arginase inhibitor L-norvaline. BMC Cardiovasc. Disord. 2009, 9, 12. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.-F.; Kuo, H.-P.; Chen, C.-T.; Hsu, J.-M.; Chou, C.-K.; Wei, Y.; Sun, H.-L.; Li, L.-Y.; Ping, B.; Huang, W.-C.; et al. IKKβ Suppression of TSC1 Links Inflammation and Tumor Angiogenesis via the mTOR Pathway. Cell 2007, 130, 440–455. [Google Scholar] [CrossRef] [Green Version]
- Inoki, K.; Zhu, T.; Guan, K.-L. TSC2 Mediates Cellular Energy Response to Control Cell Growth and Survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef] [Green Version]
- Smoly, I.; Shemesh, N.; Ziv-Ukelson, M.; Ben-Zvi, A.; Yeger-Lotem, E. An Asymmetrically Balanced Organization of Kinases versus Phosphatases across Eukaryotes Determines Their Distinct Impacts. PLoS Comput. Biol. 2017, 13, e1005221. [Google Scholar] [CrossRef] [Green Version]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.-L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nature 2002, 4, 648–657. [Google Scholar] [CrossRef]
- Manning, B.D.; Tee, A.; Logsdon, M.; Blenis, J.; Cantley, L.C. Identification of the Tuberous Sclerosis Complex-2 Tumor Suppressor Gene Product Tuberin as a Target of the Phosphoinositide 3-Kinase/Akt Pathway. Mol. Cell 2002, 10, 151–162. [Google Scholar] [CrossRef]
- Cam, M.; Bid, H.K.; Xiao, L.; Zambetti, G.P.; Houghton, P.J.; Cam, H. p53/TAp63 and AKT Regulate Mammalian Target of Rapamycin Complex 1 (mTORC1) Signaling through Two Independent Parallel Pathways in the Presence of DNA Damage. J. Biol. Chem. 2014, 289, 4083–4094. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Liu, Q.; Li, F.; Qiu, J.; Fan, H.; Ma, H.; Zhu, Y.; Wu, L.; Han, X.; Yang, Z.; et al. Apoptosis induced by PGC-1β in breast cancer cells is mediated by the mTOR pathway. Oncol. Rep. 2013, 30, 1631–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ly, C.; Arechiga, A.F.; Melo, J.V.; Walsh, C.M.; Ong, S.T. Bcr-Abl kinase modulates the translation regulators ribosomal protein S6 and 4E-BP1 in chronic myelogenous leukemia cells via the mammalian target of rapamycin. Cancer Res. 2003, 63, 5716–5722. [Google Scholar]
- Carayol, N.; Katsoulidis, E.; Sassano, A.; Altman, J.K.; Druker, B.; Platanias, L.C. Suppression of Programmed Cell Death 4 (PDCD4) Protein Expression by BCR-ABL-regulated Engagement of the mTOR/p70 S6 Kinase Pathway. J. Biol. Chem. 2008, 283, 8601–8610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markova, B.; Albers, C.; Breitenbuecher, F.; Melo, J.V.; Brümmendorf, T.H.; Heidel, F.; Lipka, D.; Duyster, J.; Huber, C.; Fischer, T. Novel pathway in Bcr-Abl signal transduction involves Akt-independent, PLC-γ1-driven activation of mTOR/p70S6-kinase pathway. Oncogene 2009, 29, 739–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.-B.; Wei, M.-X.; Han, J.-Y.; Wu, X.-Y.; Li, C.; Wang, J.; Shen, W.; Lu, P.-H. MicroRNA-451 regulates AMPK/mTORC1 signaling and fascin1 expression in HT-29 colorectal cancer. Cell. Signal. 2013, 26, 102–109. [Google Scholar] [CrossRef]
- Pinna, L.A. Protein kinase CK2: A challenge to canons. J. Cell Sci. 2002, 115, 3873–3878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruzzene, M.; Pinna, L.A. Addiction to protein kinase CK2: A common denominator of diverse cancer cells? Biochim. Biophys. Acta (BBA) Proteins Proteom. 2010, 1804, 499–504. [Google Scholar] [CrossRef]
- Litchfield, D.W. Protein kinase CK2: Structure, regulation and role in cellular decisions of life and death. Biochem. J. 2003, 369, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Salizzato, V.; Borgo, C.; Cesaro, L.; Pinna, L.A.; Donella-Deana, A. Inhibition of protein kinase CK2 by CX-5011 counteracts imatinib-resistance preventing rpS6 phosphorylation in chronic myeloid leukaemia cells: New combined therapeutic strategies. Oncotarget 2016, 7, 18204–18218. [Google Scholar] [CrossRef]
- Alcaraz, E.; Vilardell, J.; Borgo, C.; Sarró, E.; Plana, M.; Marin, O.; Pinna, L.A.; Bayascas, J.; Meseguer, A.; Salvi, M.; et al. Effects of CK2β subunit down-regulation on Akt signalling in HK-2 renal cells. PLoS ONE 2020, 15, e0227340. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Henderson, P.; Pettersson, S.; Satsangi, J.; Hupp, T.; Stevens, C. Tuberous sclerosis-2 (TSC2) regulates the stability of death-associated protein kinase-1 (DAPK) through a lysosome-dependent degradation pathway. FEBS J. 2010, 278, 354–370. [Google Scholar] [CrossRef]
- Koziczak, M.; Hynes, N.E. Cooperation between Fibroblast Growth Factor Receptor-4 and ErbB2 in Regulation of Cyclin D1 Translation. J. Biol. Chem. 2004, 279, 50004–50011. [Google Scholar] [CrossRef] [Green Version]
- Ruicci, K.M.; Pinto, N.; Khan, M.I.; Yoo, J.; Fung, K.; MacNeil, D.; Mymryk, J.S.; Barrett, J.W.; Nichols, A.C. ERK-TSC2 signalling in constitutively-active HRAS mutant HNSCC cells promotes resistance to PI3K inhibition. Oral Oncol. 2018, 84, 95–103. [Google Scholar] [CrossRef]
- Pusapati, R.V.; Daemen, A.; Wilson, C.; Sandoval, W.; Gao, M.; Haley, B.; Baudy, A.R.; Hatzivassiliou, G.; Evangelista, M.; Settleman, J. mTORC1-Dependent Metabolic Reprogramming Underlies Escape from Glycolysis Addiction in Cancer Cells. Cancer Cell 2016, 29, 548–562. [Google Scholar] [CrossRef] [Green Version]
- Jia, C.-H.; Li, M.; Liu, J.; Zhao, L.; Lin, J.; Lai, P.-L.; Zhou, X.; Zhang, Y.; Chen, Z.-G.; Li, H.-Y.; et al. IKK-β mediates hydrogen peroxide induced cell death through p85 S6K1. Cell Death Differ. 2012, 20, 248–258. [Google Scholar] [CrossRef] [Green Version]
- Findlay, G.M.; Yan, L.; Procter, J.; Mieulet, V.; Lamb, R.F. A MAP4 kinase related to Ste20 is a nutrient-sensitive regulator of mTOR signalling. Biochem. J. 2007, 403, 13–20. [Google Scholar] [CrossRef] [Green Version]
- Yan, L.; Mieulet, V.; Burgess, D.; Findlay, G.; Sully, K.; Procter, J.; Goris, J.; Janssens, V.; Morrice, N.A.; Lamb, R.F. PP2AT61ɛ Is an Inhibitor of MAP4K3 in Nutrient Signaling to mTOR. Mol. Cell 2010, 37, 633–642. [Google Scholar] [CrossRef]
- Nawroth, R.; Stellwagen, F.; Schulz, W.A.; Stoehr, R.; Hartmann, A.; Krause, B.J.; Gschwend, J.E.; Retz, M. S6K1 and 4E-BP1 Are Independent Regulated and Control Cellular Growth in Bladder Cancer. PLoS ONE 2011, 6, e27509. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.-N.; Wang, X.-K.; Wu, S.-Q.; Lu, J.; Zheng, M.; Wang, Y.-H.; Zhou, H.; Zhang, H.; Han, J. Phosphorylation of Raptor by p38β Participates in Arsenite-induced Mammalian Target of Rapamycin Complex 1 (mTORC1) Activation. J. Biol. Chem. 2011, 286, 31501–31511. [Google Scholar] [CrossRef] [Green Version]
- Alessi, D.R.; James, S.R.; Downes, C.; Holmes, A.B.; Gaffney, P.R.; Reese, C.B.; Cohen, P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Bα. Curr. Biol. 1997, 7, 261–269. [Google Scholar] [CrossRef] [Green Version]
- Scheid, M.P.; Marignani, P.A.; Woodgett, J. Multiple Phosphoinositide 3-Kinase-Dependent Steps in Activation of Protein Kinase B. Mol. Cell. Biol. 2002, 22, 6247–6260. [Google Scholar] [CrossRef] [Green Version]
- Pullen, N.; Dennis, P.B.; Andjelkovic, M.; Dufner, A.; Kozma, S.C.; Hemmings, B.A.; Thomas, G. Phosphorylation and Activation of p70 s6k by PDK1. Science 1998, 279, 707–710. [Google Scholar] [CrossRef]
- Jensen, C.J.; Buch, M.-B.; Krag, T.; Hemmings, B.A.; Gammeltoft, S.; Frödin, M. 90-kDa Ribosomal S6 Kinase Is Phosphorylated and Activated by 3-Phosphoinositide-dependent Protein Kinase-1. J. Biol. Chem. 1999, 274, 27168–27176. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.; Ma, Y.; Moore, M.; Hemmings, B.A.; Taylor, S.S. Phosphorylation and activation of cAMP-dependent protein kinase by phosphoinositide-dependent protein kinase. Proc. Natl. Acad. Sci. USA 1998, 95, 9849–9854. [Google Scholar] [CrossRef] [Green Version]
- Chou, M.M.; Hou, W.; Johnson, J.; Graham, L.K.; Lee, M.H.; Chen, C.-S.; Newton, A.C.; Schaffhausen, B.S.; Toker, A. Regulation of protein kinase C ζ by PI 3-kinase and PDK-1. Curr. Biol. 1998, 8, 1069–1078. [Google Scholar] [CrossRef] [Green Version]
- King, C.C.; Gardiner, E.M.M.; Zenke, F.T.; Bohl, B.P.; Newton, A.C.; Hemmings, B.A.; Bokoch, G.M. p21-activated Kinase (PAK1) Is Phosphorylated and Activated by 3-Phosphoinositide-dependent Kinase-1 (PDK1). J. Biol. Chem. 2000, 275, 41201–41209. [Google Scholar] [CrossRef] [Green Version]
- Balendran, A.; Casamayor, A.; Deak, M.; Paterson, A.; Gaffney, P.; Currie, R.; Downes, C.; Alessi, D.R. PDK1 acquires PDK2 activity in the presence of a synthetic peptide derived from the carboxyl terminus of PRK2. Curr. Biol. 1999, 9, 393–404. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, H.; Fujita, N.; Tsuruo, T. 3-Phosphoinositide-dependent Protein Kinase-1-mediated IκB Kinase β (IKKB) Phosphorylation Activates NF-κB Signaling. J. Biol. Chem. 2005, 280, 40965–40973. [Google Scholar] [CrossRef] [Green Version]
- Alessi, D.; Kozlowski, M.T.; Weng, Q.-P.; Morrice, N.; Avruch, J. 3-Phosphoinositide-dependent protein kinase 1 (PDK1) phosphorylates and activates the p70 S6 kinase in vivo and in vitro. Curr. Biol. 1998, 8, 69–81. [Google Scholar] [CrossRef] [Green Version]
- Kazemi, S.; Mounir, Z.; Baltzis, D.; Raven, J.F.; Wang, S.; Krishnamoorthy, J.-L.; Pluquet, O.; Pelletier, J.; Koromilas, A.E. A Novel Function of eIF2α Kinases as Inducers of the Phosphoinositide-3 Kinase Signaling Pathway. Mol. Biol. Cell 2007, 18, 3635–3644. [Google Scholar] [CrossRef]
- Wojtalla, A.; Fischer, B.; Kotelevets, N.; Mauri, F.A.; Sobek, J.; Rehrauer, H.; Wotzkow, C.; Tschan, M.P.; Seckl, M.J.; Zangemeister-Wittke, U.; et al. Targeting the Phosphoinositide 3-Kinase p110-α Isoform Impairs Cell Proliferation, Survival, and Tumor Growth in Small Cell Lung Cancer. Clin. Cancer Res. 2012, 19, 96–105. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Zavorotinskaya, T.; Dai, Y.; Niu, X.-H.; Castillo, J.; Sim, J.; Yu, J.; Wang, Y.; Langowski, J.L.; Holash, J.; et al. Pim2 is required for maintaining multiple myeloma cell growth through modulating TSC2 phosphorylation. Blood 2013, 122, 1610–1620. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, A.; Eckerdt, F.; Bell, J.; Nakano, I.; Giles, F.J.; Cheng, S.-Y.; Lulla, R.; Goldman, S.; Platanias, L.C. Targeting of glioblastoma cell lines and glioma stem cells by combined PIM kinase and PI3K-p110α inhibition. Oncotarget 2016, 7, 33192–33201. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Stevens, P.D.; Li, X.; Schmidt, M.D.; Gao, T. PHLPP-Mediated Dephosphorylation of S6K1 Inhibits Protein Translation and Cell Growth. Mol. Cell. Biol. 2011, 31, 4917–4927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, T.; Brognard, J.; Newton, A.C. The Phosphatase PHLPP Controls the Cellular Levels of Protein Kinase C. J. Biol. Chem. 2008, 283, 6300–6311. [Google Scholar] [CrossRef] [Green Version]
- Renner, A.G.; Créancier, L.; Dos Santos, C.; Fialin, C.; Récher, C.; Bailly, C.; Kruczynski, A.; Payrastre, B.; Manenti, S. A functional link between Polo-like kinase 1 and the mammalian Target-Of-Rapamycin pathway? Cell Cycle 2010, 9, 1690–1696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, J.B.; Pallas, D.C. Circumventing Cellular Control of PP2A by Methylation Promotes Transformation in an Akt-Dependent Manner. Neoplasia 2012, 14, 585–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.-R.; Chen, M.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor: New modes and prospects. Nat. Rev. Mol. Cell Biol. 2018, 19, 547–562. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Washington, A.M.; Hinton, B.T. PTEN signaling through RAF1 proto-oncogene serine/threonine kinase (RAF1)/ERK in the epididymis is essential for male fertility. Proc. Natl. Acad. Sci. USA 2014, 111, 18643–18648. [Google Scholar] [CrossRef] [Green Version]
- Steward, O.; Coulibaly, A.P.; Metcalfe, M.; Yonan, J.M.; Yee, K.M. AAVshRNA-mediated PTEN knockdown in adult neurons attenuates activity-dependent immediate early gene induction. Exp. Neurol. 2019, 326, 113098. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, N.; Ma, D.; Liu, L.; Jiang, L.; Zhou, Y.; Zeng, X.; Li, J.; Chen, Q. Receptor for activated C kinase 1 (RACK1) promotes the progression of OSCC via the AKT/mTOR pathway. Int. J. Oncol. 2016, 49, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Roux, P.; Ballif, B.A.; Anjum, R.; Gygi, S.P.; Blenis, J. Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proc. Natl. Acad. Sci. USA 2004, 101, 13489–13494. [Google Scholar] [CrossRef] [Green Version]
- Sun, E.L.; Liu, C.X.; Ma, Z.X.; Mou, X.Y.; Mu, X.A.; Ni, Y.H.; Li, X.L.; Zhang, D.; Ju, Y.R. Knockdown of human serine/threonine kinase 33 suppresses human small cell lung carcinoma by blocking RPS6/BAD signaling transduction. Neoplasma 2017, 64, 869–879. [Google Scholar] [CrossRef] [Green Version]
- Zachari, M.; Ganley, I.G. The mammalian ULK1 complex and autophagy initiation. Essays Biochem. 2017, 61, 585–596. [Google Scholar] [CrossRef] [Green Version]
- Dunlop, E.; Hunt, D.K.; Acosta-Jaquez, H.A.; Fingar, D.C.; Tee, A.R. ULK1 inhibits mTORC1 signaling, promotes multisite Raptor phosphorylation and hinders substrate binding. Autophagy 2011, 7, 737–747. [Google Scholar] [CrossRef]
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; Gelino, S.; Kohnz, R.A.; Mair, W.; Vasquez, D.S.; Joshi, A.; Gwinn, D.M.; Taylor, R.; et al. Phosphorylation of ULK1 (hATG1) by AMP-Activated Protein Kinase Connects Energy Sensing to Mitophagy. Science 2011, 331, 456–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Li, X.; Liu, S.; Guo, L.; Zhang, B.; Zhang, J.; Ye, Q. Programmed cell death-1 (PD-1) checkpoint blockade in combination with a mammalian target of rapamycin inhibitor restrains hepatocellular carcinoma growth induced by hepatoma cell-intrinsic PD-1. Hepatology 2017, 66, 1920–1933. [Google Scholar] [CrossRef] [Green Version]
- Kleffel, S.; Posch, C.; Barthel, S.R.; Mueller, H.; Schlapbach, C.; Guenova, E.; Elco, C.P.; Lee, N.; Juneja, V.R.; Zhan, Q.; et al. Melanoma Cell-Intrinsic PD-1 Receptor Functions Promote Tumor Growth. Cell 2015, 162, 1242–1256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, L.; Qian, G.; Zhang, S.; Zheng, H.; Fan, S.; Lesinski, G.B.; Owonikoko, T.K.; Ramalingam, S.S.; Sun, S.-Y. Inhibition of mTOR complex 1/p70 S6 kinase signaling elevates PD-L1 levels in human cancer cells through enhancing protein stabilization accompanied with enhanced β-TrCP degradation. Oncogene 2019, 38, 6270–6282. [Google Scholar] [CrossRef]
- Crane, C.A.; Panner, A.; Murray, J.C.; Wilson, S.; Xu, H.; Chen, L.; Simko, J.P.; Waldman, F.M.; Pieper, R.; Parsa, A.T. PI(3) kinase is associated with a mechanism of immunoresistance in breast and prostate cancer. Oncogene 2008, 28, 306–312. [Google Scholar] [CrossRef] [Green Version]
- Qiao, J.; Kang, J.; Ishola, T.A.; Rychahou, P.; Evers, B.M.; Chung, D.H. Gastrin-releasing peptide receptor silencing suppresses the tumorigenesis and metastatic potential of neuroblastoma. Proc. Natl. Acad. Sci. USA 2008, 105, 12891–12896. [Google Scholar] [CrossRef] [Green Version]
- Suber, T.L.; Nikolli, I.; O’Brien, M.E.; Londino, J.; Zhao, J.; Chen, K.; Mallampalli, R.K.; Zhao, Y. FBXO17 promotes cell proliferation through activation of Akt in lung adenocarcinoma cells. Respir. Res. 2018, 19, 206. [Google Scholar] [CrossRef] [Green Version]
- Zhou, G.; Wang, J.; Zhao, M.; Xie, T.-X.; Tanaka, N.; Sano, D.; Patel, A.A.; Ward, A.M.; Sandulache, V.; Jasser, S.A.; et al. Gain-of-Function Mutant p53 Promotes Cell Growth and Cancer Cell Metabolism via Inhibition of AMPK Activation. Mol. Cell 2014, 54, 960–974. [Google Scholar] [CrossRef] [Green Version]
- Martinez, H.D.; Hsiao, J.; Jasavala, R.J.; Hinkson, I.V.; Eng, J.; Wright, M.E. Androgen-Sensitive Microsomal Signaling Networks Coupled to the Proliferation and Differentiation of Human Prostate Cancer Cells. Genes Cancer 2011, 2, 956–978. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Jiang, L.; Wang, W.; Jiang, T. BTF3 Silencing Inhibits the Proliferation of Osteosarcoma Cells. J. Cancer 2019, 10, 1855–1861. [Google Scholar] [CrossRef] [PubMed]
- Khatri, S.; Yepiskoposyan, H.; Gallo, C.A.; Tandon, P.; Plas, D.R. FOXO3a Regulates Glycolysis via Transcriptional Control of Tumor Suppressor TSC1. J. Biol. Chem. 2010, 285, 15960–15965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brugarolas, J.; Lei, K.; Hurley, R.L.; Manning, B.D.; Reiling, J.H.; Hafen, E.; Witters, L.A.; Ellisen, L.W.; Kaelin, W.G., Jr. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004, 18, 2893–2904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorrello, N.V.; Peschiaroli, A.; Guardavaccaro, D.; Colburn, N.H.; Sherman, N.E.; Pagano, M. S6K1- and βTRCP-Mediated Degradation of PDCD4 Promotes Protein Translation and Cell Growth. Science 2006, 314, 467–471. [Google Scholar] [CrossRef]
- Hwang, S.-H.; Bang, S.; Kim, W.; Chung, J. Von Hippel–Lindau tumor suppressor (VHL) stimulates TOR signaling by interacting with phosphoinositide 3-kinase (PI3K). J. Biol. Chem. 2020, 295, 2336–2347. [Google Scholar] [CrossRef] [PubMed]
- Kim, U.; Wang, Y.; Sanford, T.; Zeng, Y.; Nishikura, K. Molecular cloning of cDNA for double-stranded RNA adenosine deaminase, a candidate enzyme for nuclear RNA editing. Proc. Natl. Acad. Sci. USA 1994, 91, 11457–11461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dou, N.; Yu, S.; Ye, X.; Yang, D.; Li, Y.; Gao, Y. Aberrant overexpression of ADAR1 promotes gastric cancer progression by activating mTOR/p70S6K signaling. Oncotarget 2016, 7, 86161–86173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheidenhelm, D.K.; Cresswell, J.; Haipek, C.A.; Fleming, T.P.; Mercer, R.W.; Gutmann, D.H. Akt-Dependent Cell Size Regulation by the Adhesion Molecule on Glia Occurs Independently of Phosphatidylinositol 3-Kinase and Rheb Signaling. Mol. Cell. Biol. 2005, 25, 3151–3162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugita, T.; Aya, H.; Ueno, M.; Ishizuka, T.; Kawashima, K. Characterization of molecularly cloned human 5-aminoimidazole-4-carboxamide ribonucleotide transformylase. J. Biochem. 1997, 122, 309–313. [Google Scholar] [CrossRef]
- Li, M.; Jin, C.; Xu, M.; Zhou, L.; Li, D.; Yin, Y. Bifunctional enzyme ATIC promotes propagation of hepatocellular carcinoma by regulating AMPK-mTOR-S6 K1 signaling. Cell Commun. Signal. 2017, 15, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, T.; Zhu, Z.; Wu, J.; She, H.; Han, R.; Xu, H.; Qin, Z.-H. DRAM1 regulates autophagy and cell proliferation via inhibition of the phosphoinositide 3-kinase-Akt-mTOR-ribosomal protein S6 pathway. Cell Commun. Signal. 2019, 17, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, M.; Sandercock, A.M.; Fraser, C.S.; Ridlova, G.; Stephens, E.; Schenauer, M.R.; Yokoi-Fong, T.; Barsky, D.; Leary, J.A.; Hershey, J.W.; et al. Mass spectrometry reveals modularity and a complete subunit interaction map of the eukaryotic translation factor eIF3. Proc. Natl. Acad. Sci. USA 2008, 105, 18139–18144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damoc, E.; Fraser, C.S.; Zhou, M.; Videler, H.; Mayeur, G.L.; Hershey, J.W.B.; Doudna, J.A.; Robinson, C.V.; Leary, J.A. Structural Characterization of the Human Eukaryotic Initiation Factor 3 Protein Complex by Mass Spectrometry. Mol. Cell. Proteom. 2007, 6, 1135–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hershey, J.W. The role of eIF3 and its individual subunits in cancer. Biochim. Biophys. Acta (BBA) Bioenerg. 2015, 1849, 792–800. [Google Scholar] [CrossRef]
- Holz, M.K.; Ballif, B.A.; Gygi, S.P.; Blenis, J. mTOR and S6K1 Mediate Assembly of the Translation Preinitiation Complex through Dynamic Protein Interchange and Ordered Phosphorylation Events. Cell 2005, 123, 569–580. [Google Scholar] [CrossRef] [Green Version]
- Schipany, K.; Rosner, M.; Ionce, L.; Hengstschläger, M.; Kovacic, B. eIF3 controls cell size independently of S6K1-activity. Oncotarget 2015, 6, 24361–24375. [Google Scholar] [CrossRef] [Green Version]
- Fuka, G.; Kantner, H.-P.; Grausenburger, R.; Inthal, A.; Bauer, E.; Krapf, G.; Kaindl, U.; Kauer, M.; Dworzak, M.N.; Stoiber-Sakaguchi, D.; et al. Silencing of ETV6/RUNX1 abrogates PI3K/AKT/mTOR signaling and impairs reconstitution of leukemia in xenografts. Leukemia 2011, 26, 927–933. [Google Scholar] [CrossRef] [PubMed]
- Tomek, K.; Wagner, R.; Varga, F.; Singer, C.F.; Karlic, H.; Grunt, T.W. Blockade of Fatty Acid Synthase Induces Ubiquitination and Degradation of Phosphoinositide-3-Kinase Signaling Proteins in Ovarian Cancer. Mol. Cancer Res. 2011, 9, 1767–1779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaistha, B.P.; Lorenz, H.; Schmidt, H.; Sipos, B.; Pawlak, M.; Gierke, B.; Kreider, R.; Lankat-Buttgereit, B.; Sauer, M.; Fiedler, L.; et al. PLAC8 Localizes to the Inner Plasma Membrane of Pancreatic Cancer Cells and Regulates Cell Growth and Disease Progression through Critical Cell-Cycle Regulatory Pathways. Cancer Res. 2015, 76, 96–107. [Google Scholar] [CrossRef] [Green Version]
- Kinsey, C.; Balakrishnan, V.; O’Dell, M.R.; Huang, J.L.; Newman, L.; Whitney-Miller, C.L.; Hezel, A.F.; Land, H. Plac8 Links Oncogenic Mutations to Regulation of Autophagy and Is Critical to Pancreatic Cancer Progression. Cell Rep. 2014, 7, 1143–1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Wang, X.; Fang, Y.; Huo, Z.; Lu, X.; Zhan, X.; Deng, X.; Peng, C.; Shen, B. Integrated expression profiles analysis reveals novel predictive biomarker in pancreatic ductal adenocarcinoma. Oncotarget 2017, 8, 52571–52583. [Google Scholar] [CrossRef]
- Tatura, M.; Schmidt, H.; Haijat, M.; Stark, M.; Rinke, A.; Diels, R.; Lawlor, R.T.; Scarpa, A.; Schrader, J.; Hackert, T.; et al. Placenta-Specific 8 Is Overexpressed and Regulates Cell Proliferation in Low-Grade Human Pancreatic Neuroendocrine Tumors. Neuroendocrinology 2019, 110, 23–34. [Google Scholar] [CrossRef]
- Mejia, L.A.; Litterman, N.; Ikeuchi, Y.; De La Torre-Ubieta, L.; Bennett, E.J.; Zhang, C.; Harper, J.W.; Bonni, A. A Novel Hap1–Tsc1 Interaction Regulates Neuronal mTORC1 Signaling and Morphogenesis in the Brain. J. Neurosci. 2013, 33, 18015–18021. [Google Scholar] [CrossRef] [Green Version]
- Han, G.-S.; Carman, G.M. Characterization of the Human LPIN1-encoded Phosphatidate Phosphatase Isoforms. J. Biol. Chem. 2010, 285, 14628–14638. [Google Scholar] [CrossRef] [Green Version]
- Finck, B.N.; Gropler, M.C.; Chen, Z.; Leone, T.C.; Croce, M.A.; Harris, T.E.; Lawrence, J.C.; Kelly, D.P. Lipin 1 is an inducible amplifier of the hepatic PGC-1α/PPARα regulatory pathway. Cell Metab. 2006, 4, 199–210. [Google Scholar] [CrossRef] [Green Version]
- Péterfy, M.; Phan, J.; Reue, K. Alternatively Spliced Lipin Isoforms Exhibit Distinct Expression Pattern, Subcellular Localization, and Role in Adipogenesis. J. Biol. Chem. 2005, 280, 32883–32889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, Y.-K.; Lee, M.-Y.; Kim, J.-W.; Kim, M.; Moon, J.-S.; Lee, Y.-J.; Ahn, Y.H.; Kim, K.-S. Lipin1 Is a Key Factor for the Maturation and Maintenance of Adipocytes in the Regulatory Network with CCAAT/Enhancer-binding Protein α and Peroxisome Proliferator-activated Receptor γ2. J. Biol. Chem. 2008, 283, 34896–34906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brohée, L.; Demine, S.; Willems, J.; Arnould, T.; Colige, A.C.; Deroanne, C.F. Lipin-1 regulates cancer cell phenotype and is a potential target to potentiate rapamycin treatment. Oncotarget 2015, 6, 11264–11280. [Google Scholar] [CrossRef] [PubMed]
- Clarke, C.J.; Mediwala, K.; Jenkins, R.W.; Sutton, C.A.; Tholanikunnel, B.G.; Hannun, Y.A. Neutral Sphingomyelinase-2 Mediates Growth Arrest by Retinoic Acid through Modulation of Ribosomal S6 Kinase. J. Biol. Chem. 2011, 286, 21565–21576. [Google Scholar] [CrossRef] [Green Version]
- Duran, A.; Amanchy, R.; Linares, J.F.; Joshi, J.; Abu-Baker, S.; Porollo, A.; Hansen, M.; Moscat, J.; Diaz-Meco, M.T. p62 Is a Key Regulator of Nutrient Sensing in the mTORC1 Pathway. Mol. Cell 2011, 44, 134–146. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, C.; Garces, R.G.; Edmonds, K.A.; Hiller, S.; Hyberts, S.G.; Marintchev, A.; Wagner, G. PDCD4 inhibits translation initiation by binding to eIF4A using both its MA3 domains. Proc. Natl. Acad. Sci. USA 2008, 105, 3274–3279. [Google Scholar] [CrossRef] [Green Version]
- Loh, P.G.; Yang, H.-S.; Walsh, M.A.; Wang, Q.; Wang, X.; Cheng, Z.; Liu, D.; Song, H. Structural basis for translational inhibition by the tumour suppressor Pdcd4. EMBO J. 2009, 28, 274–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Wang, Q.; Chen, L.; Yang, H.-S. Inhibition of p70S6K1 Activation by Pdcd4 Overcomes the Resistance to an IGF-1R/IR Inhibitor in Colon Carcinoma Cells. Mol. Cancer Ther. 2015, 14, 799–809. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Zhang, Y.; Zhu, J.; Zheng, H.; Chen, S.; Chen, L.; Yang, H.-S. IGF-1R inhibition induces MEK phosphorylation to promote survival in colon carcinomas. Signal Transduct. Target. Ther. 2020, 5, 153. [Google Scholar] [CrossRef]
- Gan, B.; Melkoumian, Z.K.; Wu, X.; Guan, K.-L.; Guan, J.-L. Identification of FIP200 interaction with the TSC1–TSC2 complex and its role in regulation of cell size control. J. Cell Biol. 2005, 170, 379–389. [Google Scholar] [CrossRef] [Green Version]
- Karbowniczek, M.; Cash, T.; Cheung, M.; Robertson, G.P.; Astrinidis, A.; Henske, E.P. Regulation of B-Raf Kinase Activity by Tuberin and Rheb Is Mammalian Target of Rapamycin (mTOR)-independent. J. Biol. Chem. 2004, 279, 29930–29937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunlop, E.; Dodd, K.M.; Seymour, L.A.; Tee, A.R. Mammalian target of rapamycin complex 1-mediated phosphorylation of eukaryotic initiation factor 4E-binding protein 1 requires multiple protein–protein interactions for substrate recognition. Cell. Signal. 2009, 21, 1073–1084. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Nakashima, A.; Guo, L.; Tamanoi, F. Specific Activation of mTORC1 by Rheb G-protein in Vitro Involves Enhanced Recruitment of Its Substrate Protein. J. Biol. Chem. 2009, 284, 12783–12791. [Google Scholar] [CrossRef] [Green Version]
- Bar-Peled, L.; Chantranupong, L.; Cherniack, A.D.; Chen, W.W.; Ottina, K.A.; Grabiner, B.C.; Spear, E.D.; Carter, S.L.; Meyerson, M.; Sabatini, D.M. A Tumor Suppressor Complex with GAP Activity for the Rag GTPases That Signal Amino Acid Sufficiency to mTORC1. Science 2013, 340, 1100–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Igarashi, M.; Guarente, L. mTORC1 and SIRT1 Cooperate to Foster Expansion of Gut Adult Stem Cells during Calorie Restriction. Cell 2016, 166, 436–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Çakir, I.; Perello, M.; Lansari, O.; Messier, N.J.; Vaslet, C.A.; Nillni, E.A. Hypothalamic Sirt1 Regulates Food Intake in a Rodent Model System. PLoS ONE 2009, 4, e8322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, K.; Herzig, S.; Kulkarni, R.N.; Montminy, M. TRB3: A tribbles Homolog That Inhibits Akt/PKB Activation by Insulin in Liver. Science 2003, 300, 1574–1577. [Google Scholar] [CrossRef] [Green Version]
- Matsushima, R.; Harada, N.; Webster, N.J.G.; Tsutsumi, Y.M.; Nakaya, Y. Effect of TRB3 on Insulin and Nutrient-stimulated Hepatic p70 S6 Kinase Activity. J. Biol. Chem. 2006, 281, 29719–29729. [Google Scholar] [CrossRef] [Green Version]
- Rosner, M.; Hengstschläger, M. Nucleocytoplasmic localization of p70 S6K1, but not of its isoforms p85 and p31, is regulated by TSC2/mTOR. Oncogene 2011, 30, 4509–4522. [Google Scholar] [CrossRef] [Green Version]
- Zoncu, R.; Bar-Peled, L.; Efeyan, A.; Wang, S.; Sancak, Y.; Sabatini, D.M. mTORC1 Senses Lysosomal Amino Acids Through an Inside-Out Mechanism That Requires the Vacuolar H + -ATPase. Science 2011, 334, 678–683. [Google Scholar] [CrossRef] [Green Version]
- Gossage, L.; Eisen, T.; Maher, E.R. VHL, the story of a tumour suppressor gene. Nat. Rev. Cancer 2014, 15, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G. The von Hippel–Lindau Tumor Suppressor Protein. Annu. Rev. Cancer Biol. 2018, 2, 91–109. [Google Scholar] [CrossRef]
- Li, T.; Liu, X.; Gong, X.; E, Q.; Zhang, X.; Zhang, X. microRNA 92b-3p regulates primordial follicle assembly by targeting TSC1 in neonatal mouse ovaries. Cell Cycle 2019, 18, 824–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godlewski, J.; Nowicki, M.O.; Bronisz, A.; Nuovo, G.; Palatini, J.; De Lay, M.; Van Brocklyn, J.; Ostrowski, M.; Chiocca, E.A.; Lawler, S.E. MicroRNA-451 Regulates LKB1/AMPK Signaling and Allows Adaptation to Metabolic Stress in Glioma Cells. Mol. Cell 2010, 37, 620–632. [Google Scholar] [CrossRef] [Green Version]
- Alexander, A.; Cai, S.-L.; Kim, J.; Nanez, A.; Sahin, M.; MacLean, K.H.; Inoki, K.; Guan, K.-L.; Shen, J.; Person, M.D.; et al. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc. Natl. Acad. Sci. USA 2010, 107, 4153–4158. [Google Scholar] [CrossRef] [Green Version]
- Moon, H.-S.; Batirel, S.; Mantzoros, C.S. Alpha linolenic acid and oleic acid additively down-regulate malignant potential and positively cross-regulate AMPK/S6 axis in OE19 and OE33 esophageal cancer cells. Metabolism 2014, 63, 1447–1454. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Chen, S.-Y.; Ross, K.N.; Balk, S.P. Androgens Induce Prostate Cancer Cell Proliferation through Mammalian Target of Rapamycin Activation and Post-transcriptional Increases in Cyclin D Proteins. Cancer Res. 2006, 66, 7783–7792. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Henske, E.P. Estrogen-Induced Activation of Mammalian Target of Rapamycin Is Mediated via Tuberin and the Small GTPase Ras Homologue Enriched in Brain. Cancer Res. 2006, 66, 9461–9466. [Google Scholar] [CrossRef] [Green Version]
- Egea-Jimenez, A.L.; Zimmermann, P. Phospholipase D and phosphatidic acid in the biogenesis and cargo loading of extracellular vesicles. J. Lipid Res. 2018, 59, 1554–1560. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.; Vilella-Bach, M.; Bachmann, R.; Flanigan, A.; Chen, J. Phosphatidic Acid-Mediated Mitogenic Activation of mTOR Signaling. Science 2001, 294, 1942–1945. [Google Scholar] [CrossRef]
- Toschi, A.; Lee, E.; Xu, L.; Garcia, A.; Gadir, N.; Foster, D.A. Regulation of mTORC1 and mTORC2 Complex Assembly by Phosphatidic Acid: Competition with Rapamycin. Mol. Cell. Biol. 2009, 29, 1411–1420. [Google Scholar] [CrossRef] [Green Version]
- Baan, B.; Dihal, A.A.; Hoff, E.; Bos, C.L.; Voorneveld, P.W.; Koelink, P.J.; Wildenberg, M.E.; Muncan, V.; Heijmans, J.; Verspaget, H.W.; et al. 5-aminosalicylic acid inhibits cell cycle progression in a phospholipase D dependent manner in colorectal cancer. Gut 2011, 61, 1708–1715. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Properties of FDA-approved small molecule protein kinase inhibitors: A 2021 update. Pharmacol. Res. 2021, 165, 105463. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Lee, K.J.; Seo, Y.; Kwon, J.-H.; Yoon, J.P.; Kang, J.Y.; Lee, H.J.; Park, S.J.; Hong, S.P.; Cheon, J.H.; et al. Effects of metformin on colorectal cancer stem cells depend on alterations in glutamine metabolism. Sci. Rep. 2018, 8, 409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, A.K.; Kumar, V.; Bishop-Bailey, D.; Jang, B.-C. AZD1208, a Pan-Pim Kinase Inhibitor, Has Anti-Growth Effect on 93T449 Human Liposarcoma Cells via Control of the Expression and Phosphorylation of Pim-3, mTOR, 4EBP-1, S6, STAT-3 and AMPK. Int. J. Mol. Sci. 2019, 20, 363. [Google Scholar] [CrossRef] [Green Version]
- O’Reilly, K.E.; Rojo, F.; She, Q.-B.; Solit, D.; Mills, G.B.; Smith, D.; Lane, H.; Hofmann, F.; Hicklin, D.J.; Ludwig, D.L.; et al. mTOR Inhibition Induces Upstream Receptor Tyrosine Kinase Signaling and Activates Akt. Cancer Res. 2006, 66, 1500–1508. [Google Scholar] [CrossRef] [Green Version]
- Lane, H.; Breuleux, M. Optimal targeting of the mTORC1 kinase in human cancer. Curr. Opin. Cell Biol. 2009, 21, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Rozengurt, E.; Soares, H.P.; Sinnet-Smith, J. Suppression of Feedback Loops Mediated by PI3K/mTOR Induces Multiple Overactivation of Compensatory Pathways: An Unintended Consequence Leading to Drug Resistance. Mol. Cancer Ther. 2014, 13, 2477–2488. [Google Scholar] [CrossRef] [Green Version]
- Kelsey, I.; Manning, B.D. mTORC1 Status Dictates Tumor Response to Targeted Therapeutics. Sci. Signal. 2013, 6, pe31. [Google Scholar] [CrossRef]
- Corton, J.M.; Gillespie, J.G.; Hawley, S.A.; Hardie, D.G. 5-Aminoimidazole-4-Carboxamide Ribonucleoside. A Specific Method for Activating AMP-Activated Protein Kinase in Intact Cells? JBIC J. Biol. Inorg. Chem. 1995, 229, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Furet, P.; Guagnano, V.; Fairhurst, R.A.; Imbach-Weese, P.; Bruce, I.; Knapp, M.; Fritsch, C.; Blasco, F.; Blanz, J.; Aichholz, R.; et al. Discovery of NVP-BYL719 a potent and selective phosphatidylinositol-3 kinase alpha inhibitor selected for clinical evaluation. Bioorganic Med. Chem. Lett. 2013, 23, 3741–3748. [Google Scholar] [CrossRef]
- Bhat, R.; Xue, Y.; Berg, S.; Hellberg, S.; Ormö, M.; Nilsson, Y.; Radesäter, A.-C.; Jerning, E.; Markgren, P.-O.; Borgegård, T.; et al. Structural Insights and Biological Effects of Glycogen Synthase Kinase 3-specific Inhibitor AR-A014418. J. Biol. Chem. 2003, 278, 45937–45945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Din, F.V.; Valanciute, A.; Houde, V.P.; Zibrova, D.; Green, K.A.; Sakamoto, K.; Alessi, D.; Dunlop, M. Aspirin Inhibits mTOR Signaling, Activates AMP-Activated Protein Kinase, and Induces Autophagy in Colorectal Cancer Cells. Gastroenterology 2012, 142, 1504–1515. [Google Scholar] [CrossRef] [Green Version]
- Keeton, E.K.; McEachern, K.; Dillman, K.S.; Palakurthi, S.; Cao, Y.; Grondine, M.R.; Kaur, S.; Wang, S.; Chen, Y.; Wu, A.; et al. AZD1208, a potent and selective pan-Pim kinase inhibitor, demonstrates efficacy in preclinical models of acute myeloid leukemia. Blood 2014, 123, 905–913. [Google Scholar] [CrossRef] [Green Version]
- Chresta, C.M.; Davies, B.R.; Hickson, I.; Harding, T.; Cosulich, S.; Critchlow, S.E.; Vincent, J.P.; Ellston, R.; Jones, D.; Sini, P.; et al. AZD8055 Is a Potent, Selective, and Orally Bioavailable ATP-Competitive Mammalian Target of Rapamycin Kinase Inhibitor with In vitro and In vivo Antitumor Activity. Cancer Res. 2009, 70, 288–298. [Google Scholar] [CrossRef] [Green Version]
- Steegmaier, M.; Hoffmann, M.; Baum, A.; Lenart, P.; Petronczki, M.; Krššák, M.; Gürtler, U.; Garin-Chesa, P.; Lieb, S.; Quant, J.; et al. BI 2536, a Potent and Selective Inhibitor of Polo-like Kinase 1, Inhibits Tumor Growth In Vivo. Curr. Biol. 2007, 17, 316–322. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Yap, J.L.; Yoshioka, M.; Lanning, M.E.; Fountain, R.N.; Raje, M.; Scheenstra, J.A.; Strovel, J.W.; Fletcher, S. BRD4 Structure–Activity Relationships of Dual PLK1 Kinase/BRD4 Bromodomain Inhibitor BI-2536. ACS Med. Chem. Lett. 2015, 6, 764–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awasthi, N.; Zhang, C.; Ruan, W.; Schwarz, M.A.; Schwarz, R.E. BMS-754807, a Small-Molecule Inhibitor of Insulin-like Growth Factor-1 Receptor/Insulin Receptor, Enhances Gemcitabine Response in Pancreatic Cancer. Mol. Cancer Ther. 2012, 11, 2644–2653. [Google Scholar] [CrossRef] [Green Version]
- Newcomb, R.W.; Newcomb, M. Selective Inhibition of Glutaminase by Bis-Thiadiazoles. U.S. Patent 6451828B1, 17 September 2002. [Google Scholar]
- Burger, M.T.; Pecchi, S.; Wagman, A.; Ni, Z.-J.; Knapp, M.; Hendrickson, T.; Atallah, G.; Pfister, K.; Zhang, Y.; Bartulis, S.; et al. Identification of NVP-BKM120 as a Potent, Selective, Orally Bioavailable Class I PI3 Kinase Inhibitor for Treating Cancer. ACS Med. Chem. Lett. 2011, 2, 774–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steliou, K.; Boosalis, M.S.; Perrine, S.P.; Sangerman, J.; Faller, D.V. Butyrate Histone Deacetylase Inhibitors. BioResearch Open Access 2012, 1, 192–198. [Google Scholar] [CrossRef] [Green Version]
- Cao, M.; Zhang, Z.; Han, S.; Lu, X. Butyrate inhibits the proliferation and induces the apoptosis of colorectal cancer HCT116 cells via the deactivation of mTOR/S6K1 signaling mediated partly by SIRT1 downregulation. Mol. Med. Rep. 2019, 19, 3941–3947. [Google Scholar] [CrossRef] [PubMed]
- Rae, C.; Haberkorn, U.; Babich, J.W.; Mairs, R.J. Inhibition of Fatty Acid Synthase Sensitizes Prostate Cancer Cells to Radiotherapy. Radiat. Res. 2015, 184, 482–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkaria, J.N.; Busby, E.C.; Tibbetts, R.; Roos, P.; Taya, Y.; Karnitz, L.M.; Abraham, R.T. Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine. Cancer Res. 1999, 59, 4375–4382. [Google Scholar]
- Saiki, S.; Sasazawa, Y.; Imamichi, Y.; Kawajiri, S.; Fujimaki, T.; Tanida, I.; Kobayashi, H.; Sato, F.; Sato, S.; Ishikawa, K.-I.; et al. Caffeine induces apoptosis by enhancement of autophagy via PI3K/Akt/mTOR/p70S6K inhibition. Autophagy 2011, 7, 176–187. [Google Scholar] [CrossRef] [Green Version]
- Gross, M.I.; Demo, S.D.; Dennison, J.B.; Chen, L.; Chernov-Rogan, T.; Goyal, B.; Janes, J.R.; Laidig, G.J.; Lewis, E.R.; Li, J.; et al. Antitumor Activity of the Glutaminase Inhibitor CB-839 in Triple-Negative Breast Cancer. Mol. Cancer Ther. 2014, 13, 890–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nam, K.H.; Yi, S.A.; Nam, G.; Noh, J.S.; Park, J.W.; Lee, M.G.; Park, J.H.; Oh, H.; Lee, J.; Lee, K.R.; et al. Identification of a novel S6K1 inhibitor, rosmarinic acid methyl ester, for treating cisplatin-resistant cervical cancer. BMC Cancer 2019, 19, 773. [Google Scholar] [CrossRef]
- Guo, M.-L.; Liao, K.; Periyasamy, P.; Yang, L.; Cai, Y.; Callen, S.; Buch, S. Cocaine-mediated microglial activation involves the ER stress-autophagy axis. Autophagy 2015, 11, 995–1009. [Google Scholar] [CrossRef] [Green Version]
- Patch, R.J.; Baumann, C.A.; Liu, J.; Gibbs, A.; Ott, H.; Lattanze, J.; Player, M.R. Identification of 2-acylaminothiophene-3-carboxamides as potent inhibitors of FLT3. Bioorganic Med. Chem. Lett. 2006, 16, 3282–3286. [Google Scholar] [CrossRef]
- Haas, S.C.; Huber, R.; Gutsch, R.; Kandemir, J.D.; Cappello, C.; Krauter, J.; Duyster, J.; Ganser, A.; Brand, K. ITD- and FL-induced FLT3 signal transduction leads to increased C/EBPβ-LIP expression and LIP/LAP ratio by different signalling modules. Br. J. Haematol. 2010, 148, 777–790. [Google Scholar] [CrossRef]
- Battistutta, R.; Cozza, G.; Pierre, F.; Papinutto, E.; Lolli, G.; Sarno, S.; O’Brien, S.E.; Siddiqui-Jain, A.; Haddach, M.; Anderes, K.; et al. Unprecedented Selectivity and Structural Determinants of a New Class of Protein Kinase CK2 Inhibitors in Clinical Trials for the Treatment of Cancer. Biochemistry 2011, 50, 8478–8488. [Google Scholar] [CrossRef]
- Rheault, T.R.; Stellwagen, J.C.; Adjabeng, G.M.; Hornberger, K.R.; Petrov, K.G.; Waterson, A.G.; Dickerson, S.H.; Mook, J.R.A.; Laquerre, S.G.; King, A.J.; et al. Discovery of Dabrafenib: A Selective Inhibitor of Raf Kinases with Antitumor Activity against B-Raf-Driven Tumors. ACS Med. Chem. Lett. 2013, 4, 358–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greger, J.G.; Eastman, S.D.; Zhang, V.; Bleam, M.R.; Hughes, A.M.; Smitheman, K.N.; Dickerson, S.H.; Laquerre, S.G.; Liu, L.; Gilmer, T.M. Combinations of BRAF, MEK, and PI3K/mTOR Inhibitors Overcome Acquired Resistance to the BRAF Inhibitor GSK2118436 Dabrafenib, Mediated by NRAS or MEK Mutations. Mol. Cancer Ther. 2012, 11, 909–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maira, S.-M.; Stauffer, F.; Brueggen, J.; Furet, P.; Schnell, C.; Fritsch, C.; Brachmann, S.; Chène, P.; De Pover, A.; Schoemaker, K.; et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potentin vivoantitumor activity. Mol. Cancer Ther. 2008, 7, 1851–1863. [Google Scholar] [CrossRef] [Green Version]
- Toledo, L.; Murga, M.; Zur, R.; Soria, R.; Rodriguez, A.; Martinez, S.; Oyarzabal, J.; Pastor, J.; Bischoff, J.R.; Fernandez-Capetillo, O. A cell-based screen identifies ATR inhibitors with synthetic lethal properties for cancer-associated mutations. Nat. Struct. Mol. Biol. 2011, 18, 721–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.P.H.; Keam, S.J.; Keating, G.M. Deferasirox. Drugs 2007, 67, 2211–2230. [Google Scholar] [CrossRef]
- Ohyashiki, J.H.; Kobayashi, C.; Hamamura, R.; Okabe, S.; Tauchi, T.; Ohyashiki, K. The oral iron chelator deferasirox represses signaling through the mTOR in myeloid leukemia cells by enhancing expression of REDD1. Cancer Sci. 2009, 100, 970–977. [Google Scholar] [CrossRef]
- Zhang, D.; Li, J.; Wang, F.; Hu, J.; Wang, S.; Sun, Y. 2-Deoxy-D-glucose targeting of glucose metabolism in cancer cells as a potential therapy. Cancer Lett. 2014, 355, 176–183. [Google Scholar] [CrossRef]
- Mattern, J.; Büchler, M.W.; Herr, I. Cell Cycle Arrest by Glucocorticoids May Protect Normal Tissue and Solid Tumors from Cancer Therapy. Cancer Biol. Ther. 2007, 6, 1341–1350. [Google Scholar] [CrossRef] [Green Version]
- Okuzumi, T.; Fiedler, D.; Zhang, C.; Gray, D.C.; Aizenstein, B.; Hoffman, R.; Shokat, K.M. Inhibitor hijacking of Akt activation. Nat. Chem. Biol. 2009, 5, 484–493. [Google Scholar] [CrossRef]
- Hsieh, A.C.; Liu, Y.; Edlind, M.P.; Ingolia, N.T.; Janes, M.R.; Sher, A.; Shi, E.Y.; Stumpf, C.; Christensen, C.; Bonham, M.J.; et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature 2012, 485, 55–61. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Chen, K.; Kobayashi, S.; Timm, D.; Liang, Q. Resveratrol Attenuates Doxorubicin-Induced Cardiomyocyte Death via Inhibition of p70 S6 Kinase 1-Mediated Autophagy. J. Pharmacol. Exp. Ther. 2011, 341, 183–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sedrani, R.; Cottens, S.; Kallen, J.; Schuler, W. Chemical modification of rapamycin: The discovery of SDZ RAD. Transplant. Proc. 1998, 30, 2192–2194. [Google Scholar] [CrossRef]
- Wang, X.; Yue, P.; Tao, H.; Sun, S.-Y. Inhibition of p70S6K does not mimic the enhancement of Akt phosphorylation by rapamycin. Heliyon 2017, 3, e00378. [Google Scholar] [CrossRef] [Green Version]
- Lerner, E.C.; Qian, Y.; Blaskovich, M.A.; Fossum, R.D.; Vogt, A.; Sun, J.; Cox, A.D.; Der, C.; Hamilton, A.D.; Sebti, S.M. Ras CAAX Peptidomimetic FTI-277 Selectively Blocks Oncogenic Ras Signaling by Inducing Cytoplasmic Accumulation of Inactive Ras-Raf Complexes. J. Biol. Chem. 1995, 270, 26802–26806. [Google Scholar] [CrossRef] [Green Version]
- Cohen, M.S.; Zhang, C.; Shokat, K.M.; Taunton, J. Structural Bioinformatics-Based Design of Selective, Irreversible Kinase Inhibitors. Science 2005, 308, 1318–1321. [Google Scholar] [CrossRef] [Green Version]
- Insel, P.A. Forskolin as a Tool for Examining Adenylyl Cyclase Expression, Regulation, and G Protein Signaling. Cell. Mol. Neurobiol. 2003, 23, 305–314. [Google Scholar] [CrossRef]
- Purkey, H.E.; Robarge, K.; Chen, J.; Chen, Z.; Corson, L.B.; Ding, C.Z.; DiPasquale, A.G.; Dragovich, P.S.; Eigenbrot, C.; Evangelista, M.; et al. Cell Active Hydroxylactam Inhibitors of Human Lactate Dehydrogenase with Oral Bioavailability in Mice. ACS Med. Chem. Lett. 2016, 7, 896–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilmartin, A.G.; Faitg, T.H.; Richter, M.; Groy, A.; Seefeld, M.A.; Darcy, M.G.; Peng, X.; Federowicz, K.; Yang, J.; Zhang, S.-Y.; et al. Allosteric Wip1 phosphatase inhibition through flap-subdomain interaction. Nat. Chem. Biol. 2014, 10, 181–187. [Google Scholar] [CrossRef]
- Heinrich, M.C.; Griffith, D.J.; Druker, B.J.; Wait, C.L.; Ott, K.A.; Zigler, A.J. Inhibition of c-kit receptor tyrosine kinase activity by STI 571, a selective tyrosine kinase inhibitor. Blood 2000, 96, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.E.; Braun, A.P.; Wu, Y.; Lu, T.; Wu, Y.; Schulman, H.; Sung, R.J. KN-93, an Inhibitor of Multifunctional Ca++/Calmodulin-Dependent Protein Kinase, Decreases Early Afterdepolarizations in Rabbit Heart. J. Pharm. Exp. Ther. 1998, 287, 996–1006. [Google Scholar]
- García-Martínez, J.M.; Moran, J.; Clarke, R.G.; Gray, A.; Cosulich, S.C.; Chresta, C.M.; Alessi, D.R. Ku-0063794 is a specific inhibitor of the mammalian target of rapamycin (mTOR). Biochem. J. 2009, 421, 29–42. [Google Scholar] [CrossRef] [Green Version]
- Nyfeler, B.; Bergman, P.; Triantafellow, E.; Wilson, C.J.; Zhu, Y.; Radetich, B.; Finan, P.M.; Klionsky, D.J.; Murphy, L.O. Relieving Autophagy and 4EBP1 from Rapamycin Resistance. Mol. Cell. Biol. 2011, 31, 2867–2876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, P.S.; Melton, D.A. A molecular mechanism for the effect of lithium on development. Proc. Natl. Acad. Sci. USA 1996, 93, 8455–8459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stambolic, V.; Ruel, L.; Woodgett, J.R. Lithium inhibits glycogen synthase kinase-3 activity and mimics Wingless signalling in intact cells. Curr. Biol. 1996, 6, 1664–1669. [Google Scholar] [CrossRef] [Green Version]
- Mulvihill, M.J.; Cooke, A.; Rosenfeld-Franklin, M.; Buck, E.; Foreman, K.; Landfair, D.; O’Connor, M.; Pirritt, C.; Sun, Y.; Yao, Y.; et al. Discovery of OSI-906: A selective and orally efficacious dual inhibitor of the IGF-1 receptor and insulin receptor. Futur. Med. Chem. 2009, 1, 1153–1171. [Google Scholar] [CrossRef] [PubMed]
- Garcia, P.D.; Langowski, J.L.; Wang, Y.; Chen, M.; Castillo, J.; Fanton, C.; Ison, M.; Zavorotinskaya, T.; Dai, Y.; Lu, J.; et al. Pan-PIM Kinase Inhibition Provides a Novel Therapy for Treating Hematologic Cancers. Clin. Cancer Res. 2014, 20, 1834–1845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Njoroge, F.G.; Taveras, A.G.; Kelly, J.; Remiszewski, S.; Mallams, A.K.; Wolin, R.; Afonso, A.; Cooper, A.B.; Rane, D.F.; Liu, Y.-T.; et al. (+)-4-[2-[4-(8-Chloro-3,10-dibromo-6,11-dihydro-5H-benzo[5,6]cyclohepta[1,2-b]- pyridin-11(R)-yl)-1-piperidinyl]-2-oxo-ethyl]-1-piperidinecarboxamide (SCH-66336): A Very Potent Farnesyl Protein Transferase Inhibitor as a Novel Antitumor Agent. J. Med. Chem. 1998, 41, 4890–4902. [Google Scholar] [CrossRef]
- Basso, A.D.; Mirza, A.; Liu, G.; Long, B.J.; Bishop, W.R.; Kirschmeier, P. The Farnesyl Transferase Inhibitor (FTI) SCH66336 (lonafarnib) Inhibits Rheb Farnesylation and mTOR Signaling. J. Biol. Chem. 2005, 280, 31101–31108. [Google Scholar] [CrossRef] [Green Version]
- Reipas, K.M.; Law, J.H.; Couto, N.; Islam, S.; Li, Y.; Li, H.; Cherkasov, A.; Jung, K.; Cheema, A.; Jones, S.; et al. Luteolin is a novel p90 ribosomal S6 kinase (RSK) inhibitor that suppresses Notch4 signaling by blocking the activation of Y-box binding protein-1 (YB-1). Oncotarget 2013, 4, 329–345. [Google Scholar] [CrossRef] [PubMed]
- Chaussade, C.; Rewcastle, G.W.; Kendall, J.D.; Denny, W.A.; Cho, K.; Grønning, L.M.; Chong, M.L.; Anagnostou, S.H.; Jackson, S.P.; Daniele, N.; et al. Evidence for functional redundancy of class IA PI3K isoforms in insulin signalling. Biochem. J. 2007, 404, 449–458. [Google Scholar] [CrossRef] [Green Version]
- Gharbi, S.I.; Zvelebil, M.J.; Shuttleworth, S.J.; Hancox, T.; Saghir, N.; Timms, J.F.; Waterfield, M.D. Exploring the specificity of the PI3K family inhibitor LY294002. Biochem. J. 2007, 404, 15–21. [Google Scholar] [CrossRef] [Green Version]
- Davidson, D.; Amrein, L.; Panasci, L.; Aloyz, R. Small Molecules, Inhibitors of DNA-PK, Targeting DNA Repair, and Beyond. Front. Pharmacol. 2013, 4, 5. [Google Scholar] [CrossRef] [Green Version]
- Egan, L.J.; Mays, D.C.; Huntoon, C.J.; Bell, M.P.; Pike, M.G.; Sandborn, W.J.; Lipsky, J.J.; McKean, D.J. Inhibition of Interleukin-1-stimulated NF-κB RelA/p65 Phosphorylation by Mesalamine Is Accompanied by Decreased Transcriptional Activity. J. Biol. Chem. 1999, 274, 26448–26453. [Google Scholar] [CrossRef] [Green Version]
- Rousseaux, C.; Lefebvre, B.; Dubuquoy, L.; Lefebvre, P.; Romano, O.; Auwerx, J.; Metzger, D.; Wahli, W.; Desvergne, B.; Naccari, G.C.; et al. Intestinal antiinflammatory effect of 5-aminosalicylic acid is dependent on peroxisome proliferator–activated receptor-γ. J. Exp. Med. 2005, 201, 1205–1215. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Hirai, H.; Sootome, H.; Nakatsuru, Y.; Miyama, K.; Taguchi, S.; Tsujioka, K.; Ueno, Y.; Hatch, H.; Majumder, P.K.; Pan, B.-S.; et al. MK-2206, an Allosteric Akt Inhibitor, Enhances Antitumor Efficacy by Standard Chemotherapeutic Agents or Molecular Targeted Drugs In vitro and In vivo. Mol. Cancer Ther. 2010, 9, 1956–1967. [Google Scholar] [CrossRef] [Green Version]
- Weïwer, M.; Spoonamore, J.; Wei, J.; Guichard, B.; Ross, N.T.; Masson, K.; Silkworth, W.; Dandapani, S.; Palmer, M.; Scherer, C.A.; et al. A Potent and Selective Quinoxalinone-Based STK33 Inhibitor Does Not Show Synthetic Lethality in KRAS-Dependent Cells. ACS Med. Chem. Lett. 2012, 3, 1034–1038. [Google Scholar] [CrossRef]
- Weisberg, E.; Catley, L.; Wright, R.D.; Moreno, D.; Banerji, L.; Ray, A.; Manley, P.W.; Mestan, J.; Fabbro, D.; Jiang, J.; et al. Beneficial effects of combining nilotinib and imatinib in preclinical models of BCR-ABL+ leukemias. Blood 2006, 109, 2112–2120. [Google Scholar] [CrossRef] [Green Version]
- Holmes, C.F.; Luu, H.A.; Carrier, F.; Schmitz, F.J. Inhibition of protein phosphatases-1 and -2A with acanthifolicin. FEBS Lett. 1990, 270, 216–218. [Google Scholar] [CrossRef] [Green Version]
- Knight, S.D.; Adams, N.D.; Burgess, J.L.; Chaudhari, A.M.; Darcy, M.G.; Donatelli, C.A.; Luengo, J.I.; Newlander, K.A.; Parrish, C.A.; Ridgers, L.H.; et al. Discovery of GSK2126458, a Highly Potent Inhibitor of PI3K and the Mammalian Target of Rapamycin. ACS Med. Chem. Lett. 2010, 1, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Zhai, X.; Yang, Y.; Wan, J.; Zhu, R.; Wu, Y. Inhibition of LDH-A by oxamate induces G2/M arrest, apoptosis and increases radiosensitivity in nasopharyngeal carcinoma cells. Oncol. Rep. 2013, 30, 2983–2991. [Google Scholar] [CrossRef] [Green Version]
- Muramatsu, H.; Sumitomo, M.; Morinaga, S.; Kajikawa, K.; Kobayashi, I.; Nishikawa, G.; Kato, Y.; Watanabe, M.; Zennami, K.; Kanao, K.; et al. Targeting lactate dehydrogenase-A promotes docetaxel-induced cytotoxicity predominantly in castration-resistant prostate cancer cells. Oncol. Rep. 2019, 42, 224–230. [Google Scholar] [CrossRef]
- Chang, C.-I.; Liao, J.C.; Kuo, L. Arginase modulates nitric oxide production in activated macrophages. Am. J. Physiol. Circ. Physiol. 1998, 274, H342–H348. [Google Scholar] [CrossRef]
- Sebolt-Leopold, J.S.; Dudley, D.T.; Herrera, R.; Van Becelaere, K.; Wiland, A.; Gowan, R.C.; Tecle, H.; Barrett, S.D.; Bridges, A.; Przybranowski, S.; et al. Blockade of the MAP kinase pathway suppresses growth of colon tumors in vivo. Nat. Med. 1999, 5, 810–816. [Google Scholar] [CrossRef]
- Leontieva, O.V.; Blagosklonny, M.V. S6K in geroconversion. Cell Cycle 2013, 12, 3249–3252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakamoto, K.; Göransson, O.; Hardie, D.G.; Alessi, D.R. Activity of LKB1 and AMPK-related kinases in skeletal muscle: Effects of contraction, phenformin, and AICAR. Am. J. Physiol. Metab. 2004, 287, E310–E317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raynaud, F.I.; Eccles, S.A.; Patel, S.; Alix, S.; Box, G.; Chuckowree, I.; Folkes, A.; Gowan, S.; Brandon, A.D.H.; Di Stefano, F.; et al. Biological properties of potent inhibitors of class I phosphatidylinositide 3-kinases: From PI-103 through PI-540, PI-620 to the oral agent GDC-0941. Mol. Cancer Ther. 2009, 8, 1725–1738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, Y.W.; Kang, H.J.; Kim, H.J.; Hwang, J.S.; Wang, A.; Bae, I. Inhibition of constitutively activated phosphoinositide 3-kinase/AKT pathway enhances antitumor activity of chemotherapeutic agents in breast cancer susceptibility gene 1-defective breast cancer cells. Mol. Carcinog. 2012, 52, 667–675. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Z.; Amran, S.I.; Thompson, P.E.; Jennings, I.G. Isoform-Selective Inhibition of Phosphoinositide 3-Kinase: Identification of a New Region of Nonconserved Amino Acids Critical for p110α Inhibition. Mol. Pharmacol. 2011, 80, 657–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knight, Z.; Gonzalez, B.; Feldman, M.E.; Zunder, E.R.; Goldenberg, D.D.; Williams, O.; Loewith, R.; Stokoe, D.; Balla, A.; Toth, B.; et al. A Pharmacological Map of the PI3-K Family Defines a Role for p110α in Insulin Signaling. Cell 2006, 125, 733–747. [Google Scholar] [CrossRef] [Green Version]
- Pearce, L.R.; Alton, G.R.; Richter, D.T.; Kath, J.C.; Lingardo, L.; Chapman, J.; Hwang, C.; Alessi, D.R. Characterization of PF-4708671, a novel and highly specific inhibitor of p70 ribosomal S6 kinase (S6K1). Biochem. J. 2010, 431, 245–255. [Google Scholar] [CrossRef] [Green Version]
- Amaral, C.L.; Freitas, L.B.; Tamura, R.E.; Tavares, M.R.; Pavan, I.; Bajgelman, M.C.; Simabuco, F.M. S6Ks isoforms contribute to viability, migration, docetaxel resistance and tumor formation of prostate cancer cells. BMC Cancer 2016, 16, 602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabbro, D.; Buchdunger, E.; Wood, J.; Mestan, J.; Hofmann, F.; Ferrari, S.; Mett, H.; O’Reilly, T.; Meyer, T. Inhibitors of Protein KinasesCGP 41251, a Protein Kinase Inhibitor with Potential as an Anticancer Agent. Pharmacol. Ther. 1999, 82, 293–301. [Google Scholar] [CrossRef]
- Castagna, M.; Takai, Y.; Kaibuchi, K.; Sano, K.; Kikkawa, U.; Nishizuka, Y. Direct activation of calcium-activated, phospholipid-dependent protein kinase by tumor-promoting phorbol esters. J. Biol. Chem. 1982, 257, 7847–7851. [Google Scholar] [CrossRef]
- Martin, P.M.; Aeder, S.; Chrestensen, C.; Sturgill, T.W.; Hussaini, I.M. Phorbol 12-myristate 13-acetate and serum synergize to promote rapamycin-insensitive cell proliferation via protein kinase C-eta. Oncogene 2006, 26, 407–414. [Google Scholar] [CrossRef] [Green Version]
- Glennon, R.; Dukat, M.; Westkaemper, R.B.; Ismaiel, A.M.; Izzarelli, D.G.; Parker, E.M. The binding of propranolol at 5-hydroxytryptamine1D beta T355N mutant receptors may involve formation of two hydrogen bonds to asparagine. Mol. Pharmacol. 1996, 49, 198–206. [Google Scholar] [PubMed]
- Grkovich, A.; Johnson, C.A.; Buczynski, M.W.; Dennis, E.A. Lipopolysaccharide-induced Cyclooxygenase-2 Expression in Human U937 Macrophages Is Phosphatidic Acid Phosphohydrolase-1-dependent. J. Biol. Chem. 2006, 281, 32978–32987. [Google Scholar] [CrossRef] [Green Version]
- Albert, D.; Pergola, C.; Koeberle, A.; Dodt, G.; Steinhilber, D.; Werz, O. The role of diacylglyceride generation by phospholipase D and phosphatidic acid phosphatase in the activation of 5-lipoxygenase in polymorphonuclear leukocytes. J. Leukoc. Biol. 2008, 83, 1019–1027. [Google Scholar] [CrossRef]
- Edwards, S.R.; Wandless, T.J. The Rapamycin-binding Domain of the Protein Kinase Mammalian Target of Rapamycin Is a Destabilizing Domain. J. Biol. Chem. 2007, 282, 13395–13401. [Google Scholar] [CrossRef] [Green Version]
- Noh, W.-C.; Mondesire, W.H.; Peng, J.; Jinjiang, D.; Zhang, H.; Dong, J.; Mills, G.B.; Hung, M.-C.; Meric-Bernstam, F. Determinants of Rapamycin Sensitivity in Breast Cancer Cells. Clin. Cancer Res. 2004, 10, 1013–1023. [Google Scholar] [CrossRef] [Green Version]
- Calamini, B.; Ratia, K.; Malkowski, M.; Cuendet, M.; Pezzuto, J.M.; Santarsiero, B.D.; Mesecar, A.D. Pleiotropic mechanisms facilitated by resveratrol and its metabolites. Biochem. J. 2010, 429, 273–282. [Google Scholar] [CrossRef] [Green Version]
- Howitz, K.T.; Bitterman, K.J.; Cohen, H.Y.; Lamming, D.W.; Lavu, S.; Wood, J.G.; Zipkin, R.E.; Chung, P.; Kisielewski, A.; Zhang, L.-L.; et al. Small Molecule Activators of SirtuinsextendSaccharomyces Cerevisiaelifespan. Nature 2003, 425, 191–196. [Google Scholar] [CrossRef]
- Armour, S.M.; Baur, J.; Hsieh, S.N.; Land-Bracha, A.; Thomas, S.M.; Sinclair, D.A. Inhibition of mammalian S6 kinase by resveratrol suppresses autophagy. Aging 2009, 1, 515–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, X.; Liu, Q.; Wang, M.; Liang, M.; Yang, X.; Xu, X.; Zou, H.; Qiu, J. Activation of Sirt1 by Resveratrol Inhibits TNF-α Induced Inflammation in Fibroblasts. PLoS ONE 2011, 6, e27081. [Google Scholar] [CrossRef]
- Coghlan, M.P.; Culbert, A.A.; Cross, D.A.; Corcoran, S.L.; Yates, J.W.; Pearce, N.J.; Rausch, O.L.; Murphy, G.J.; Carter, P.S.; Cox, L.R.; et al. Selective small molecule inhibitors of glycogen synthase kinase-3 modulate glycogen metabolism and gene transcription. Chem. Biol. 2000, 7, 793–803. [Google Scholar] [CrossRef] [Green Version]
- Huynh, H.; Soo, K.C.; Chow, P.; Tran, E. Targeted inhibition of the extracellular signal-regulated kinase kinase pathway with AZD6244 (ARRY-142886) in the treatment of hepatocellular carcinoma. Mol. Cancer Ther. 2007, 6, 138–146. [Google Scholar] [CrossRef] [Green Version]
- Atefi, M.; Von Euw, E.; Attar, N.; Ng, C.; Chu, C.; Guo, D.; Nazarian, R.; Chmielowski, B.; Glaspy, J.A.; Comin-Anduix, B.; et al. Reversing Melanoma Cross-Resistance to BRAF and MEK Inhibitors by Co-Targeting the AKT/mTOR Pathway. PLoS ONE 2011, 6, e28973. [Google Scholar] [CrossRef]
- Chen, L.S.; Redkar, S.; Taverna, P.; Cortes, J.E.; Gandhi, V. Mechanisms of cytotoxicity to Pim kinase inhibitor, SGI-1776, in acute myeloid leukemia. Blood 2011, 118, 693–702. [Google Scholar] [CrossRef] [Green Version]
- Siddiqui-Jain, A.; Drygin, D.; Streiner, N.; Chua, P.; Pierre, F.; O’Brien, S.E.; Bliesath, J.; Omori, M.; Huser, N.; Ho, C.; et al. CX-4945, an Orally Bioavailable Selective Inhibitor of Protein Kinase CK2, Inhibits Prosurvival and Angiogenic Signaling and Exhibits Antitumor Efficacy. Cancer Res. 2010, 70, 10288–10298. [Google Scholar] [CrossRef] [Green Version]
- Milne, J.C.; Lambert, P.D.; Schenk, S.; Carney, D.P.; Smith, J.J.; Gagne, D.J.; Jin, L.; Boss, O.; Perni, R.B.; Vu, C.B.; et al. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature 2007, 450, 712–716. [Google Scholar] [CrossRef] [Green Version]
- Thoreen, C.C.; Kang, S.A.; Chang, J.W.; Liu, Q.; Zhang, J.; Gao, Y.; Reichling, L.J.; Sim, T.; Sabatini, D.M.; Gray, N.S. An ATP-competitive Mammalian Target of Rapamycin Inhibitor Reveals Rapamycin-resistant Functions of mTORC1. J. Biol. Chem. 2009, 284, 8023–8032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Chang, J.W.; Wang, J.; Kang, S.A.; Thoreen, C.C.; Markhard, A.; Hur, W.; Zhang, J.; Sim, T.; Sabatini, D.M.; et al. Discovery of 1-(4-(4-Propionylpiperazin-1-yl)-3-(trifluoromethyl)phenyl)-9-(quinolin-3-yl)benzo[h][1,6]naphthyridin-2(1H)-one as a Highly Potent, Selective Mammalian Target of Rapamycin (mTOR) Inhibitor for the Treatment of Cancer. J. Med. Chem. 2010, 53, 7146–7155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apsel, B.; Blair, J.; Gonzalez, B.; Nazif, T.; Feldman, M.; Aizenstein, B.; Hoffman, R.; Williams, R.; Shokat, K.M.; Knight, Z. Targeted polypharmacology: Discovery of dual inhibitors of tyrosine and phosphoinositide kinases. Nat. Chem. Biol. 2008, 4, 691–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakai, T.; Yamaguchi, T.; Kakefuda, R.; Tajima, N.; Sowa, Y. Antitumor activities of JTP-74057 (GSK1120212), a novel MEK1/2 inhibitor, on colorectal cancer cell lines in vitro and in vivo. Int. J. Oncol. 2011, 39, 23–31. [Google Scholar] [CrossRef] [Green Version]
- Duncia, J.V.; Santella, J.B.; Higley, C.; Pitts, W.; Wityak, J.; Frietze, W.E.; Rankin, F.; Sun, J.-H.; Earl, R.A.; Tabaka, A.; et al. MEK inhibitors: The chemistry and biological activity of U0126, its analogs, and cyclization products. Bioorganic Med. Chem. Lett. 1998, 8, 2839–2844. [Google Scholar] [CrossRef]
- Bollag, G.; Hirth, P.; Tsai, J.; Zhang, J.; Ibrahim, P.N.; Cho, H.; Spevak, W.; Zhang, C.; Zhang, Y.; Habets, G.; et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 2010, 467, 596–599. [Google Scholar] [CrossRef] [PubMed]
- Kobori, M.; Yang, Z.; Gong, D.; Heissmeyer, V.; Zhu, H.; Jung, Y.-K.; Gakidis, M.A.M.; Rao, A.; Sekine, T.; Ikegami, F.; et al. Wedelolactone suppresses LPS-induced caspase-11 expression by directly inhibiting the IKK Complex. Cell Death Differ. 2003, 11, 123–130. [Google Scholar] [CrossRef] [Green Version]
- Yu, K.; Toral-Barza, L.; Shi, C.; Zhang, W.-G.; Lucas, J.; Shor, B.; Kim, J.; Verheijen, J.; Curran, K.; Malwitz, D.J.; et al. Biochemical, Cellular, and In vivo Activity of Novel ATP-Competitive and Selective Inhibitors of the Mammalian Target of Rapamycin. Cancer Res. 2009, 69, 6232–6240. [Google Scholar] [CrossRef] [Green Version]
- Condliffe, A.; Davidson, K.; Anderson, K.E.; Ellson, C.D.; Crabbe, T.; Okkenhaug, K.; Vanhaesebroeck, B.; Turner, M.; Webb, L.; Wymann, M.P.; et al. Sequential activation of class IB and class IA PI3K is important for the primed respiratory burst of human but not murine neutrophils. Blood 2005, 106, 1432–1440. [Google Scholar] [CrossRef] [Green Version]
- Cisterna, B.; Necchi, D.; Prosperi, E.; Biggiogera, M. Small ribosomal subunits associate with nuclear myosin and actin in transit to the nuclear pores. FASEB J. 2006, 20, 1901–1903. [Google Scholar] [CrossRef]
- Schmidt, C.; Lipsius, E.; Kruppa, J. Nuclear and nucleolar targeting of human ribosomal protein S6. Mol. Biol. Cell 1995, 6, 1875–1885. [Google Scholar] [CrossRef] [Green Version]
- Rosner, M.; Fuchs, C.; Dolznig, H.; Hengstschläger, M. Different cytoplasmic/nuclear distribution of S6 protein phosphorylated at S240/244 and S235/236. Amino Acids 2010, 40, 595–600. [Google Scholar] [CrossRef]
- Rosner, M.; Schipany, K.; Hengstschläger, M. Phosphorylation of nuclear and cytoplasmic pools of ribosomal protein S6 during cell cycle progression. Amino Acids 2012, 44, 1233–1240. [Google Scholar] [CrossRef]
- Lam, Y.W.; Lamond, A.I.; Mann, M.; Andersen, J. Analysis of Nucleolar Protein Dynamics Reveals the Nuclear Degradation of Ribosomal Proteins. Curr. Biol. 2007, 17, 749–760. [Google Scholar] [CrossRef] [Green Version]
- Xiao, H.; Wang, H.; Silva, E.; Thompson, J.; Guillou, A.; Yates, J.R.; Buchon, N.; Franc, N.C. The Pallbearer E3 Ligase Promotes Actin Remodeling via RAC in Efferocytosis by Degrading the Ribosomal Protein S6. Dev. Cell 2014, 32, 19–30. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Chen, H.-P.; Duan, H.-F.; Gao, L.-H.; Shao, Y.; Chen, K.-Y.; Wang, Y.-L.; Lan, F.-H.; Hu, X.-W. Aggregation of Ribosomal Protein S6 at Nucleolus Is Cell Cycle-Controlled and Its Function in Pre-rRNA Processing Is Phosphorylation Dependent. J. Cell. Biochem. 2015, 117, 1649–1657. [Google Scholar] [CrossRef] [PubMed]
- Richter, W.; Zang, K.; Issinger, O.-G. Influence of hyperthermia on the phosphorylation of ribosomal protein S6 from human skin fibroblasts and meningioma cells. FEBS Lett. 1983, 153, 262–266. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, I.M.; Burdon, R.H.; Leader, D.P. Heat shock causes diverse changes in the phosphorylation of the ribosomal proteins of mammalian cells. FEBS Lett. 1984, 169, 267–273. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.-S.; Jang, C.-Y.; Kim, H.D.; Lee, J.Y.; Ahn, B.-Y.; Kim, J. Interaction of Hsp90 with Ribosomal Proteins Protects from Ubiquitination and Proteasome-dependent Degradation. Mol. Biol. Cell 2006, 17, 824–833. [Google Scholar] [CrossRef] [PubMed]
- Robledo, S.; Idol, R.A.; Crimmins, D.L.; Ladenson, J.H.; Mason, P.J.; Bessler, M. The role of human ribosomal proteins in the maturation of rRNA and ribosome production. RNA 2008, 14, 1918–1929. [Google Scholar] [CrossRef] [Green Version]
- Volarević, S.; Stewart, M.J.; Ledermann, B.; Zilberman, F.; Terracciano, L.; Montini, E.; Grompe, M.; Kozma, S.C.; Thomas, G. Proliferation, But Not Growth, Blocked by Conditional Deletion of 40 S Ribosomal Protein S6. Science 2000, 288, 2045–2047. [Google Scholar] [CrossRef]
- Šulić, S.; Panić, L.; Barkić, M.; Merćep, M.; Uzelac, M.; Volarević, S. Inactivation of S6 ribosomal protein gene in T lymphocytes activates a p53-dependent checkpoint response. Genes Dev. 2005, 19, 3070–3082. [Google Scholar] [CrossRef] [Green Version]
- Panić, L.; Tamarut, S.; Sticker-Jantscheff, M.; Barkić, M.; Solter, D.; Uzelac, M.; Grabušić, K.; Volarević, S. Ribosomal Protein S6 Gene Haploinsufficiency Is Associated with Activation of a p53-Dependent Checkpoint during Gastrulation. Mol. Cell. Biol. 2006, 26, 8880–8891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, P.J.; Duncan, R.; McConkey, E.H. Phosphorylation of Ribosomal Protein S6. Relationship to Protein Synthesis in HeLa Cells. JBIC J. Biol. Inorg. Chem. 1981, 120, 523–527. [Google Scholar] [CrossRef]
- Duncan, R.; McConkey, E.H. Preferential Utilization of Phosphorylated 40-S Ribosomal Subunits during Initiation Complex Formation. JBIC J. Biol. Inorg. Chem. 2005, 123, 535–538. [Google Scholar] [CrossRef]
- Thomas, G. The effect of serum, EGF, PGF2α and insulin on S6 phosphorylation and the initiation of protein and DNA synthesis. Cell 1982, 30, 235–242. [Google Scholar] [CrossRef]
- Hagner, P.R.; Mazan-Mamczarz, K.; Dai, B.; Balzer, E.M.; Corl, S.; Martin, S.S.; Zhao, X.F.; Gartenhaus, R.B. Ribosomal protein S6 is highly expressed in non-Hodgkin lymphoma and associates with mRNA containing a 5′ terminal oligopyrimidine tract. Oncogene 2010, 30, 1531–1541. [Google Scholar] [CrossRef] [Green Version]
- Fumagalli, S.; Di Cara, A.; Neb-Gulati, A.; Natt, F.; Schwemberger, S.; Hall, J.; Babcock, G.F.; Bernardi, R.; Pandolfi, P.P.; Thomas, G. Absence of nucleolar disruption after impairment of 40S ribosome biogenesis reveals an rpL11-translation-dependent mechanism of p53 induction. Nature 2009, 11, 501–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeon, Y.-J.; Kim, I.K.; Hong, S.-H.; Nan, H.; Kim, H.-J.; Lee, H.-J.; Masuda, E.S.; Meyuhas, O.; Oh, B.-H.; Jung, Y.-K. Ribosomal protein S6 is a selective mediator of TRAIL-apoptotic signaling. Oncogene 2008, 27, 4344–4352. [Google Scholar] [CrossRef] [Green Version]
- Pian, J.P.; Huang, T.-L.; Tsai, P.-C.; Shi, J.-P.; Cu, H.; Pan, B.-T. A 32 kDa protein?whose phosphorylation correlates with oncogenic Ras-induced cell cycle arrest in activatedXenopus egg extracts?is identified as ribosomal protein S6. J. Cell. Physiol. 2004, 201, 305–319. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Bayle, J.H.; Olson, D.; Levine, A.J. The p53-mdm-2 autoregulatory feedback loop. Genes Dev. 1993, 7, 1126–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Besson, A.; Dowdy, S.F.; Roberts, J.M. CDK Inhibitors: Cell Cycle Regulators and Beyond. Dev. Cell 2008, 14, 159–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granot, Z.; Swisa, A.; Magenheim, J.; Stolovich-Rain, M.; Fujimoto, W.; Manduchi, E.; Miki, T.; Lennerz, J.K.; Stoeckert, C.J.; Meyuhas, O.; et al. LKB1 Regulates Pancreatic β Cell Size, Polarity, and Function. Cell Metab. 2009, 10, 296–308. [Google Scholar] [CrossRef] [Green Version]
- Pende, M.; Kozma, S.C.; Jaquet, M.; Oorschot, V.; Burcelin, R.; Le Marchand-Brustel, Y.; Klumperman, J.; Thorens, B.; Thomas, G.H. Hypoinsulinaemia, glucose intolerance and diminished β-cell size in S6K1-deficient mice. Nature 2000, 408, 994–997. [Google Scholar] [CrossRef]
- Olea-Flores, M.; Juárez-Cruz, J.C.; Zuñiga-Eulogio, M.D.; Acosta, E.; García-Rodríguez, E.; Zacapala-Gomez, A.E.; Mendoza-Catalán, M.A.; Ortiz-Ortiz, J.; Ortuño-Pineda, C.; Navarro-Tito, N. New Actors Driving the Epithelial—Mesenchymal Transition in Cancer: The Role of Leptin. Biomolecules 2020, 10, 1676. [Google Scholar] [CrossRef]
- Lane, H.A.; Wood, J.M.; McSheehy, P.M.; Allegrini, P.R.; Boulay, A.; Brueggen, J.; Littlewood-Evans, A.; Maira, S.-M.; Martiny-Baron, G.; Schnell, C.R.; et al. mTOR Inhibitor RAD001 (Everolimus) Has Antiangiogenic/Vascular Properties Distinct from a VEGFR Tyrosine Kinase Inhibitor. Clin. Cancer Res. 2009, 15, 1612–1622. [Google Scholar] [CrossRef] [Green Version]
- Argentiero, A.; Solimando, A.G.; Krebs, M.; Leone, P.; Susca, N.; Brunetti, O.; Racanelli, V.; Vacca, A.; Silvestris, N. Anti-angiogenesis and Immunotherapy: Novel Paradigms to Envision Tailored Approaches in Renal Cell-Carcinoma. J. Clin. Med. 2020, 9, 1594. [Google Scholar] [CrossRef] [PubMed]
- Solimando, A.G.; De Summa, S.; Vacca, A.; Ribatti, D. Cancer-Associated Angiogenesis: The Endothelial Cell as a Checkpoint for Immunological Patrolling. Cancers 2020, 12, 3380. [Google Scholar] [CrossRef]
- Crighton, D.; Wilkinson, S.; O’Prey, J.; Syed, N.; Smith, P.; Harrison, P.R.; Gasco, M.; Garrone, O.; Crook, T.; Ryan, K.M. DRAM, a p53-Induced Modulator of Autophagy, Is Critical for Apoptosis. Cell 2006, 126, 121–134. [Google Scholar] [CrossRef] [Green Version]
- Deng, W.; Gopal, Y.N.V.; Scott, A.; Chen, G.; Woodman, S.E.; Davies, M.A. Role and therapeutic potential of PI3K-mTOR signaling in de novo resistance to BRAF inhibition. Pigment. Cell Melanoma Res. 2011, 25, 248–258. [Google Scholar] [CrossRef]
- Grasso, S.; Tristante, E.; Saceda, M.; Carbonell, P.; Mayor-López, L.; Carballo-Santana, M.; Carrasco-García, E.; Rocamora-Reverte, L.; García-Morales, P.; Carballo, F.; et al. Resistance to Selumetinib (AZD6244) in Colorectal Cancer Cell Lines is Mediated by p70S6K and RPS6 Activation. Neoplasia 2014, 16, 845–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gambardella, V.; Gimeno-Valiente, F.; Tarazona, N.; Ciarpaglini, C.M.; Roda, D.; Fleitas, T.; Tolosa, P.; Cejalvo, J.M.; Huerta, M.; Roselló, S.; et al. NRF2 through RPS6 Activation Is Related to Anti-HER2 Drug Resistance in HER2-Amplified Gastric Cancer. Clin. Cancer Res. 2018, 25, 1639–1649. [Google Scholar] [CrossRef]
- Marosvári, D.; Nagy, N.; Kriston, C.; Deák, B.; Hajdu, M.; Bödör, C.; Csala, I.; Bagó, A.G.; Szallasi, Z.; Sebestyén, A.; et al. Discrepancy Between Low Levels of mTOR Activity and High Levels of P-S6 in Primary Central Nervous System Lymphoma May Be Explained by PAS Domain-Containing Serine/Threonine-Protein Kinase-Mediated Phosphorylation. J. Neuropathol. Exp. Neurol. 2018, 77, 268–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elkabets, M.; Pazarentzos, E.; Juric, D.; Sheng, Q.; Pelossof, R.A.; Brook, S.; Benzaken, A.O.; Rodon, J.; Morse, N.; Yan, J.J.; et al. AXL Mediates Resistance to PI3Kα Inhibition by Activating the EGFR/PKC/mTOR Axis in Head and Neck and Esophageal Squamous Cell Carcinomas. Cancer Cell 2015, 27, 533–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elkabets, M.; Vora, S.; Juric, D.; Morse, N.; Mino-Kenudson, M.; Muranen, T.; Tao, J.; Campos, A.B.; Rodon, J.; Ibrahim, Y.H.; et al. mTORC1 Inhibition Is Required for Sensitivity to PI3K p110α Inhibitors in PIK3CA -Mutant Breast Cancer. Sci. Transl. Med. 2013, 5, 196ra99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosch, A.; Li, Z.; Bergamaschi, A.; Ellis, H.; Toska, E.; Prat, A.; Tao, J.J.; Spratt, D.E.; Viola-Villegas, N.T.; Castel, P.; et al. PI3K inhibition results in enhanced estrogen receptor function and dependence in hormone receptor–positive breast cancer. Sci. Transl. Med. 2015, 7, 283ra51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirashita, Y.; Tsukamoto, Y.; Yanagihara, K.; Fumoto, S.; Hijiya, N.; Nakada, C.; Uchida, T.; Matsuura, K.; Kodama, M.; Okimoto, T.; et al. Reduced phosphorylation of ribosomal protein S6 is associated with sensitivity to MEK inhibition in gastric cancer cells. Cancer Sci. 2016, 107, 1919–1928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang-Kolodji, G.; Mumenthaler, S.M.; Mehta, A.; Ji, L.; Tripathy, D. Phosphorylated ribosomal S6 (p-rpS6) as a post-treatment indicator of HER2 signalling targeted drug resistance. Biomarkers 2015, 20, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.J.; Yi, Y.W.; Bin Hong, Y.; Kim, H.J.; Jang, Y.-J.; Seong, Y.-S.; Bae, I. HER2 confers drug resistance of human breast cancer cells through activation of NRF2 by direct interaction. Sci. Rep. 2014, 4, 7201. [Google Scholar] [CrossRef] [Green Version]
- Yi, Y.W.; Oh, S. Comparative analysis of NRF2-responsive gene expression in AcPC-1 pancreatic cancer cell line. Genes Genom. 2014, 37, 97–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duong, H.-Q.; Yi, Y.W.; Kang, H.J.; Bin Hong, Y.; Tang, W.; Wang, A.; Seong, Y.-S.; Bae, I. Inhibition of NRF2 by PIK-75 augments sensitivity of pancreatic cancer cells to gemcitabine. Int. J. Oncol. 2013, 44, 959–969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryan, H.K.; Olayanju, A.; Goldring, C.E.; Park, B.K. The Nrf2 cell defence pathway: Keap1-dependent and -independent mechanisms of regulation. Biochem. Pharmacol. 2012, 85, 705–717. [Google Scholar] [CrossRef] [Green Version]
- Ma, Q. Role of Nrf2 in Oxidative Stress and Toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [Green Version]
- Ma, Q. Transcriptional responses to oxidative stress: Pathological and toxicological implications. Pharmacol. Ther. 2010, 125, 376–393. [Google Scholar] [CrossRef]
- Mitsuishi, Y.; Motohashi, H.; Yamamoto, M. The Keap1–Nrf2 system in cancers: Stress response and anabolic metabolism. Front. Oncol. 2012, 2, 200. [Google Scholar] [CrossRef] [Green Version]
- Niture, S.; Khatri, R.; Jaiswal, A.K. Regulation of Nrf2—An update. Free. Radic. Biol. Med. 2013, 66, 36–44. [Google Scholar] [CrossRef] [Green Version]
- Penning, T.M. Aldo-Keto Reductase Regulation by the Nrf2 System: Implications for Stress Response, Chemotherapy Drug Resistance, and Carcinogenesis. Chem. Res. Toxicol. 2016, 30, 162–176. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Dinh, T.N.; Kappeler, K.; Tsaprailis, G.; Chen, Q.M. La Autoantigen Mediates Oxidant Induced De Novo Nrf2 Protein Translation. Mol. Cell. Proteom. 2012, 11, m111.015032. [Google Scholar] [CrossRef] [Green Version]
- Guerra, C.; Schuhmacher, A.J.; Cañamero, M.; Grippo, P.J.; Verdaguer, L.; Pérez-Gallego, L.; Dubus, P.; Sandgren, E.P.; Barbacid, M. Chronic Pancreatitis Is Essential for Induction of Pancreatic Ductal Adenocarcinoma by K-Ras Oncogenes in Adult Mice. Cancer Cell 2007, 11, 291–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, L.J.; Yang, L.-X. ν-H2AX—A Novel Biomarker for DNA Double-Strand Breaks. Vivo Athens Greece 2008, 22, 305–309. [Google Scholar]
- Fernandez-Capetillo, O.; Chen, H.-T.; Celeste, A.; Ward, I.; Romanienko, P.J.; Morales, J.; Naka, K.; Xia, Z.; Camerini-Otero, R.D.; Motoyama, N.; et al. DNA damage-induced G2–M checkpoint activation by histone H2AX and 53BP1. Nat. Curell Biol. 2002, 4, 993–997. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.W.; Park, J.-S.; Kwak, S.-J.; Seong, Y.-S. Co-treatment with BEZ235 Enhances Sensitivity of BRCA1-negative Breast Cancer Cells to Olaparib. Anticancer. Res. 2015, 35, 3829–3838. [Google Scholar] [PubMed]
- Berger, N.D.; Stanley, F.; Moore, S.; Goodarzi, A.A. ATM-dependent pathways of chromatin remodelling and oxidative DNA damage responses. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160283. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Zhang, F.; Xiang, T.; Chen, Q.; Pandita, T.K.; Huang, Y.; Hu, M.C.; Yang, Q. Phosphorylation of ribosomal protein S6 confers PARP inhibitor resistance in BRCA1-deficient cancers. Oncotarget 2014, 5, 3375–3385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prakash, R.; Zhang, Y.; Feng, W.; Jasin, M. Homologous Recombination and Human Health: The Roles of BRCA1, BRCA2, and Associated Proteins. Cold Spring Harb. Perspect. Biol. 2015, 7, a016600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zinzalla, V.; Stracka, D.; Oppliger, W.; Hall, M.N. Activation of mTORC2 by Association with the Ribosome. Cell 2011, 144, 757–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magnusson, R.; Gustafsson, M.; Cedersund, G.; Strålfors, P.; Nyman, E. Cross-talks via mTORC2 can explain enhanced activation in response to insulin in diabetic patients. Biosci. Rep. 2017, 37, BSR20160514. [Google Scholar] [CrossRef] [Green Version]
- Ha, D.H.; Kim, H.-K.; Lee, J.; Kwon, H.H.; Park, G.-H.; Yang, S.H.; Jung, J.Y.; Choi, H.; Lee, J.H.; Sung, S.; et al. Mesenchymal Stem/Stromal Cell-Derived Exosomes for Immunomodulatory Therapeutics and Skin Regeneration. Cells 2020, 9, 1157. [Google Scholar] [CrossRef] [PubMed]
- Dodig, S.; Čepelak, I.; Pavić, I. Hallmarks of senescence and aging. Biochem. Med. 2019, 29, 483–497. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Blagosklonny, M.V. Aging: ROS or TOR. Cell Cycle 2008, 7, 3344–3354. [Google Scholar] [CrossRef] [PubMed]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Darzynkiewicz, Z. Attenuation of Replication Stress–Induced Premature Cellular Senescence to Assess Anti-Aging Modalities. Curr. Protoc. Cytom. 2014, 69, 9–47. [Google Scholar] [CrossRef]
- Kang, H.T.; Lee, K.B.; Kim, S.Y.; Choi, H.R.; Park, S.C. Autophagy Impairment Induces Premature Senescence in Primary Human Fibroblasts. PLoS ONE 2011, 6, e23367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narla, A.; Ebert, B.L. Ribosomopathies: Human disorders of ribosome dysfunction. Blood 2010, 115, 3196–3205. [Google Scholar] [CrossRef] [PubMed]
- McGowan, K.A.; Pang, W.W.; Bhardwaj, R.; Perez, M.G.; Pluvinage, J.V.; Glader, B.E.; Malek, R.; Mendrysa, S.M.; Weissman, I.L.; Park, C.Y.; et al. Reduced ribosomal protein gene dosage and p53 activation in low-risk myelodysplastic syndrome. Blood 2011, 118, 3622–3633. [Google Scholar] [CrossRef] [Green Version]
- Keel, S.B.; Phelps, S.; Sabo, K.M.; O’Leary, M.N.; Kirn-Safran, C.B.; Abkowitz, J.L. Establishing Rps6 hemizygous mice as a model for studying how ribosomal protein haploinsufficiency impairs erythropoiesis. Exp. Hematol. 2012, 40, 290–294. [Google Scholar] [CrossRef] [Green Version]
- Pirbhoy, P.S.; Farris, S.; Steward, O. Synaptic activation of ribosomal protein S6 phosphorylation occurs locally in activated dendritic domains. Learn. Mem. 2016, 23, 255–269. [Google Scholar] [CrossRef] [Green Version]
- Pirbhoy, P.S.; Farris, S.; Steward, O. Synaptically driven phosphorylation of ribosomal protein S6 is differentially regulated at active synapses versus dendrites and cell bodies by MAPK and PI3K/mTOR signaling pathways. Learn. Mem. 2017, 24, 341–357. [Google Scholar] [CrossRef] [Green Version]
- Biever, A.; Valjent, E.; Puighermanal, E. Ribosomal Protein S6 Phosphorylation in the Nervous System: From Regulation to Function. Front. Mol. Neurosci. 2015, 8, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lecue, I.I.; Diez-Alarcia, R.; Morentin, B.; Meana, J.J.; Callado, L.F.; Urigüen, L. Ribosomal Protein S6 Hypofunction in Postmortem Human Brain Links mTORC1-Dependent Signaling and Schizophrenia. Front. Pharmacol. 2020, 11, 344. [Google Scholar] [CrossRef] [Green Version]
- Parnell, G.; Gatt, P.N.; McKay, F.C.; Schibeci, S.; Krupa, M.; Powell, J.; Visscher, P.; Montgomery, G.; Lechner-Scott, J.; Broadley, S.; et al. Ribosomal protein S6 mRNA is a biomarker upregulated in multiple sclerosis, downregulated by interferon treatment, and affected by season. Mult. Scler. J. 2013, 20, 675–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S. Regulation of Ribosomal Proteins on Viral Infection. Cells 2019, 8, 508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaerlein, M.; Horak, I. Phosphorylation of ribosomal proteins in HeLa cells infected with vaccinia virus. Nature 1976, 259, 150–151. [Google Scholar] [CrossRef]
- Ballestas, M.; Kaye, K.M. The latency-associated nuclear antigen, a multifunctional protein central to Kaposi’s sarcoma-associated herpesvirus latency. Futur. Microbiol. 2011, 6, 1399–1413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Dittmer, D.P. Ribosomal Protein S6 Interacts with the Latency-Associated Nuclear Antigen of Kaposi’s Sarcoma-Associated Herpesvirus. J. Virol. 2011, 85, 9495–9505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwun, H.J.; da Silva, S.R.; Shah, I.M.; Blake, N.; Moore, P.; Chang, Y. Kaposi’s Sarcoma-Associated Herpesvirus Latency-Associated Nuclear Antigen 1 Mimics Epstein-Barr Virus EBNA1 Immune Evasion through Central Repeat Domain Effects on Protein Processing. J. Virol. 2007, 81, 8225–8235. [Google Scholar] [CrossRef] [Green Version]
- Kuang, E.; Fu, B.; Liang, Q.; Myoung, J.; Zhu, F. Phosphorylation of Eukaryotic Translation Initiation Factor 4B (EIF4B) by Open Reading Frame 45/p90 Ribosomal S6 Kinase (ORF45/RSK) Signaling Axis Facilitates Protein Translation during Kaposi Sarcoma-associated Herpesvirus (KSHV) Lytic Replication. J. Biol. Chem. 2011, 286, 41171–41182. [Google Scholar] [CrossRef] [Green Version]
- Park, S.M.; Paek, K.Y.; Hong, K.Y.; Jang, C.J.; Cho, S.; Park, J.H.; Kim, J.H.; Jan, E.; Jang, S.K. Translation-competent 48S complex formation on HCV IRES requires the RNA-binding protein NSAP1. Nucleic Acids Res. 2011, 39, 7791–7802. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.-Y.; Su, W.-C.; Jeng, K.-S.; Chang, T.-H.; Lai, M.M.C. Attenuation of 40S Ribosomal Subunit Abundance Differentially Affects Host and HCV Translation and Suppresses HCV Replication. PLOS Pathog. 2012, 8, e1002766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haesen, D. Cellular inhibitors of Protein Phosphatase PP2A in Cancer. Biomed. Res. 2012, 23, 197–211. [Google Scholar]
- Park, H.-J.; Lee, K.-W.; Oh, S.; Yan, R.; Zhang, J.; Beach, T.G.; Adler, C.H.; Voronkov, M.; Braithwaite, S.P.; Stock, J.B.; et al. Protein Phosphatase 2A and Its Methylation Modulating Enzymes LCMT-1 and PME-1 Are Dysregulated in Tauopathies of Progressive Supranuclear Palsy and Alzheimer Disease. J. Neuropathol. Exp. Neurol. 2017, 77, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Talmage, D.; Blenis, J.; Benjamin, T.L. Polyomavirus middle T antigen induces ribosomal protein S6 phosphorylation through pp60c-src-dependent and -independent pathways. Mol. Cell. Biol. 1988, 8, 2309–2315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrabi, S.; Gjoerup, O.V.; Kean, J.A.; Roberts, T.M.; Schaffhausen, B. Protein phosphatase 2A regulates life and death decisions via Akt in a context-dependent manner. Proc. Natl. Acad. Sci. USA 2007, 104, 19011–19016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glennon, E.K.; Austin, L.S.; Arang, N.; Kain, H.S.; Mast, F.; Vijayan, K.; Aitchison, J.D.; Kappe, S.H.; Kaushansky, A. Alterations in Phosphorylation of Hepatocyte Ribosomal Protein S6 Control Plasmodium Liver Stage Infection. Cell Rep. 2019, 26, 3391–3399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, T.M.; Espina, V.; Senina, S.; Woodson, C.; Brahms, A.; Carey, B.D.; Lin, S.-C.; Lundberg, L.; Pinkham, C.; Baer, A.; et al. Rapamycin modulation of p70 S6 kinase signaling inhibits Rift Valley fever virus pathogenesis. Antivir. Res. 2017, 143, 162–175. [Google Scholar] [CrossRef] [Green Version]
- Jakubowicz, T.; Leader, D.P. Activation of a ribosomal protein S6 kinase in mouse fibroblasts during infection with herpesvirus. JBIC J. Biol. Inorg. Chem. 1987, 168, 371–376. [Google Scholar] [CrossRef]
- Wang, Y.; Weiss, L.M.; Orlofsky, A. Intracellular parasitism withToxoplasma gondiistimulates mammalian-target-of-rapamycin-dependent host cell growth despite impaired signalling to S6K1 and 4E-BP1. Cell. Microbiol. 2009, 11, 983–1000. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.H.; Kim, Y.-S.; Lee, S.; An, S. The effect of three-dimensional cultured adipose tissue-derived mesenchymal stem cell–conditioned medium and the antiaging effect of cosmetic products containing the medium. Biomed. Dermatol. 2019, 4, 1. [Google Scholar] [CrossRef]
- Son, O.; Kim, S.; Shin, Y.-J.; Kim, W.-Y.; Koh, H.-J.; Cheon, C.-I. Identification of nucleosome assembly protein 1 (NAP1) as an interacting partner of plant ribosomal protein S6 (RPS6) and a positive regulator of rDNA transcription. Biochem. Biophys. Res. Commun. 2015, 465, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Romero, E.B.; Martinez, J.; Heckmann, B.L.; Green, D.R. The clearance of dead cells by efferocytosis. Nat. Rev. Mol. Cell Biol. 2020, 21, 398–414. [Google Scholar] [CrossRef] [PubMed]
- Sender, R.; Milo, R. The distribution of cellular turnover in the human body. Nat. Med. 2021, 27, 45–48. [Google Scholar] [CrossRef]
- Fliedner, T.M.; Graessle, D.H. Hematopoietic cell renewal systems: Mechanisms of coping and failing after chronic exposure to ionizing radiation. Radiat. Environ. Biophys. 2007, 47, 63–69. [Google Scholar] [CrossRef]
- Silva, E.; Au-Yeung, H.W.; Van Goethem, E.; Burden, J.; Franc, N.C. Requirement for a Drosophila E3-Ubiquitin Ligase in Phagocytosis of Apoptotic Cells. Immunity 2007, 27, 585–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kratz, A.-S.; Richter, K.T.; Schlosser, Y.T.; Schmitt, M.; Shumilov, A.; Delecluse, H.-J.; Hoffmann, I. Fbxo28 promotes mitotic progression and regulates topoisomerase IIα-dependent DNA decatenation. Cell Cycle 2016, 15, 3419–3431. [Google Scholar] [CrossRef] [Green Version]
- Wittenberg, A.D.; Azar, S.; Klochendler, A.; Stolovich-Rain, M.; Avraham, S.; Birnbaum, L.; Gallimidi, A.B.; Katz, M.; Dor, Y.; Meyuhas, O. Phosphorylated Ribosomal Protein S6 Is Required for Akt-Driven Hyperplasia and Malignant Transformation, but Not for Hypertrophy, Aneuploidy and Hyperfunction of Pancreatic β-Cells. PLoS ONE 2016, 11, e0149995. [Google Scholar] [CrossRef] [Green Version]
- Polyak, K.; Lee, M.-H.; Erdjument-Bromage, H.; Koff, A.; Roberts, J.M.; Tempst, P.; Massague, J. Cloning of p27Kip1, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell 1994, 78, 59–66. [Google Scholar] [CrossRef]
- Toyoshima, H.; Hunter, T. p27, a novel inhibitor of G1 cyclin-Cdk protein kinase activity, is related to p21. Cell 1994, 78, 67–74. [Google Scholar] [CrossRef]
- Hsieh, A.C.; Costa, M.; Zollo, O.; Davis, C.; Feldman, M.E.; Testa, J.R.; Meyuhas, O.; Shokat, K.M.; Ruggero, D. Genetic Dissection of the Oncogenic mTOR Pathway Reveals Druggable Addiction to Translational Control via 4EBP-eIF4E. Cancer Cell 2010, 17, 249–261. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Liu, J.; Mi, X.; Liang, Y.; Li, J.; Huang, C. The N-terminal region of p27 inhibits HIF-1α protein translation in ribosomal protein S6-dependent manner by regulating PHLPP-Ras-ERK-p90RSK axis. Cell Death Dis. 2014, 5, e1535. [Google Scholar] [CrossRef] [PubMed]
- James, M.K.; Ray, A.; Leznova, D.; Blain, S.W. Differential Modification of p27 Kip1 Controls Its Cyclin D-cdk4 Inhibitory Activity. Mol. Cell. Biol. 2008, 28, 498–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, A.; James, M.K.; Larochelle, S.; Fisher, R.P.; Blain, S.W. p27 Kip1 Inhibits Cyclin D-Cyclin-Dependent Kinase 4 by Two Independent Modes. Mol. Cell. Biol. 2009, 29, 986–999. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Zeng, J.; Sun, F.; Tong, X.; Meng, G.; Wu, C.; Ding, X.; Liu, L.; Han, M.; Lu, C.; et al. p27 inhibits CDK6/CCND1 complex formation resulting in cell cycle arrest and inhibition of cell proliferation. Cell Cycle 2018, 17, 2335–2348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Nakano, T.; Wick, S.; Dubay, M.; Brizuela, L. Mechanism of Cdk2/Cyclin E Inhibition by p27 and p27 Phosphorylation. Biochemistry 1999, 38, 8713–8722. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Okada, M.; Nagai, K.; Fukada, Y. Suprachiasmatic Nucleus Circadian Oscillatory Protein, a Novel Binding Partner of K-Ras in the Membrane Rafts, Negatively Regulates MAPK Pathway. J. Biol. Chem. 2003, 278, 14920–14925. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Stevens, P.D.; Liu, J.; Yang, H.; Wang, W.; Wang, C.; Zeng, Z.; Schmidt, M.D.; Yang, M.; Lee, E.Y.; et al. PHLPP Is a Negative Regulator of RAF1, Which Reduces Colorectal Cancer Cell Motility and Prevents Tumor Progression in Mice. Gastroenterology 2014, 146, 1301–1312. [Google Scholar] [CrossRef] [Green Version]
- Gao, T.; Furnari, F.; Newton, A.C. PHLPP: A Phosphatase that Directly Dephosphorylates Akt, Promotes Apoptosis, and Suppresses Tumor Growth. Mol. Cell 2005, 18, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Brognard, J.; Sierecki, E.; Gao, T.; Newton, A.C. PHLPP and a Second Isoform, PHLPP2, Differentially Attenuate the Amplitude of Akt Signaling by Regulating Distinct Akt Isoforms. Mol. Cell 2007, 25, 917–931. [Google Scholar] [CrossRef]
- Chu, I.; Sun, J.; Arnaout, A.; Kahn, H.; Hanna, W.; Narod, S.; Sun, P.; Tan, C.-K.; Hengst, L.; Slingerland, J. p27 Phosphorylation by Src Regulates Inhibition of Cyclin E-Cdk2. Cell 2007, 128, 281–294. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, A.K.; De Melo, J.M.L.; Mørup, N.; Tritsaris, K.; Pedersen, S.F. Tumor microenvironment conditions alter Akt and Na+/H+ exchanger NHE1 expression in endothelial cells more than hypoxia alone: Implications for endothelial cell function in cancer. BMC Cancer 2017, 17, 542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grundy, M.; Jones, T.; Elmi, L.; Hall, M.; Graham, A.; Russell, N.; Pallis, M. Early changes in rpS6 phosphorylation and BH3 profiling predict response to chemotherapy in AML cells. PLoS ONE 2018, 13, e0196805. [Google Scholar] [CrossRef] [Green Version]
- Yanai, A.; Inoue, N.; Yagi, T.; Nishimukai, A.; Miyagawa, Y.; Murase, K.; Imamura, M.; Enomoto, Y.; Takatsuka, Y.; Watanabe, T.; et al. Activation of mTOR/S6K But Not MAPK Pathways Might Be Associated with High Ki-67, ER+, and HER2− Breast Cancer. Clin. Breast Cancer 2014, 15, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, Y.; Wang, Y. Retracted: MicroRNA-99b suppresses human cervical cancer cell activity by inhibiting the PI3K/AKT/mTOR signaling pathway. J. Cell. Physiol. 2018, 234, 9577–9591. [Google Scholar] [CrossRef]
- Kim, S.-H.; Jang, Y.; Chau, G.C.; Pyo, S.; Um, S.H. Prognostic significance and function of phosphorylated ribosomal protein S6 in esophageal squamous cell carcinoma. Mod. Pathol. 2012, 26, 327–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Z.; Zheng, Y.; Zhang, M.; Wang, J.; Yu, G.; Fang, W. Reciprocal expression of p-AMPKa and p-S6 is strongly associated with the prognosis of gastric cancer. Tumor Biol. 2015, 37, 4803–4811. [Google Scholar] [CrossRef]
- Shirakawa, Y.; Hide, T.; Yamaoka, M.; Ito, Y.; Ito, N.; Ohta, K.; Shinojima, N.; Mukasa, A.; Saito, H.; Jono, H. Ribosomal protein S6 promotes stem-like characters in glioma cells. Cancer Sci. 2020, 111, 2041–2051. [Google Scholar] [CrossRef] [PubMed]
- Molinolo, A.A.; Hewitt, S.; Amornphimoltham, P.; Keelawat, S.; Rangdaeng, S.; García, A.M.; Raimondi, A.R.; Jufe, R.; Itoiz, M.; Gao, Y.; et al. Dissecting the Akt/Mammalian Target of Rapamycin Signaling Network: Emerging Results from the Head and Neck Cancer Tissue Array Initiative. Clin. Cancer Res. 2007, 13, 4964–4973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corcoran, R.B.; Rothenberg, S.M.; Hata, A.N.; Faber, A.C.; Piris, A.; Nazarian, R.M.; Brown, R.D.; Godfrey, J.T.; Winokur, D.; Walsh, J.; et al. TORC1 Suppression Predicts Responsiveness to RAF and MEK Inhibition in BRAF- Mutant Melanoma. Sci. Transl. Med. 2013, 5, 196ra98. [Google Scholar] [CrossRef] [Green Version]
- Chaisuparat, R.; Rojanawatsirivej, S.; Yodsanga, S. Ribosomal Protein S6 Phosphorylation is Associated with Epithelial Dysplasia and Squamous Cell Carcinoma of the Oral Cavity. Pathol. Oncol. Res. 2012, 19, 189–193. [Google Scholar] [CrossRef] [PubMed]
- De Vicente, J.C.; Peña, I.; Rodríguez-Santamarta, T.; Lequerica-Fernández, P.; Bsc, L.S.; Allonca, E.; García-Pedrero, J.M. Phosphorylated ribosomal protein S6 correlation with p21 expression and inverse association with tumor size in oral squamous cell carcinoma. Head Neck 2017, 39, 1876–1887. [Google Scholar] [CrossRef]
- Liu, Z.; Yun, R.; Yu, X.; Hu, H.; Huang, G.; Tan, B.; Chen, T. Overexpression of Notch3 and pS6 Is Associated with Poor Prognosis in Human Ovarian Epithelial Cancer. Mediat. Inflamm. 2016, 2016, 5953498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komori, Y.; Yada, K.; Ohta, M.; Uchida, H.; Iwashita, Y.; Fukuzawa, K.; Kashima, K.; Yokoyama, S.; Inomata, M.; Kitano, S. Mammalian target of rapamycin signaling activation patterns in pancreatic neuroendocrine tumors. J. Hepato-Biliary-Pancreat. Sci. 2013, 21, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Knoll, M.; Macher-Goeppinger, S.; Kopitz, J.; Duensing, S.; Pahernik, S.; Hohenfellner, M.; Schirmacher, P.; Roth, W. The ribosomal protein S6 in renal cell carcinoma: Functional relevance and potential as biomarker. Oncotarget 2015, 7, 418–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwenofu, O.H.; Lackman, R.D.; Staddon, A.P.; Goodwin, D.G.; Haupt, H.M.; Brooks, J.S.J. Phospho-S6 ribosomal protein: A potential new predictive sarcoma marker for targeted mTOR therapy. Mod. Pathol. 2007, 21, 231–237. [Google Scholar] [CrossRef] [Green Version]
- Pinto, A.P.; Degen, M.; Barron, P.; Crum, C.P.; Rheinwald, J.G. Phosphorylated S6 as an immunohistochemical biomarker of vulvar intraepithelial neoplasia. Mod. Pathol. 2013, 26, 1498–1507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Järås, M.; Miller, P.; Chu, L.P.; Puram, R.V.; Fink, E.C.; Schneider, R.K.; Al-Shahrour, F.; Peña-Martínez, P.; Breyfogle, L.J.; Hartwell, K.A.; et al. Csnk1a1 inhibition has p53-dependent therapeutic efficacy in acute myeloid leukemia. J. Exp. Med. 2014, 211, 605–612. [Google Scholar] [CrossRef]
- Peters, J.M.; McKay, R.M.; McKay, J.P.; Graff, J.M. Casein kinase I transduces Wnt signals. Nature 1999, 401, 345–350. [Google Scholar] [CrossRef]
- Cuperjani, F.; Gashi, L.; Kurshumliu, F.; Dreshaj, S.; Selimi, F. Relationship between Ribosomal Protein S6-pS240 Expression and other Prognostic Factors in Non-Special Type Invasive Breast Cancer. Breast Care 2018, 14, 171–175. [Google Scholar] [CrossRef] [PubMed]
- You, K.S.; Yi, Y.W.; Kwak, S.-J.; Seong, Y.-S. Inhibition of RPTOR overcomes resistance to EGFR inhibition in triple-negative breast cancer cells. Int. J. Oncol. 2018, 52, 828–840. [Google Scholar] [CrossRef] [Green Version]
- Arteaga, C.L.; Truica, C. Challenges in the development of anti-epidermal growth factor receptor therapies in breast cancer. Semin. Oncol. 2004, 31, 3–8. [Google Scholar] [CrossRef]
- Nakai, K.; Hung, M.-C.; Yamaguchi, H. A perspective on anti-EGFR therapies targeting triple-negative breast cancer. Am. J. Cancer Res. 2016, 6, 1609–1623. [Google Scholar] [PubMed]
- You, K.; Yi, Y.; Cho, J.; Seong, Y.-S. Dual Inhibition of AKT and MEK Pathways Potentiates the Anti-Cancer Effect of Gefitinib in Triple-Negative Breast Cancer Cells. Cancers 2021, 13, 1205. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.W.; Hong, W.; Kang, H.J.; Kim, H.J.; Zhao, W.; Wang, A.; Seong, Y.; Bae, I. Inhibition of the PI 3K/ AKT pathway potentiates cytotoxicity of EGFR kinase inhibitors in triple-negative breast cancer cells. J. Cell. Mol. Med. 2013, 17, 648–656. [Google Scholar] [CrossRef] [PubMed]
- You, K.; Yi, Y.; Cho, J.; Park, J.-S.; Seong, Y.-S. Potentiating Therapeutic Effects of Epidermal Growth Factor Receptor Inhibition in Triple-Negative Breast Cancer. Pharmaceuticals 2021, 14, 589. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-C.; Bajikar, S.S.; Jamal, L.; Atkins, K.A.; Janes, K.A. A time- and matrix-dependent TGFBR3–JUND–KRT5 regulatory circuit in single breast epithelial cells and basal-like premalignancies. Nat. Cell Biol. 2014, 16, 345–356. [Google Scholar] [CrossRef] [Green Version]
- Corradetti, M.N.; Inoki, K.; Bardeesy, N.; DePinho, R.A.; Guan, K.-L. Regulation of the TSC pathway by LKB1: Evidence of a molecular link between tuberous sclerosis complex and Peutz-Jeghers syndrome. Genes Dev. 2004, 18, 1533–1538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK Phosphorylation of Raptor Mediates a Metabolic Checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llanos, S.; Pedrero, J.M.G.; Morgado-Palacin, L.; Rodrigo, J.P.; Serrano, M. Stabilization of p21 by mTORC1/4E-BP1 predicts clinical outcome of head and neck cancers. Nat. Commun. 2016, 7, 10438. [Google Scholar] [CrossRef]
- McDonald, J.M.; Pelloski, C.E.; LeDoux, A.; Sun, M.; Raso, G.; Komaki, R.; Wistuba, I.I.; Bekele, B.N.; Aldape, K. Elevated Phospho-S6 Expression Is Associated with Metastasis in Adenocarcinoma of the Lung. Clin. Cancer Res. 2008, 14, 7832–7837. [Google Scholar] [CrossRef] [Green Version]
- Bailis, W.; Pear, W.S. Notch and PI3K: How is the road traveled? Blood 2012, 120, 1349–1350. [Google Scholar] [CrossRef] [Green Version]
- Sabatini, D.M.; Erdjument-Bromage, H.; Lui, M.; Tempst, P.; Snyder, S.H. RAFT1: A mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell 1994, 78, 35–43. [Google Scholar] [CrossRef]
- Pinto, A.P.; Lin, M.-C.; Mutter, G.L.; Sun, D.; Villa, L.L.; Crum, C.P. Allelic Loss in Human Papillomavirus-Positive and -Negative Vulvar Squamous Cell Carcinomas. Am. J. Pathol. 1999, 154, 1009–1015. [Google Scholar] [CrossRef] [Green Version]
- Pinto, A.P.; Miron, A.; Yassin, Y.; Monte, N.; Woo, T.Y.C.; Mehra, K.K.; Medeiros, F.; Crum, C.P. Differentiated vulvar intraepithelial neoplasia contains Tp53 mutations and is genetically linked to vulvar squamous cell carcinoma. Mod. Pathol. 2010, 23, 404–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsubota, Y.; Ogawa, T.; Oyanagi, J.; Nagashima, Y.; Miyazaki, K. Expression of laminin γ2 chain monomer enhances invasive growth of human carcinoma cells in vivo. Int. J. Cancer 2010, 127, 2031–2041. [Google Scholar] [CrossRef]
- Pyke, C.; Rømer, J.; Kallunki, P.; Lund, L.R.; Ralfkiaer, E.; Danø, K.; Tryggvason, K. The gamma 2 chain of kalinin/laminin 5 is preferentially expressed in invading malignant cells in human cancers. Am. J. Pathol. 1994, 145, 782–791. [Google Scholar] [PubMed]
- Ono, Y.; Nakanishi, Y.; Ino, Y.; Niki, T.; Yamada, T.; Yoshimura, K.; Saikawa, M.; Nakajima, T.; Hirohashi, S. Clinicopathologic Significance of Laminin-5 Γ2 Chain Expression in Squamous Cell Carcinoma of the Tongue. Cancer 1999, 85, 2315–2321. [Google Scholar] [CrossRef]
- Spigel, D.R.; Burstein, H.J. Trastuzumab Regimens for HER2-Overexpressing Metastatic Breast Cancer. Clin. Breast Cancer 2003, 4, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Angulo, A.M.; Litton, J.K.; Broglio, K.R.; Meric-Bernstam, F.; Rakkhit, R.; Cardoso, F.; Peintinger, F.; Hanrahan, E.O.; Sahin, A.; Guray, M.; et al. High Risk of Recurrence for Patients with Breast Cancer Who Have Human Epidermal Growth Factor Receptor 2–Positive, Node-Negative Tumors 1 cm or Smaller. J. Clin. Oncol. 2009, 27, 5700–5706. [Google Scholar] [CrossRef] [PubMed]
- Gianni, L.; Dafni, U.; Gelber, R.D.; Azambuja, E.; Muehlbauer, S.; Goldhirsch, A.; Untch, M.; Smith, I.; Baselga, J.; Jackisch, C.; et al. Treatment with trastuzumab for 1 year after adjuvant chemotherapy in patients with HER2-positive early breast cancer: A 4-year follow-up of a randomised controlled trial. Lancet Oncol. 2011, 12, 236–244. [Google Scholar] [CrossRef]
- Markham, A.; Keam, S.J. Selumetinib: First Approval. Drugs 2020, 80, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Wagle, N.; Emery, C.; Berger, M.F.; Davis, M.J.; Sawyer, A.; Pochanard, P.; Kehoe, S.M.; Johannessen, C.M.; MacConaill, L.E.; Hahn, W.C.; et al. Dissecting Therapeutic Resistance to RAF Inhibition in Melanoma by Tumor Genomic Profiling. J. Clin. Oncol. 2011, 29, 3085–3096. [Google Scholar] [CrossRef] [Green Version]
- Pilié, P.G.; Gay, C.M.; Byers, L.A.; O’Connor, M.J.; Yap, T.A. PARP Inhibitors: Extending Benefit Beyond BRCA-Mutant Cancers. Clin. Cancer Res. 2019, 25, 3759–3771. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Wang, J.; Mishail, D.; Wang, C.-Y. Recent advancements in PARP inhibitors-based targeted cancer therapy. Precis. Clin. Med. 2020, 3, 187–201. [Google Scholar] [CrossRef]
- Lu, X.; Ma, J.; Chu, J.; Shao, Q.; Zhang, Y.; Lu, G.; Li, J.; Huang, X.; Li, W.; Li, Y.; et al. MiR-129-5p Sensitizes the Response of Her-2 Positive Breast Cancer to Trastuzumab by Reducing Rps6. Cell. Physiol. Biochem. 2017, 44, 2346–2356. [Google Scholar] [CrossRef] [PubMed]
- Sulaiman, A.; McGarry, S.; Lam, K.M.; El-Sahli, S.; Chambers, J.; Kaczmarek, S.; Li, L.; Addison, C.; Dimitroulakos, J.; Arnaout, A.; et al. Co-inhibition of mTORC1, HDAC and ESR1α retards the growth of triple-negative breast cancer and suppresses cancer stem cells. Cell Death Dis. 2018, 9, 815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, I.; Chen, H.; Maddalo, G.; Tuominen, R.; Rebecca, V.W.; Herlyn, M.; Hansson, J.; Davies, M.A.; Brage, S.E. Inhibiting insulin and mTOR signaling by afatinib and crizotinib combination fosters broad cytotoxic effects in cutaneous malignant melanoma. Cell Death Dis. 2020, 11, 882. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Sheng, X.; Willson, A.K.; Roque, D.R.; Stine, J.; Guo, H.; Jones, H.M.; Zhou, C.; Bae-Jump, V.L. Glutamine promotes ovarian cancer cell proliferation through the mTOR/S6 pathway. Endocr. -Relat. Cancer 2015, 22, 577–591. [Google Scholar] [CrossRef] [PubMed]
- Potratz, J.C.; Saunders, D.; Wai, D.H.; Ng, T.L.; McKinney, S.E.; Carboni, J.M.; Gottardis, M.M.; Triche, T.J.; Jürgens, H.; Pollak, M.N.; et al. Synthetic Lethality Screens Reveal RPS6 and MST1R as Modifiers of Insulin-like Growth Factor-1 Receptor Inhibitor Activity in Childhood Sarcomas. Cancer Res. 2010, 70, 8770–8781. [Google Scholar] [CrossRef] [Green Version]
- Luan, Q.-X.; Zhang, B.-G.; Li, X.-J.; Guo, M.-Y. MiR-129-5p is downregulated in breast cancer cells partly due to promoter H3K27m3 modification and regulates epithelial-mesenchymal transition and multi-drug resistance. Eur. Rev. Med Pharmacol. Sci. 2016, 20, 4257–4265. [Google Scholar]
- Cuesta, R.; Gupta, M.; Schneider, R.J. Chapter 7 The Regulation of Protein Synthesis in Cancer. Prog. Mol. Biol. Transl. Sci. 2009, 90, 255–292. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Mukherjee, S.; Tucker-Burden, C.; Ross, J.L.; Chau, M.J.; Kong, J.; Brat, D.J. TRIM8 regulates stemness in glioblastoma through PIAS3-STAT3. Mol. Oncol. 2017, 11, 280–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, M.; Shanmugam, M.K.; Bhardwaj, V.; Goel, A.; Gupta, R.; Sharma, A.; Baligar, P.; Kumar, A.P.; Goh, B.C.; Wang, L.; et al. The pleiotropic role of transcription factor STAT3 in oncogenesis and its targeting through natural products for cancer prevention and therapy. Med. Res. Rev. 2020, 41, 1291–1336. [Google Scholar] [CrossRef] [PubMed]
- Arvanitakis, L.; Mesri, E.; Nador, R.; Said, J.; Asch, A.; Knowles, D.; Cesarman, E. Establishment and characterization of a primary effusion (body cavity-based) lymphoma cell line (BC-3) harboring kaposi’s sarcoma-associated herpesvirus (KSHV/HHV-8) in the absence of Epstein-Barr virus. Blood 1996, 88, 2648–2654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.-K.; Park, K.-G. Targeting Glutamine Metabolism for Cancer Treatment. Biomol. Ther. 2018, 26, 19–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Secondary structure and post–translational modification of RPS6. The structural information was adopted from UniProt Knowledgebase entry P62753 (RS6_HUMAN).
Figure 1.
Secondary structure and post–translational modification of RPS6. The structural information was adopted from UniProt Knowledgebase entry P62753 (RS6_HUMAN).

Figure 2.
Schematic diagram of the RPS6 regulatory pathway. Arrows indicate unidirectional regulation.
Figure 2.
Schematic diagram of the RPS6 regulatory pathway. Arrows indicate unidirectional regulation.

Figure 3.
Extra-ribosomal functions of RPS6.

Figure 4.
Cancers associated with high levels of RPS6 or p-RPS6. A variety of cancers has been reported to be associated with high levels of RPS6 or p-RPS6. The image of the human body adopted from https://www.pikpng.com/downpngs/iwmhwRb_female-chest-anatomy-diagram-female-human-anatomy-human/, accessed on 1 June 2021.
Figure 4.
Cancers associated with high levels of RPS6 or p-RPS6. A variety of cancers has been reported to be associated with high levels of RPS6 or p-RPS6. The image of the human body adopted from https://www.pikpng.com/downpngs/iwmhwRb_female-chest-anatomy-diagram-female-human-anatomy-human/, accessed on 1 June 2021.

Figure 5.
The effects of RPS6-KD in cancer cells.

Figure 6.
Putative signaling pathways downstream of RPS6.

Table 1.
Selected examples of the extracellular proteins regulating RPS6 phosphorylation.
| Protein | Effects |
|---|---|
| BDNF (brain-derived neurotrophic factor) |
|
| C3 |
|
| CG (chorionic gonadotropin) |
|
| EGF (epidermal growth factor) |
|
| FGF8 (fibroblast growth factor 8) |
|
| FGF18 (fibroblast growth factor 18) |
|
| Insulin |
|
| IFNα (interferon α) |
|
| IFNα2 (interferon α2) |
|
| IFNβ (interferon β) | |
| IFNν (interferon ν) |
|
| MK (midkine) |
|
| NGF (nerve growth factor) |
|
| OSM (oncostatin M) |
|
| PDGF-BB (platelet-derived growth factor-BB) |
|
| Serum | |
| TNFα (tumor necrosis factor α) |
|
| WNT3A (WNT family member 3A) |
|
Table 3.
Selected examples of the membrane proteins regulating RPS6 phosphorylation.
| Protein | Effects |
|---|---|
| GRPR (gastrin-releasing peptide receptor) |
|
| PD-1 (programmed cell death protein 1) |
|
| PD-L1 (programmed death ligand 1; B7-H1, CD274) |
|
Table 4.
Selected examples of the transcription factors regulating RPS6 phosphorylation.
| Protein | Effects |
|---|---|
| AR (androgen receptor) |
|
| BTF3 (basic transcription factor 3) |
|
| FOXO3 (Forkhead box protein O3) |
|
| FOXO17 (Forkhead box protein O17) |
|
| MYC |
|
| mtp53P151S |
|
Table 6.
miRNAs regulating RPS6 level.
| miRNA | Effects |
|---|---|
| miR-92b-3p |
|
| miR-451 |
|
Table 7.
Selected examples of the natural ligands and stimuli regulating RPS6 phosphorylation.
| Natural Ligand or Stimulus | Effects |
|---|---|
| Alpha-linoleic acid (ALA) |
|
| Amino acids | |
| ATRA (all-trans-retinoic acid) |
|
| DHT (5α-dihydrotestosterone; androgen) |
|
| E2 (17β estradiol) |
|
| Hydrogen peroxide (H2O2) | |
| Hypoxia |
|
| Mannitol |
|
| Oleic acid (OA) |
|
| Phosphatidic acid (PA) |
Table 9.
Anticancer effects of RPS6 depletion in human cancer cells.
| Cancer Type | Cell Lines | Knockdown by | Effects of RPS6-KD | Ref |
|---|---|---|---|---|
| Breast cancer, trastuzumab-resistant | JIMT-1 SK-BR-3 | miR-129-5p mimic/siRNA |
| [476] |
| Breast cancer, triple-negative (TNBC) | HS578T MDA-MB-231 | siRNA |
| [21] |
| MDA-MB-231 SUMM149PT | siRNA |
| [477] | |
| Cervical carcinoma | HeLa | Antisense & shRNA |
| [340] |
| Esophageal squamous cell carcinoma (ESCC) | TE8 TE10 | siRNA |
| [436] |
| Gastric cancer, HER2-amplified | OE19 NCI-N87 | siRNA |
| [354] |
| Glioblastoma multiforme (GBM) | U251MG | siRNA |
| [438] |
| Hepatocellular carcinoma (HCC) | SK-HEP-1 | shRNA |
| [340] |
| Leukemia, T cell | Jurkat | antisense |
| [340] |
| Lung cancer (adenocarcinoma) | A549 | siRNA |
| [339] |
| Lung cancer (adenocarcinoma) | A549 | shRNA |
| [37] |
| Lung cancer, non-small cell (NSCLC) | SK-MES-1 H1650 | shRNA |
| [38] |
| Lung cancer (squamous cell carcinoma) | H520 | shRNA |
| [37] |
| Lymphoma, primary effusion lymphoma (PEL) | BC-3 | shRNA |
| [399] |
| Lymphoma, non-Hodgkin | OCI-LY3 SUDHL-6 | shRNA |
| [338] |
| Melanoma, cutaneous malignant (CMM) | A375 A375VR4 SK-MEL-2 3918 | siRNA |
| [478] |
| Melanoma, cutaneous malignant (CMM), kinase inhibitor-resistant | A375-DR A375-TR | siRNA |
| [39] |
| Ovarian cancer | HEY | siRNA |
| [479] |
| Ovarian cancer | SKOV-3 HO8910 | siRNA |
| [40] |
| Sarcoma, Ewing | TC32 TC71 | siRNA |
| [480] |
| Sarcoma, alveolar Rhabdomyosarcoma | Rh18 Rh30 | siRNA |
| [480] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Yi, Y.W.; You, K.S.; Park, J.-S.; Lee, S.-G.; Seong, Y.-S. Ribosomal Protein S6: A Potential Therapeutic Target against Cancer? Int. J. Mol. Sci. 2022, 23, 48. https://doi.org/10.3390/ijms23010048
AMA Style
Yi YW, You KS, Park J-S, Lee S-G, Seong Y-S. Ribosomal Protein S6: A Potential Therapeutic Target against Cancer? International Journal of Molecular Sciences. 2022; 23(1):48. https://doi.org/10.3390/ijms23010048
Chicago/Turabian StyleYi, Yong Weon, Kyu Sic You, Jeong-Soo Park, Seok-Geun Lee, and Yeon-Sun Seong. 2022. "Ribosomal Protein S6: A Potential Therapeutic Target against Cancer?" International Journal of Molecular Sciences 23, no. 1: 48. https://doi.org/10.3390/ijms23010048
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.