mTOR Inhibitors in Advanced Biliary Tract Cancers


1
Division of Hematology-Oncology, Department of Internal Medicine, Chang Gung Memorial Hospital, Linkou branch, Chang Gung University, Taoyuan 333, Taiwan
2
School of Medicine, National Yang-Ming University, Taipei 112, Taiwan
3
Department of Oncology, Taipei Veterans General Hospital, Taipei 112, Taiwan
4
Department of General Surgery and Liver Research Center, Chang Gung Memorial Hospital, Linkou branch, Chang Gung University, Taoyuan 333, Taiwan
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2019, 20(3), 500; https://doi.org/10.3390/ijms20030500
Submission received: 6 January 2019
/
Revised: 19 January 2019
/
Accepted: 22 January 2019
/
Published: 24 January 2019
(This article belongs to the Special Issue mTOR in Human Diseases)
Abstract
:Patients with advanced biliary tract cancers (BTCs), including cholangiocarcinoma (CCA), have poor prognosis so novel treatment is warranted for advanced BTC. In current review, we discuss the limitations of current treatment in BTC, the importance of mTOR signalling in BTC, and the possible role of mTOR inhibitors as a future treatment in BTC. Chemotherapy with gemcitabine-based chemotherapy is still the standard of care and no targeted therapy has been established in advanced BTC. PI3K/AKT/mTOR signaling pathway linking to several other pathways and networks regulates cancer proliferation and progression. Emerging evidences reveal mTOR activation is associated with tumorigenesis and drug-resistance in BTC. Rapalogs, such as sirolimus and everolimus, partially inhibit mTOR complex 1 (mTORC1) and exhibit anti-cancer activity in vitro and in vivo in BTC. Rapalogs in clinical trials demonstrate some activity in patients with advanced BTC. New-generation mTOR inhibitors against ATP-binding pocket inhibit both TORC1 and TORC2 and demonstrate more potent anti-tumor effects in vitro and in vivo, however, prospective clinical trials are warranted to prove its efficacy in patients with advanced BTC.
1. Introduction of Bile Duct Cancers
Bile duct cancers (BTCs) including intrahepatic/extrahepatic cholangiocarcinoma (CCA), gallbladder cancer (GBC), and Ampullar Vater cancer, are the malignant neoplasms arising from epithelial cells of bile ducts [1]. CCA was considered as primary liver cancer and, currently, the term CCA has been used for bile duct cancers arising from intrahepatic and extrahepatic bile system, excluding the malignancies of gallbladder and Ampulla of Vater.
The estimated annual cases of primary liver cancers including intrahepatic CAA is 42,220 in the United States [2] and around 15% of them are intrahepatic CCA according to Surveillance, Epidemiology, and End Results (SEER) program [3,4]. Estimated 12,190 cases of gallbladder and other biliary cancers are diagnosed annually in the United States [2]. Although they are uncommon and relatively rare, the patients with BTC have a poor prognosis because most of them are locally advanced at presentation and high recurrence rate for the early stage after curative surgery [5,6]. The efficacy of systemic treatment is limited, therefore, novel agents are warranted for the patients with biliary tract cancers.
2. Current Evidences of Systemic Treatment for Advanced Bile Duct Cancers
2.1. In the Era of Chemotherapy
Systemic chemotherapy is the standard treatment in biliary duct cancers based on a randomized study which showed fluorouracil (FU)-based systemic chemotherapy provided longer overall survival (6 versus 2.5 months) than best supportive care alone in 90 eligible patients with pancreatic (n = 53) or biliary cancer (n = 37) [7]. Therefore, chemotherapy with FU-based regimens proved the efficacy of chemotherapy and became the standard of care for patients with advanced BTC in 1996. A later study in patients with advanced pancreatic cancer showed gemcitabine-treated patients experienced better clinical benefit response compositing of measurements of pain, Karnofsky performance status, and body weight (23.8% vs. 4.8%, p = 0.0022) and longer overall survival (OS, 5.65 and 4.41 months, p = 0.0025) than 5-FU-treated patients [8], gemcitabine was also wildly used in patients with advanced BTC. Subsequently, chemotherapy with FU, and gemcitabine, with or without platinum has been studied, but the optimal chemotherapy regimen has been debated for more than a decade.
In 2007, pooled phase II studies by Eckel et al. showed superior response rates (RRs) and tumor control rates (TCRs) of gemcitabine- or platinum-based regimens and highest RRs and TCRs was found in the gemcitabine/platinum combination subgroup so this study concluded that gemcitabine/platinum combination represented the provisional standard for chemotherapy [9] even lack of direct comparison of gemcitabine and 5-FU in these patients. In 2010, ABC-02 trial, the first randomized phase III study in advanced BTC, reported that gemcitabine plus cisplatin has better TCRs (81.4% versus 71.8%, p = 0.049), median progression-free survival (PFS, 8.0 months versus 5.0 months, p < 0.001) and median OS (11.7 months versus 8.1 months, p < 0.001) than gemcitabine alone [10] so the combination of gemcitabine and cisplatin has been considered the standard of care as the first-line treatment in patients with advanced BTC and widely used in clinical practice [11]. This regimen has not been compared head to head with other gemcitabine-based combinations except gemcitabine plus TS-1 which demonstrated non-inferiority in the Japanese phase III FUGA-BT study [12]. This study enrolled a total of 354 patients with chemotherapy-naïve advanced BTC and a preliminary report presented at the 2018 American Society of Clinical Oncology (ASCO) Gastrointestinal Cancers Symposium showed the combination of gemcitabine/TS-1 was non-inferior in terms of median OS (15.1 versus 13.4 months), median PFS (6.8 versus 5.8 months), and objective RRs (30% versus 32%) so that this combination can be considered as another standard treatment in patients with advanced BTC.
2.2. Development of Targeted Therapy in Advanced BTC
Few prospective trials have been undertaken of first-line chemotherapy and targeted therapy in advanced BTC. Molecularly targeted agents targeting vascular endothelial growth factor (VEGF) or epidermal growth factor receptor (EGFR) were investigated in advanced BTC. Although the addition of bevacizumab [13] or cetuximab [14] to chemotherapy showed promising clinical results in phase II trials, randomized study [15,16] failed to demonstrate additional activity of cetuximab when it combined with chemotherapy. In a study of pooled trials published during January 2000 to January 2014, the authors concluded that triplet combinations of gemcitabine/FU/platinum and gemcitabine-based chemotherapy plus targeted therapy (predominantly targeting EGFR) are most effective concerning TCRs and survivals [17]. However, gemcitabine-based chemotherapy is still the standard of care in advanced BTC and the use of additional targeted therapy is questionable.
2.3. Immune Checkpoints Inhibitors
The immune checkpoints inhibitors against cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), programmed cee death protein-1(PD-1), or programmed death-ligand 1 (PD-Ll) have been developed to show efficacy in a variety of cancers. Nakamura et al. found that the poorest prognosis for BTC patients was in those with significant enrichment of hypermutated tumors and elevated expression of immune checkpoint molecules such as CTLA-4 and IDO but which are associated with favourable clinical response to anti-PD-L1 treatment [18]. In this study, 45.2% of patients showed an increase in the expression of immune checkpoint molecules. In Keynote-026 (NCT02054806), a phase 1b trial to evaluate the safety and efficacy of pembrolizumab in advanced pre-treated BTC patients, Bang et al. [19] reported interim results that 8 out of 23 PD-L1-positive patients (35%) had PD and SD and some of them had disease control lasting for 40+ weeks. A number of immunotherapy studies are currently recruiting and ongoing [20].
In addition, based on data from the patients with microsatellite instability-high (MSI-H) or deficient mismatch repair (dMMR) cancers enrolled across uncontrolled, multi-cohort, multi-center, single-arm clinical trials, in May 2017, the US FDA approved pembrolizumab for treatment of a variety of advanced MSI-H or dMMR solid tumors (including BTC) [21] so the patients with advanced BTC harboring MSI-H or dMMR are candidates for immune checkpoint inhibitors.
3. Molecular Alterations in Cholangiocarcinoma
A variety of molecular alterations involving both oncogenes (e.g., RAS [22,23,24], BRAF [25], ERBB2/HER2, EGFR [26], and PIK3CA (phosphoinositide 3-kinase, catalytic, α-polypeptide) [27]) and tumor suppressor genes (e.g., p53 [23], SMAD4 [28], and CDKN2A [29]) have been described in invasive BTC [30]. Most of the genetic alterations involve phosphoinositide 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) through MAPKinase activation or p53 suppression resulting in activation of mTOR. p53 negatively regulates the PI3K/AKT/mTOR pathway via its upregulation of phosphatase and tensin homolog (PTEN), TSC2, AMP-activated protein kinase (AMPK), and other proteins [31].
In addition, gene expression profiling of BTC compared with normal biliary epithelium has identified upregulated ribosomal protein S6 kinase, 70kD (RPS6K encoding p70-S6K) and eukaryotic translation initiation factor 4E (EIF4E), which are two important downstream mediators of AKT/mTOR signaling pathway, as well as the potential drug target insulin-like growth factor 1 receptor (IGF1-R) [32]. The collective evidences of genetic studies in BTC detailed above suggest mTOR plays a central and critical role in invasive BTC, therefore, targeting mTOR pathway by mTOR inhibitors could be envisioned as a novel treatment in advanced BTC (Figure 1).
4. mTOR Pathway in Cancers
4.1. mTOR, Its Complexes and Downstream Regulations in Cancers
The serine/threonine kinase mTOR, a member in a family of protein kinase called PI3K-related kinases, integrates intracellular and extracellular signal transduction leading to regulation of in a variety of cellular functions such as cell cycle progression, cell metabolism, cell proliferation, survival [33,34,35,36]. The mTOR pathway is dysregulated in various cancers including cholangiocarcinoma [37,38], making mTOR an important target for the development of new anticancer drugs [39,40].
The mTOR exists in two structurally and functionally distinct complexes, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2), which regulated by and regulate distinct signaling pathways resulting from different complex co-factors. Both complexes contain mTOR and a protein, called mLST8, that associates with its kinase domain. It is considered that the functional differences between mTORC1 and mTORC2 result from the other core components such as Raptor in mTORC1 and a complex of Rictor and mSIN1 in mTORC2.
mTORC1 is the downstream of the two proto-oncogenes kinase pathways, PI3K/AKT as well as RAS/RAF/MEK/ERK, through inhibition of TSC2 and PRAS40, both are negative regulators of mTORC1 [41,42,43,44,45,46]. mTORC1 is the upstream of two distinct pathways which control translation of specific subsets of mRNA. One involves p70-S6K, and another pathway is related with eukaryotic initiation factor 4E binding protein-1 (4E-BP1) [47]. The PI3K/AKT/mTOR signaling cascade is central to cell survival, apoptosis, metabolism, motility, and angiogenesis [48].
In response to PI3K/AKT signaling activation, mTOR rapidly phosphorylates both downstream substrates, p70-S6K and 4E-BP1, the latter leading to release of EIF4E, resulting in initiation of translation. This pathway was also found to be up-regulated using tissue microarrays in CCA [27] and is a key pathway for CCA drug development [30]. mTOR can be inhibited by using the macrolide rapamycin. However, a subset of biliary cancers will be possibly resistant to mTOR inhibitors as the downstream activation bypass mTOR regulation. Therefore, in a study of gene expression comparing BTC and normal biliary epithelium identified two genes involving mTOR pathway, p70-S6K and EIF4E, which are differentially up-regulated in BTC so this study provides alternative downstream targets for inhibition [47].
In contrast, mTORC2 contains Rictor in place of Raptor so it phosphorylates a distinct set of substrates [49]. AKT/mTORC2 forms a positive feedback loop that AKT phosphorylates SIN1 at Tyrosine 86 which enhances mTORC2 kinase activity to phosphorylate and catalyse AKT(Serine 473) leading to AKT activation to control various cellular processes [50,51]. mTORC2 is tumorigenic and is reported to promote cancer via formation of lipids essential for growth and energy production in hepatocellular carcinoma model [52,53,54].
4.2. Upstream Regulation of mTOR in Cancers
4.2.1. The Physiological Regulation of mTOR Pathway
PI3Ks are a family of intracellular signal transducers and regulate a crucial signal transduction system linking multiple receptors and oncogenes to many essential cellular functions including cell survival, proliferation, and differentiation [55]. Upon signals from various growth factors and cytokines stimulating receptor tyrosine kinases (RTKs) and G protein-coupled receptors (GPCRs), PI3Ks transduce the signals into intracellular messages via activating the serine/threonine kinase AKT followed by downstream effector pathways.
Several classes of PI3K kinases have been identified in mammalian cells, and only class I PI3K can function as second-messenger being implicated in oncogenesis. The class I PI3K kinase consists of two main subunits, p85 and p110, which mediate regulatory and catalytic activity of kinase respectively [56]. Three different genes, PIK3CA, PIK3CB, and PIK3CD, encode three specific p110 isoforms, p110α, β, and δ, respectively [57], and activating missense mutations of PIK3CA have been found as oncogenic in a variety of cancers [58]. PIK3CB mutation is rare but has been reported to be activating and oncogenic [59].
The PI3K kinases activated by RTKs phosphorylate the 3′-hydroxyl group of phosphatidylinositol (4,5)-bisphosphate (PIP2) to generate phosphatidylinositol (3,4,5)-trisphosphate (PIP3) [60], which is an important second messenger that transduces signals through AKT to downstream activators of cellular growth and survival [61]. PTEN is a phosphatase which negatively regulates PIP3 activity by dephosphorylation [62].
AMPK activity can be regulated by the cellular energy level through the balance in ATP/AMP ratio, so low ratio under nutrient deprivation can activate AMPK followed by mTOR inhibition via TSC1/2 activation [63]. p53 was reported as a substrate of AMPK which activates p53 phosphorylation on serine 15 required to initiate AMPK-dependent cell cycle arrest [64]. In addition, AMPK, TSC2, and PTEN were also regulated by p53 [65]. Furthermore, MAPKinase pathway activates ERK/RSK which regulate mTOR via TSC-2 suppression [66]. Therefore, those pathways are tightly regulated to affect mTOR activities leading to the balance of cell survival and death (Figure 1).
4.2.2. Alterations of mTOR Pathway in Cancers
IGF1-R is a receptor on the cell surface and stimulated through the binding with IGF1 resulting in activation of PI3K/AKT/mTOR pathway. IGF1-R overexpression was reported to be associated with more aggressive phenotypes of cancer [67]. PTEN is a negative regulator of PI3K so is considered as a tumor suppressor in tumorigenesis [62]. Dysregulation of the above genes or proteins leads to mTOR activation resulting in tumor progression and survival in BTC.
PIK3CA mutations are commonly found in cancers such colon, breast, gastric, and brain cancers, but such mutations are rarely found in BTC [18,27] and are associated with poor prognosis [68]. Although not high rate of PIK3CA activating mutations, immunohistochemical evaluation of downstream PIK3CA targets EIF4E and 4E-BP1 suggests that additional mechanisms may play positively regulation in mTOR pathway in cancers. In addition, PTEN downregulation was reported to be associated with mTOR activation in BTC [69]. Expression profiling of BTC compared with normal biliary epithelium has identified upregulated AKT/mTOR signaling components, including the potential drug target IGF1-R [47]. Expression of IGF1-R and its ligands are seen in the majority of GBCs and metastases providing a targeted candidate for therapeutic strategies to interfering with IGF pathway [70]. Therefore, treating BTC cell lines with a small-molecule inhibitor of the IGF1-R was suggested and showed the efficacy of targeting this pathway [71].
5. mTOR Inhibitors
5.1. Rapalogs, First-Generation of mTOR Inhibitors
Rapalogs include rapamycin, also known as sirolimus, and its analogues such as everolimus, temsirolimus are all highly specific allosteric inhibitors of mTOR with the same mechanism of action [72,73]. Rapalogs binding to the intracellular protein FKBP12 forms a drug-protein complex. This FKBP12–rapalog complex binds to the FKBP12–rapamycin binding (FRB) domain of mTOR, which is located at just N-terminal next to the kinase domain [74,75]. Binding of FKBP12–rapalog complex to the FRB domain interferes the association of mTOR and Raptor in mTORC1 so that inhibits mTORC1 signaling within minutes at low doses of rapalogs. In contrast, higher doses or prolonged use of rapalogs can sequester mTOR from mTORC2 to block mTORC2 signaling [27]. Although rapalogs are highly specific to mTOR, it is well known that rapalogs can only partially inhibit the functions of mTORC1 [76,77]. For example, rapamycin highly inhibits S6K activity in all settings but does not inhibit 4E-BP1 which is also a direct substrate of mTORC1 [76]. Therefore, the sensitivity to rapalogs cannot determine whether the cellular processes are mTORC1-dependent or mTORC1-independent.
5.2. Second-Generation mTOR Inhibitors
For the limitations of rapalogs in mTOR inhibition, a number of second-generation mTOR inhibitors have been developed. Like most kinase inhibitors, second-generation mTOR inhibitors were designed to directly target the ATP-binding pocket of the mTOR kinase domain so these new generation mTOR inhibitors can inhibit both mTORC1 and mTORC2. The next important question is whether these compounds display superior anti-cancer activity via inhibition of both mTORC1 and mTORC2 and whether such treatments can be tolerated at the effective doses because of off-target effects on the evolutionarily related protein kinases [78]. Currently, several compounds such as AZD-2014, MLN0128 (INK128, TAK228), OSI-027, and GDC-0349 have been investigated in clinical trials to prove the clinical significance in cancer treatments. Furthermore, NVP-BEZ235, LY3023414, and PF-04691502 are dual PI3K/mTOR inhibitors and have been investigated in clinical trials.
AZD-2014, a dual mTORC1/2 inhibitor, showed superior activity than everolimus in vitro and in vivo in renal cell carcinoma [79] but demonstrate inferior efficacy in patients with renal cell carcinoma [80]. Therefore, although preliminary studies showed promising efficacy of dual mTOR inhibitors in various of cancers [81,82], the clinical significance should still be investigated in clinical trials to prove their activities in cancer treatment [83].
6. Sustained mTORC1/2 Signaling Activation as a Driver of Resistance to Anti-Cancer Treatment
Several studies in different cancer types have already shown that sustained mTORC1 signaling under certain targeted therapy is strongly associated with primary and acquired resistance to such treatment so mTORC1 inhibition seems to be an effective therapeutic strategy in combination with other targeted agents even the efficacy is limited as single-use [84,85]. mTORC1 activation has been also reported to be resistant to various anti-cancer treatments including chemotherapy, targeted therapy, and hormonal treatment. On the contrary, mTOR inhibition by rapalogs was shown sensitization to anti-cancer treatments [86].
mTORC2 activation and AKT phosphorylation have also been found to escape MAPKinase inhibition by sorafenib in CCA cells. Therefore, prevention of escape by suppressing mTORC2 activity may lead to promising new approaches in CCA therapy [87].
7. Preclinical Studies of mTOR Inhibitors in BTC
7.1. The Rationale of mTOR Inhibitors Alone or in Combination with Chemotherapeutic Agents in Cholangiocarcinoma
As discussed above, a number of genetic alterations directly or indirectly involving PI3K/AKT/mTOR activation were reported in advanced BTC [30]. In addition, gene expression profiling of invasive BTC has showed upregulation of downstream mediators in mTOR pathway, RPS6K and EIF4E as well as IGF1-R [32]. These genetic studies in BTC suggest mTOR plays an important role in invasive BTC, therefore, mTOR inhibitors targeting mTOR pathway could be considered as a reasonable therapeutic strategy.
Furthermore, in a preclinical study to investigate the functional role and mechanism of miR-199a-3p in the regulation of cisplatin sensitivities in CCA, Li et al. demonstrated that miR-199a-3p enhances cisplatin activity in CCA cell lines (GBC-SD and RBE) via both inhibiting the mTOR signaling pathway and decreasing the expression of MDR1. In this study, mTOR suppression by siRNA or miR-199a-3p potentiates cisplatin sensitivity of CCA cell lines indicating mTOR pathway regulates cisplatin activity in CCA although the exact mechanism is unclear [88].
Ling et al. found metformin increases AMPK phosphorylation and inhibits the activation of mTORC1 complex and can sensitize sorafenib, 5-FU, and As2O3 but not gemcitabine in cholangiocarcinoma cell lines (RBE and HCCC-9810) [89]. Wandee et al. found metformin sensitizes cisplatin in CCA cell lines (KKU-100 and KKU-452) via AMPK activation and AKT/mTOR/p70-S6K suppression [90]. Lyu et al. demonstrated Fyn was associated with AMPK/mTOR regulation [91] and was overexpressed in CCA cell lines. Furthermore, Fyn knockdown in CCA cell lines induces AMPK phosphorylation, followed by inhibiting downstream mTOR phosphorylation leading to inhibition of migration and invasion [92].
Above studies have shown mTOR pathway is crucial for regulation of tumor growth and sensitivities to anti-cancer drugs in CCA.
7.2. Preclinical Studies of Rapalogs in BTC
Everolimus exhibits in vitro multiple effects in a CCA cell line (RMCCA-1). Everolimus at low concentrations reduced in vitro invasion and migration and high concentrations exhibited cytotoxic effects such as suppression of cell proliferation and induction of apoptosis [93]. Everolimus was also found to inhibit the secretion of proinflammatory cytokines by cancer-associated myofibroblasts (CAFs) and inhibits proliferation of CCA cells (HuCCT1 and TFK1) at low concentrations [94]. Both studies confirmed the previous hypothesis that mTOR plays important role in CCA and mTOR inhibitors exert anticancer effects via mTOR inhibition. In addition, rapamycin was found to initiate AKT activation in CCA and inhibition of AKT by salubrinal potentiates the in vitro and in vivo efficacy of rapamycin in CCA both [95]. In terms of combination of rapalogs and cytotoxic agents, our group reported gemcitabine plus everolimus combination showed synergistic effect in the CCA cells in vitro and in vivo [96].
7.3. New Generation mTOR Inhibitors in BTC
Zhang et al. established a novel mouse model of intrahepatic CCA exhibiting activated AKT/mTOR cascade and found both mTORC1 and mTORC2 signalings are required for AKT/YapS127A-induced cholangiocarcinogenesis [97]. MLN0128, a second generation, ATP-competitive mTOR inhibitor, suppress cell growth and induce apoptosis in vitro and in vivo via suppression of both mTORC1 and mTORC2 signaling. An important finding in this study was that MLN0128 had better therapeutic efficacy than gemcitabine/oxaliplatin combination (one of the standard chemotherapy regimen) as well as everolimus in the treatment of AKT/YapS127A intrahepatic CCA model. In addition, the same group reported that the combination of palbociclib, a CDK4/6 inhibitor, and MLN0128 demonstrated a pronounced, synergistic growth inhibition in intrahepatic CCA cell lines and in AKT/YapS127A mice [98].
7.4. Dual PI3K/mTOR Inhibitors in BTC
New dual inhibitors targeted to PI3K/mTOR such as NVP-BEZ235, which exerts strong antiproliferative properties against primary cultures of intrahepatic CCA subtypes with differential drug sensitivity, have been developed [99]. In addition, our group identified both HSP90 overexpression and loss of PTEN were poor prognostic factors in patients with intrahepatic CCA. Thus, the combination of the HSP90 inhibitor (NVP-AUY922) and the PI3K/mTOR inhibitor (NVP-BEZ235) in CCA were evaluated and showed synergistic effects in vitro and in vivo. This combination not only inhibited the PI3K/AKT/mTOR pathway but also induced reactive oxygen species (ROS), which may enhance the vicious cycle of endoplasmic reticulum (ER) stress. Our data suggest the simultaneous targeting of the PI3K/mTOR and HSP pathways could be a novel and active therapeutic strategy for advanced CCA [100].
7.5. Other Indirect Inhibition of mTOR Pathway
VEGF can induce phosphorylation of both VEGFR1 and VEGFR2 but only VEGFR2 played a role in the promoting anti-apoptotic cell growth through activating a PI3K/AKT/mTOR signaling pathway. Apatinib, a VEGFR2-specific inhibitor, was reported to inhibit the anti-apoptosis induced by VEGF signaling, and promoted cell death in vitro and delayed tumor growth in vivo [101].
8. mTOR Inhibitors in Clinical Setting
There are two settings for mTOR inhibitors used in the patients with advanced cholangiocarcinoma. Firstly, mTOR inhibitors could be used alone or in combination with other agents in the patients with advanced cholangiocarcinoma refractory to standard treatments. Secondly, mTOR inhibitors could be used in combination with standard treatment in patients with treatment-naïve advanced cholangiocarcinoma to investigate the possibly better response, progression-free survival and overall survival than conventional standard treatment. The combination of mTOR inhibitors with standard treatment aims to overcome resistance and potentiate the cytotoxicity of chemotherapy. Published clinical studies of mTOR inhibitors in advanced BTC were summarized in Table 1.
8.1. Clinical Studies of Everolimus in Advanced BTC
Bian et al. reported a 31-year-old male patient was diagnosed as stage IV intrahepatic CCA with PIK3CA mutation (E545G), which may result in activating mTOR pathway so patient received everolimus and achieved partial response (PR) after 2-month everolimus and at least 6-month PFS [106]. Larger series of everolimus in advanced BTC were studied. A phase I study reported that everolimus achieved 50% disease-control-rate (DCR) in a subgroup of 22 advanced BTC patients [107]. A phase II ITMO study in Italy enrolled 39 patients with previously chemotherapy-treated advanced BTC and the DCR was 44.7%, and the RR was 5.1%. Among two patients who experienced response, one patient showed a PR at 2 months and another patient showed a complete response (CR) sustained up to 8 months. The median PFS and OS were 3.2 (CI: 1.8–4.0) and 7.7 (CI: 5.5–13.2) months respectively [108]. In another phase II study to evaluate the activity of everolimus in 10 patients with PIK3CA amplification/mutation or PTEN loss refractory solid cancer, one patient with CCA with PTEN loss experienced disease control [109]. Recently, another phase II the RADiChol study published to evaluate the efficacy of everolimus as first-line treatment in treatment naïve advanced BTC, 27 patients enrolled showed DCR at 12 weeks was 48% and PFS and OS were 5.5 (2.2–10.0) and 9.5 (5.5–16.6) months, respectively [110]. Three (12%) of 25 patients evaluable for response experienced PR and 15 patients had stable diseases (SD). In addition, the authors performed immunohistochemistry (IHC) staining of PI3K/AKT/mTOR and found no association between IHC and clinical outcomes [110].
8.2. Clinical Studies of Sirolimus in Advanced BTC
In terms of other mTOR inhibitors, sirolimus, Rizell reported a cohort of sirolimus used in patients with hepatocellular carcinoma (n = 21) and iCCA (n = 9). Three (33%) of nine patients with intrahepatic CCA achieved SD after sirolimus treatment and others experienced progressive disease [111]. In a pilot study enrolling patients with PIK3CA mutant/amplified refractory solid cancer, sirolimus failed to demonstrate the clinical benefit in a patient with hilar cholangiocarcinoma (PIK3CA E545K mutation) who experienced disease progression following the second cycle of sirolimus with PFS of 0.9 months [112].
mTOR inhibitors alone, either sirolimus or everolimus showed some activity in advanced BTC with acceptable toxicities in treatment-naïve or pre-treated advanced BTC. The DCR (~50%) and survivals are compatible with the current standard of care. The use of mTOR inhibitors should be validated by larger randomized controlled trial (RCT) studies particularly in treatment naïve patients. For refractory BTC patients, mTOR inhibitors provide limited disease control which might benefit some patients whose BTCs are refractory to standard treatment.
The only published study investigating the combination of everolimus and chemotherapy was performed to determine the maximally tolerated dose (MTD) of different combinations [113]. The MTD for Cohort I of the two-drug combination was everolimus 5 mg on Monday/Wednesday/Friday and gemcitabine 800 mg/m2. Cohort II was to determine the MTC when cisplatin was added in everolimus/gemcitabine as a three-drug combination and cohort III was evaluation the activity of everolimus 5 mg on Monday/Wednesday/Friday, gemcitabine 600 mg/m2, cisplatin 12.5 mg/m2, 60% of 10 CCA and GBC carcinoma in cohort 3 experienced SD. The hematological DLT (mainly thrombocytopenia) limited the dosage used in three-drug combination and resulted in limited response rate. However, everolimus/gemcitabine could be an interesting regimen which demonstrated 2 CRs in this two-drug combination.
8.3. Clinical Studies of New Generation mTOR Inhibitors in Advanced BTC
Although new generation mTOR inhibitors and dual PI3K/mTOR inhibitors targeting both mTORC1 and mTORC2 showed anticancer activities in BTC (discussed in Section 8.2 and Section 8.3), no clinical trials of these agents in advanced BTC were reported. All of these compounds are being investigated under early clinical trials to evaluate the efficacy in various refractory solid or hematologic cancers. Therefore, more and more results of new generation mTOR inhibitors will be released and published in the near future.
9. Summary of mTOR Inhibitors in BTC
In conclusion, mTOR signaling pathway connecting with several other pathways and networks regulates cancer proliferation and progression. Activation of mTOR is associated with drug-resistance in BTC. Rapalogs partially inhibit mTORC1 and exhibit anti-cancer activity. Rapalogs in clinical trials demonstrate some activity in patients with advanced BTC. New-generation mTOR inhibitors against ATP-binding pocket inhibit both TORC1 and TORC2 and demonstrate more potent anti-tumor effects in vitro and in vivo. Prospective clinical trials are warranted to prove its efficacy in patients with advanced BTC.
Funding
This research was supported by CMRPG310231, CMRPG310241, MOST107-2314-B-182A-134-MY3, NMRPG3H6211, CRRPG3F0031-3, and MOST105-2314-B-182A-MY2, NMRPG3F6021-2 to C.-N.Y.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Patel, T. Cholangiocarcinoma. Nat. Clin. Pract. Gastroenterol. Hepatol. 2006, 3, 33–42. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef]
- Patel, T. Increasing incidence and mortality of primary intrahepatic cholangiocarcinoma in the United States. Hepatology 2001, 33, 1353–1357. [Google Scholar] [CrossRef] [Green Version]
- Shaib, Y.H.; Davila, J.A.; McGlynn, K.; El-Serag, H.B. Rising incidence of intrahepatic cholangiocarcinoma in the United States: A true increase? J. Hepatol. 2004, 40, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Jarnagin, W.R.; Fong, Y.; DeMatteo, R.P.; Gonen, M.; Burke, E.C.; Bodniewicz, B.J.; Youssef, B.M.; Klimstra, D.; Blumgart, L.H. Staging, resectability, and outcome in 225 patients with hilar cholangiocarcinoma. Ann. Surg. 2001, 234, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Valle, J.W.; Wasan, H.; Johnson, P.; Jones, E.; Dixon, L.; Swindell, R.; Baka, S.; Maraveyas, A.; Corrie, P.; Falk, S.; et al. Gemcitabine alone or in combination with cisplatin in patients with advanced or metastatic cholangiocarcinomas or other biliary tract tumours: A multicentre randomised phase II study—The UK ABC-01 Study. Br. J. Cancer 2009, 101, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Glimelius, B.; Hoffman, K.; Sjoden, P.O.; Jacobsson, G.; Sellstrom, H.; Enander, L.K.; Linne, T.; Svensson, C. Chemotherapy improves survival and quality of life in advanced pancreatic and biliary cancer. Ann. Oncol. 1996, 7, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Burris, H.A., 3rd; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Cripps, M.C.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P.; et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: A randomized trial. J. Clin. Oncol. 1997, 15, 2403–2413. [Google Scholar] [CrossRef]
- Eckel, F.; Schmid, R.M. Chemotherapy in advanced biliary tract carcinoma: A pooled analysis of clinical trials. Br. J. Cancer 2007, 96, 896–902. [Google Scholar] [CrossRef]
- Valle, J.; Wasan, H.; Palmer, D.H.; Cunningham, D.; Anthoney, A.; Maraveyas, A.; Madhusudan, S.; Iveson, T.; Hughes, S.; Pereira, S.P.; et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N. Engl. J. Med. 2010, 362, 1273–1281. [Google Scholar] [CrossRef]
- Wu, C.E.; Hsu, H.C.; Shen, W.C.; Lin, Y.C.; Wang, H.M.; Chang, J.W.; Chen, J.S. Chemotherapy with gemcitabine plus cisplatin in patients with advanced biliary tract carcinoma at Chang Gung Memorial Hospital: A retrospective analysis. Chang Gung Med. J. 2012, 35, 420–427. [Google Scholar] [PubMed]
- Ueno, M.; Morizane, C.; Okusaka, T.; Mizusawa, J.; Katayama, H.; Ikeda, M. Randomized phase III study of gemcitabine plus S-1 combination therapy versus gemcitabine plus cisplatin combination therapy in advanced biliary tract cancer: A Japan Clinical Oncology Group study (JCOG1113, FUGA-BT). J. Clin. Oncol. 2018, 36, 205. [Google Scholar] [CrossRef]
- Zhu, A.X.; Meyerhardt, J.A.; Blaszkowsky, L.S.; Kambadakone, A.; Muzikansky, A.; Zheng, H.; Clark, J.W.; Abrams, T.A.; Chan, J.A.; Enzinger, P.C.; et al. Efficacy and safety of gemcitabine, oxaliplatin, and bevacizumab in advanced biliary-tract cancers and correlation of changes in 18-fluorodeoxyglucose PET with clinical outcome: A phase 2 study. Lancet Oncol. 2010, 11, 48–54. [Google Scholar] [CrossRef]
- Rubovszky, G.; Lang, I.; Ganofszky, E.; Horvath, Z.; Juhos, E.; Nagy, T.; Szabo, E.; Szentirmay, Z.; Budai, B.; Hitre, E. Cetuximab, gemcitabine and capecitabine in patients with inoperable biliary tract cancer: A phase 2 study. Eur. J. Cancer 2013, 49, 3806–3812. [Google Scholar] [CrossRef] [PubMed]
- Malka, D.; Cervera, P.; Foulon, S.; Trarbach, T.; de la Fouchardiere, C.; Boucher, E.; Fartoux, L.; Faivre, S.; Blanc, J.F.; Viret, F.; et al. Gemcitabine and oxaliplatin with or without cetuximab in advanced biliary-tract cancer (BINGO): A randomised, open-label, non-comparative phase 2 trial. Lancet Oncol. 2014, 15, 819–828. [Google Scholar] [CrossRef]
- Chen, J.S.; Hsu, C.; Chiang, N.J.; Tsai, C.S.; Tsou, H.H.; Huang, S.F.; Bai, L.Y.; Chang, I.C.; Shiah, H.S.; Ho, C.L.; et al. A KRAS mutation status-stratified randomized phase II trial of gemcitabine and oxaliplatin alone or in combination with cetuximab in advanced biliary tract cancer. Ann. Oncol. 2015, 26, 943–949. [Google Scholar] [CrossRef] [Green Version]
- Eckel, F.; Schmid, R.M. Chemotherapy and targeted therapy in advanced biliary tract carcinoma: A pooled analysis of clinical trials. Chemotherapy 2014, 60, 13–23. [Google Scholar] [CrossRef]
- Nakamura, H.; Arai, Y.; Totoki, Y.; Shirota, T.; Elzawahry, A.; Kato, M.; Hama, N.; Hosoda, F.; Urushidate, T.; Ohashi, S.; et al. Genomic spectra of biliary tract cancer. Nat. Genet. 2015, 47, 1003–1010. [Google Scholar] [CrossRef]
- Bang, Y.J.; Doi, T.; De Braud, F.; Piha-Paul, S.; Hollebecque, A.; Razak, A.R.A.; Lin, C.C.; Ott, P.A.; He, A.R.; Yuan, S.S.; et al. Safety and efficacy of pembrolizumab (MK-3475) in patients (pts) with advanced biliary tract cancer: Interim results of KEYNOTE-028. Eur. J. Cancer 2015, 51, S112. [Google Scholar] [CrossRef]
- Goldstein, D.; Lemech, C.; Valle, J. New molecular and immunotherapeutic approaches in biliary cancer. ESMO Open 2017, 2, e000152. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanada, K.; Tsuchida, A.; Iwao, T.; Eguchi, N.; Sasaki, T.; Morinaka, K.; Matsubara, K.; Kawasaki, Y.; Yamamoto, S.; Kajiyama, G. Gene mutations of K-ras in gallbladder mucosae and gallbladder carcinoma with an anomalous junction of the pancreaticobiliary duct. Am. J. Gastroenterol. 1999, 94, 1638–1642. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.T.; Kim, J.; Jang, Y.H.; Lee, W.J.; Ryu, J.K.; Park, Y.K.; Kim, S.W.; Kim, W.H.; Yoon, Y.B.; Kim, C.Y. Genetic alterations in gallbladder adenoma, dysplasia and carcinoma. Cancer Lett. 2001, 169, 59–68. [Google Scholar] [CrossRef]
- Tannapfel, A.; Benicke, M.; Katalinic, A.; Uhlmann, D.; Kockerling, F.; Hauss, J.; Wittekind, C. Frequency of p16(INK4A) alterations and k-ras mutations in intrahepatic cholangiocarcinoma of the liver. Gut 2000, 47, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Tannapfel, A.; Sommerer, F.; Benicke, M.; Katalinic, A.; Uhlmann, D.; Witzigmann, H.; Hauss, J.; Wittekind, C. Mutations of the BRAF gene in cholangiocarcinoma but not in hepatocellular carcinoma. Gut 2003, 52, 706–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leone, F.; Cavalloni, G.; Pignochino, Y.; Sarotto, I.; Ferraris, R.; Piacibello, W.; Venesio, T.; Capussotti, L.; Risio, M.; Aglietta, M. Somatic mutations of epidermal growth factor receptor in bile duct and gallbladder carcinoma. Clin. Cancer Res. 2006, 12, 1680–1685. [Google Scholar] [CrossRef] [PubMed]
- Riener, M.O.; Bawohl, M.; Clavien, P.A.; Jochum, W. Rare PIK3CA hotspot mutations in carcinomas of the biliary tract. Genes Chromosomes Cancer 2008, 47, 363–367. [Google Scholar] [CrossRef]
- Argani, P.; Shaukat, A.; Kaushal, M.; Wilentz, R.E.; Su, G.H.; Sohn, T.A.; Yeo, G.J.; Cameron, J.L.; Kern, S.E.; Hruban, R.H. Differing rates of loss of Dpc4 expression and of p53 overexpression among carcinomas of the proximal and distal bile ducts—Evidence for biologic distinction. Cancer 2001, 91, 1332–1341. [Google Scholar] [CrossRef]
- Ueki, T.; Hsing, A.W.; Gao, Y.T.; Wang, B.S.; Shen, M.C.; Cheng, J.; Deng, J.; Fraumeni, J.F., Jr.; Rashid, A. Alterations of p16 and prognosis in biliary tract cancers from a population-based study in China. Clin. Cancer Res. 2004, 10, 1717–1725. [Google Scholar] [CrossRef]
- Hezel, A.F.; Deshpande, V.; Zhu, A.X. Genetics of biliary tract cancers and emerging targeted therapies. J. Clin. Oncol. 2010, 28, 3531–3540. [Google Scholar] [CrossRef]
- Ekshyyan, O.; Anandharaj, A.; Nathan, C.A.O. Dual PI3K/mTOR Inhibitors: Does p53 Modulate Response? Clin. Cancer Res. 2013, 19, 3719–3721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansel, D.E.; Rahman, A.; Hidalgo, M.; Thuluvath, P.J.; Lillemoe, K.D.; Schulick, R.; Ku, J.L.; Park, J.G.; Miyazaki, K.; Ashfaq, R.; et al. Identification of novel cellular targets in biliary tract cancers using global gene expression technology. Am. J. Pathol. 2003, 163, 217–229. [Google Scholar] [CrossRef]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR signaling in growth and metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Crino, P.B. The mTOR signalling cascade: Paving new roads to cure neurological disease. Nat. Rev. Neurol. 2016, 12, 379–392. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Liu, W.; Hu, X.; Dorrance, A.; Garzon, R.; Houghton, P.J.; Shen, C. Regulation of CHK1 by mTOR contributes to the evasion of DNA damage barrier of cancer cells. Sci. Rep. 2017, 7, 1535. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.Y.; Hong, S.M.; Choi, B.Y.; Cho, H.; Yu, E.; Hewitt, S.M. The expression of phospho-AKT, phospho-mTOR, and PTEN in extrahepatic cholangiocarcinoma. Clin. Cancer Res. 2009, 15, 660–667. [Google Scholar] [CrossRef]
- Ma, X.M.; Blenis, J. Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 2009, 10, 307–318. [Google Scholar] [CrossRef]
- Zhou, H.Y.; Huang, S.L. Current development of the second generation of mTOR inhibitors as anticancer agents. Chin. J. Cancer 2012, 31, 8–18. [Google Scholar] [CrossRef]
- Faivre, S.; Kroemer, G.; Raymond, E. Current development of mTOR inhibitors as anticancer agents. Nat. Rev. Drug Discov. 2006, 5, 671–688. [Google Scholar] [CrossRef]
- Mendoza, M.C.; Er, E.E.; Blenis, J. The Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef]
- Kim, L.C.; Cook, R.S.; Chen, J. mTORC1 and mTORC2 in cancer and the tumor microenvironment. Oncogene 2017, 36, 2191–2201. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Thoreen, C.C.; Peterson, T.R.; Lindquist, R.A.; Kang, S.A.; Spooner, E.; Carr, S.A.; Sabatini, D.M. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol. Cell 2007, 25, 903–915. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.F.; Harris, T.E.; Roth, R.A.; Lawrence, J.C. PRAS40 regulates mTORC1 kinase activity by functioning as a direct inhibitor of substrate binding. J. Biol. Chem. 2007, 282, 20036–20044. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Li, Y.; Xu, T.; Guan, K.L. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Gene Dev. 2003, 17, 1829–1834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.X.; Manning, B.D. A complex interplay between Akt, TSC2 and the two mTOR complexes. Biochem. Soc. T 2009, 37, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Schulick, R. Identification of Novel Cellular Targets in Biliary Tract Cancers Using Global Gene Expression Technology (vol 163, pg 217, 2003). Am. J. Pathol. 2017, 187, 936. [Google Scholar]
- Markman, B.; Dienstmann, R.; Tabernero, J. Targeting the PI3K/Akt/mTOR pathway--beyond rapalogs. Oncotarget 2010, 1, 530–543. [Google Scholar]
- Wang, X.M.; Proud, C.G. mTORC2 is a tyrosine kinase. Cell Res. 2016, 26, 1–2. [Google Scholar] [CrossRef]
- Yang, G.; Murashige, D.S.; Humphrey, S.J.; James, D.E. A Positive Feedback Loop between Akt and mTORC2 via SIN1 Phosphorylation. Cell Rep. 2015, 12, 937–943. [Google Scholar] [CrossRef] [Green Version]
- Sarbassov, D.D.; Ali, S.M.; Sengupta, S.; Sheen, J.H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 2006, 22, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Guri, Y.; Colombi, M.; Dazert, E.; Hindupur, S.K.; Roszik, J.; Moes, S.; Jenoe, P.; Heim, M.H.; Riezman, I.; Riezman, H.; et al. mTORC2 Promotes Tumorigenesis via Lipid Synthesis. Cancer Cell 2017, 32, 807–823. [Google Scholar] [CrossRef] [PubMed]
- Guertin, D.A.; Stevens, D.M.; Saitoh, M.; Kinkel, S.; Crosby, K.; Sheen, J.H.; Mullholland, D.J.; Magnuson, M.A.; Wu, H.; Sabatini, D.M. mTOR Complex 2 Is Required for the Development of Prostate Cancer Induced by Pten Loss in Mice. Cancer Cell 2009, 15, 148–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guri, Y.; Hall, M.N. mTOR Signaling Confers Resistance to Targeted Cancer Drugs. Trends Cancer 2016, 2, 688–697. [Google Scholar] [CrossRef]
- Liu, P.; Cheng, H.; Roberts, T.M.; Zhao, J.J. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Discov. 2009, 8, 627–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitman, M.; Downes, C.P.; Keeler, M.; Keller, T.; Cantley, L. Type I phosphatidylinositol kinase makes a novel inositol phospholipid, phosphatidylinositol-3-phosphate. Nature 1988, 332, 644–646. [Google Scholar] [CrossRef] [PubMed]
- Owonikoko, T.K.; Khuri, F.R. Targeting the PI3K/AKT/mTOR pathway: Biomarkers of success and tribulation. Am. Soc. Clin. Oncol. Educ. Book 2013. [Google Scholar] [CrossRef] [PubMed]
- Samuels, Y.; Waldman, T. Oncogenic mutations of PIK3CA in human cancers. Curr. Top. Microbiol. Immunol. 2010, 347, 21–41. [Google Scholar] [PubMed]
- Whale, A.D.; Colman, L.; Lensun, L.; Rogers, H.L.; Shuttleworth, S.J. Functional characterization of a novel somatic oncogenic mutation of PIK3CB. Signal Transduct. Target. Ther. 2017, 2, 17063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czech, M.P. PIP2 and PIP3: Complex roles at the cell surface. Cell 2000, 100, 603–606. [Google Scholar] [CrossRef]
- Cantley, L.C.; Neel, B.G. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase AKT pathway. Proc. Natl. Acad. Sci. USA 1999, 96, 4240–4245. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, A.; Pandolfi, P.P. The PTEN-PI3K pathway: Of feedbacks and cross-talks. Oncogene 2008, 27, 5527–5541. [Google Scholar] [CrossRef] [PubMed]
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, R.G.; Plas, D.R.; Kubek, S.; Buzzai, M.; Mu, J.; Xu, Y.; Birnbaum, M.J.; Thompson, C.B. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 2005, 18, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Hu, W.; de Stanchina, E.; Teresky, A.K.; Jin, S.; Lowe, S.; Levine, A.J. The regulation of AMPK beta1, TSC2, and PTEN expression by p53: Stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. 2007, 67, 3043–3053. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Chen, Z.; Erdjument-Bromage, H.; Tempst, P.; Pandolfi, P.P. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell 2005, 121, 179–193. [Google Scholar] [CrossRef] [PubMed]
- Lopez, T.; Hanahan, D. Elevated levels of IGF-1 receptor convey invasive and metastatic capability in a mouse model of pancreatic islet tumorigenesis. Cancer Cell 2002, 1, 339–353. [Google Scholar] [CrossRef] [Green Version]
- Roa, I.; Garcia, H.; Game, A.; de Toro, G.; de Aretxabala, X.; Javle, M. Somatic Mutations of PI3K in Early and Advanced Gallbladder Cancer Additional Options for an Orphan Cancer. J. Mol. Diagn. 2016, 18, 388–394. [Google Scholar] [CrossRef]
- Petzold, J.; Lederer, E.; Reihs, R.; Ernst, C.; Bettermann, K.; Halbwedl, I.; Lax, S.; Park, Y.N.; Kim, K.S.; Kiesslich, T.; et al. Pten inactivation and alteration of the pi3k/AKT/MTOR pathway in biliary tract cancer. Anticancer Res. 2014, 34, 5942–5943. [Google Scholar]
- Kornprat, P.; Rehak, P.; Ruschoff, J.; Langner, C. Expression of IGF-I, IGF-II, and IGF-IR in gallbladder carcinoma. A systematic analysis including primary and corresponding metastatic tumours. J. Clin. Pathol. 2006, 59, 202–206. [Google Scholar] [CrossRef] [Green Version]
- Wolf, S.; Lorenz, J.; Mossner, J.; Wiedmann, M. Treatment of biliary tract cancer with NVP-AEW541: Mechanisms of action and resistance. World J. Gastroenterol. 2010, 16, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Guertin, D.A.; Sabatini, D.M. The Pharmacology of mTOR Inhibition. Sci. Signal. 2009, 2, pe24. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Kim, S.G.; Blenis, J. Rapamycin: One Drug, Many Effects. Cell Metab. 2014, 19, 373–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilella-Bach, M.; Nuzzi, P.; Fang, Y.; Chen, J. The FKBP12-rapamycin-binding domain is required for FKBP12-rapamycin-associated protein kinase activity and G1 progression. J. Biol. Chem. 1999, 274, 4266–4272. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Chen, J.; Schreiber, S.L.; Clardy, J. Structure of the FKBP12-rapamycin complex interacting with the binding domain of human FRAP. Science 1996, 273, 239–242. [Google Scholar] [CrossRef]
- Choo, A.Y.; Yoon, S.O.; Kim, S.G.; Roux, P.P.; Blenis, J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc. Natl. Acad. Sci. USA 2008, 105, 17414–17419. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.A.; Pacold, M.E.; Cervantes, C.L.; Lim, D.; Lou, H.J.; Ottina, K.; Gray, N.S.; Turk, B.E.; Yaffe, M.B.; Sabatini, D.M. mTORC1 phosphorylation sites encode their sensitivity to starvation and rapamycin. Science 2013, 341, 1236566. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Mao, J.H.; Qian, L.; Zhu, H.; Gu, D.H.; Pan, X.D.; Yi, F.; Ji, D.M. Pre-clinical evaluation of AZD-2014, a novel mTORC1/2 dual inhibitor, against renal cell carcinoma. Cancer Lett. 2015, 357, 468–475. [Google Scholar] [CrossRef]
- Liu, Q.S.; Kirubakaran, S.; Hur, W.; Niepel, M.; Westover, K.; Thoreen, C.C.; Wang, J.H.; Ni, J.; Patricelli, M.P.; Vogel, K.; et al. Kinome-wide Selectivity Profiling of ATP-competitive Mammalian Target of Rapamycin (mTOR) Inhibitors and Characterization of Their Binding Kinetics. J. Biol. Chem. 2012, 287, 9742–9752. [Google Scholar] [CrossRef] [Green Version]
- Powles, T.; Wheater, M.; Din, O.; Geldart, T.; Boleti, E.; Stockdale, A.; Sundar, S.; Robinson, A.; Ahmed, I.; Wimalasingham, A.; et al. A Randomised Phase 2 Study of AZD2014 Versus Everolimus in Patients with VEGF-Refractory Metastatic Clear Cell Renal Cancer. Eur. Urol. 2016, 69, 450–456. [Google Scholar] [CrossRef]
- Petrossian, K.; Nguyen, D.; Lo, C.; Kanaya, N.; Somlo, G.; Cui, Y.X.; Huang, C.S.; Chen, S.A. Use of dual mTOR inhibitor MLN0128 against everolimus-resistant breast cancer. Breast Cancer Res. Treat. 2018, 170, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Vandamme, T.; Beyens, M.; de Beeck, K.O.; Dogan, F.; van Koetsveld, P.M.; Pauwels, P.; Mortier, G.; Vangestel, C.; de Herder, W.; Van Camp, G.; et al. Long-term acquired everolimus resistance in pancreatic neuroendocrine tumours can be overcome with novel PI3K-AKT-mTOR inhibitors. Br. J. Cancer 2016, 114, 650–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, P.; Ferreira, M.; Dubey, S.; Zaiss, M.; Harper-Wynne, C.; Makris, A.; Brown, V.; Kristeleit, H.; Patel, G.; Perello, A.; et al. MANTA: A randomized phase II study of fulvestrant in combination with the dual mTOR inhibitor AZD2014 or everolimus or fulvestrant alone in estrogen receptor-positive advanced or metastatic breast cancer. Cancer Res. 2018, 78. Abstract GS2-07. [Google Scholar] [CrossRef]
- Kelsey, I.; Manning, B.D. mTORC1 status dictates tumor response to targeted therapeutics. Sci. Signal. 2013, 6, pe31. [Google Scholar] [CrossRef] [PubMed]
- Ilagan, E.; Manning, B.D. Emerging role of mTOR in the response to cancer therapeutics. Trends Cancer 2016, 2, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.H.; Liu, L.Z. Role of mTOR in anticancer drug resistance: Perspectives for improved drug treatment. Drug Resist. Updates 2008, 11, 63–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokoi, K.; Kobayashi, A.; Motoyama, H.; Kitazawa, M.; Shimizu, A.; Notake, T.; Yokoyama, T.; Matsumura, T.; Takeoka, M.; Miyagawa, S.I. Survival pathway of cholangiocarcinoma via AKT/mTOR signaling to escape RAF/MEK/ERK pathway inhibition by sorafenib. Oncol. Rep. 2018, 39, 843–850. [Google Scholar] [CrossRef]
- Li, Q.; Xia, X.; Ji, J.; Ma, J.; Tao, L.; Mo, L.; Chen, W. MiR-199a-3p enhances cisplatin sensitivity of cholangiocarcinoma cells by inhibiting mTOR signaling pathway and expression of MDR1. Oncotarget 2017, 8, 33621–33630. [Google Scholar] [CrossRef] [Green Version]
- Ling, S.; Feng, T.; Ke, Q.; Fan, N.; Li, L.; Li, Z.; Dong, C.; Wang, C.; Xu, F.; Li, Y.; et al. Metformin inhibits proliferation and enhances chemosensitivity of intrahepatic cholangiocarcinoma cell lines. Oncol. Rep. 2014, 31, 2611–2618. [Google Scholar] [CrossRef] [Green Version]
- Wandee, J.; Prawan, A.; Senggunprai, L.; Kongpetch, S.; Tusskorn, O.; Kukongviriyapan, V. Metformin enhances cisplatin induced inhibition of cholangiocarcinoma cells via AMPK-mTOR pathway. Life Sci. 2018, 207, 172–183. [Google Scholar] [CrossRef]
- Ahn, C.S.; Han, J.A.; Lee, H.S.; Lee, S.; Pai, H.S. The PP2A regulatory subunit Tap46, a component of the TOR signaling pathway, modulates growth and metabolism in plants. Plant Cell 2011, 23, 185–209. [Google Scholar] [CrossRef] [PubMed]
- Lyu, S.C.; Han, D.D.; Li, X.L.; Ma, J.; Wu, Q.; Dong, H.M.; Bai, C.; He, Q. Fyn knockdown inhibits migration and invasion in cholangiocarcinoma through the activated AMPK/mTOR signaling pathway. Oncol. Lett. 2018, 15, 2085–2090. [Google Scholar] [CrossRef] [PubMed]
- Moolthiya, P.; Tohtong, R.; Keeratichamroen, S.; Leelawat, K. Role of mTOR inhibitor in cholangiocarcinoma cell progression. Oncol. Lett. 2014, 7, 854–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heits, N.; Heinze, T.; Bernsmeier, A.; Kerber, J.; Hauser, C.; Becker, T.; Kalthoff, H.; Egberts, J.H.; Braun, F. Influence of mTOR-inhibitors and mycophenolic acid on human cholangiocellular carcinoma and cancer associated fibroblasts. BMC Cancer 2016, 16, 322. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.F.; Zhang, C.Y.; Zhou, H.; Xiao, B.; Cheng, Y.; Wang, J.J.; Yao, F.L.; Duan, C.Y.; Chen, R.; Liu, Y.P.; et al. Synergistic antitumor activity of the combination of salubrinal and rapamycin against human cholangiocarcinoma cells. Oncotarget 2016, 7, 85492–85501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, G.G.; Lin, K.J.; Wang, F.; Chen, T.C.; Yen, T.C.; Yeh, T.S. Synergistic antiproliferative effects of an mTOR inhibitor (rad001) plus gemcitabine on cholangiocarcinoma by decreasing choline kinase activity. Dis. Models Mech. 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.S.; Song, X.H.; Cao, D.; Xu, Z.; Fan, B.A.; Che, L.; Hu, J.J.; Chen, B.; Dong, M.J.; Pilo, M.G.; et al. Pan-mTOR inhibitor MLN0128 is effective against intrahepatic cholangiocarcinoma in mice. J. Hepatol. 2017, 67, 1194–1203. [Google Scholar] [CrossRef]
- Song, X.; Liu, X.; Wang, H.; Wang, J.; Qiao, Y.; Cigliano, A.; Utpatel, K.; Ribback, S.; Pilo, M.G.; Serra, M.; et al. Combined CDK4/6 and Pan-mTOR Inhibition Is Synergistic Against Intrahepatic Cholangiocarcinoma. Clin. Cancer Res. 2018. [Google Scholar] [CrossRef]
- Fraveto, A.; Cardinale, V.; Bragazzi, M.C.; Giuliante, F.; De Rose, A.M.; Grazi, G.L.; Napoletano, C.; Semeraro, R.; Lustri, A.M.; Costantini, D.; et al. Sensitivity of Human Intrahepatic Cholangiocarcinoma Subtypes to Chemotherapeutics and Molecular Targeted Agents: A Study on Primary Cell Cultures. PLoS ONE 2015, 10, e0142124. [Google Scholar] [CrossRef]
- Chen, M.H.; Chiang, K.C.; Cheng, C.T.; Huang, S.C.; Chen, Y.Y.; Chen, T.W.; Yeh, T.S.; Jan, Y.Y.; Wang, H.M.; Weng, J.J.; et al. Antitumor activity of the combination of an HSP90 inhibitor and a PI3K/mTOR dual inhibitor against cholangiocarcinoma. Oncotarget 2014, 5, 2372–2389. [Google Scholar] [CrossRef] [Green Version]
- Peng, H.; Zhang, Q.Y.; Li, J.L.; Zhang, N.; Hua, Y.P.; Xu, L.X.; Deng, Y.B.; Lai, J.M.; Peng, Z.W.; Peng, B.G.; et al. Apatinib inhibits VEGF signaling and promotes apoptosis in intrahepatic cholangiocarcinoma. Oncotarget 2016, 7, 17220–17229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.E.; Koay, T.S.; Esfandiari, A.; Ho, Y.H.; Lovat, P.; Lunec, J. ATM Dependent DUSP6 Modulation of p53 Involved in Synergistic Targeting of MAPK and p53 Pathways with Trametinib and MDM2 Inhibitors in Cutaneous Melanoma. Cancers 2018, 11, 3. [Google Scholar] [CrossRef]
- Andersen, N.J.; Boguslawski, E.B.; Kuk, C.Y.; Chambers, C.M.; Duesbery, N.S. Combined inhibition of MEK and mTOR has a synergic effect on angiosarcoma tumorgrafts. Int. J. Oncol. 2015, 47, 71–80. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.E.; Esfandiari, A.; Ho, Y.H.; Wang, N.; Mahdi, A.K.; Aptullahoglu, E.; Lovat, P.; Lunec, J. Targeting negative regulation of p53 by MDM2 and WIP1 as a therapeutic strategy in cutaneous melanoma. Br. J. Cancer 2018, 118, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Laroche, A.; Chaire, V.; Algeo, M.P.; Karanian, M.; Fourneaux, B.; Italiano, A. MDM2 antagonists synergize with PI3K/mTOR inhibition in well-differentiated/dedifferentiated liposarcomas. Oncotarget 2017, 8, 53968–53977. [Google Scholar] [CrossRef] [PubMed]
- Bian, J.L.; Wang, M.M.; Tong, E.J.; Sun, J.; Li, M.; Miao, Z.B.; Li, Y.L.; Zhu, B.H.; Xu, J.J. Benefit of everolimus in treatment of an intrahepatic cholangiocarcinoma patient with a PIK3CA mutation. World J. Gastroenterol. 2017, 23, 4311–4316. [Google Scholar] [CrossRef] [PubMed]
- Verzoni, E.; Pusceddu, S.; Buzzoni, R.; Garanzini, E.; Damato, A.; Biondani, P.; Testa, I.; Grassi, P.; Bajetta, E.; DeBraud, F.; et al. Safety profile and treatment response of everolimus in different solid tumors: An observational study. Future Oncol. 2014, 10, 1611–1617. [Google Scholar] [CrossRef] [PubMed]
- Buzzoni, R.; Pusceddu, S.; Bajetta, E.; De Braud, F.; Platania, M.; Iannacone, C.; Cantore, M.; Mambrini, A.; Bertolini, A.; Alabiso, O.; et al. Activity and safety of RAD001 (everolimus) in patients affected by biliary tract cancer progressing after prior chemotherapy: A phase II ITMO study. Ann. Oncol. 2014, 25, 1597–1603. [Google Scholar] [CrossRef]
- Kim, S.T.; Lee, J.; Park, S.H.; Park, J.O.; Park, Y.S.; Kang, W.K.; Lim, H.Y. Prospective phase II trial of everolimus in PIK3CA amplification/mutation and/or PTEN loss patients with advanced solid tumors refractory to standard therapy. BMC Cancer 2017, 17, 211. [Google Scholar] [CrossRef]
- Lau, D.K.; Tay, R.Y.; Yeung, Y.H.; Chionh, F.; Mooi, J.; Murone, C.; Skrinos, E.; Price, T.J.; Mariadason, J.M.; Tebbutt, N.C. Phase II study of everolimus (RAD001) monotherapy as first-line treatment in advanced biliary tract cancer with biomarker exploration: The RADiChol Study. Br. J. Cancer 2018, 118, 966–971. [Google Scholar] [CrossRef]
- Rizell, M.; Andersson, M.; Cahlin, C.; Hafstrom, L.; Olausson, M.; Lindner, P. Effects of the mTOR inhibitor sirolimus in patients with hepatocellular and cholangiocellular cancer. Int. J. Clin. Oncol. 2008, 13, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.S.; Lee, J.; Park, S.H.; Park, J.O.; Park, Y.S.; Lim, H.Y.; Kang, W.K.; Kim, S.T. Pilot study of sirolimus in patients with PIK3CA mutant/amplified refractory solid cancer. Mol. Clin. Oncol. 2017, 7, 27–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costello, B.A.; Borad, M.J.; Qi, Y.; Kim, G.P.; Northfelt, D.W.; Erlichman, C.; Alberts, S.R. Phase I trial of everolimus, gemcitabine and cisplatin in patients with solid tumors. Investig. New Drugs 2014, 32, 710–716. [Google Scholar] [CrossRef] [PubMed]
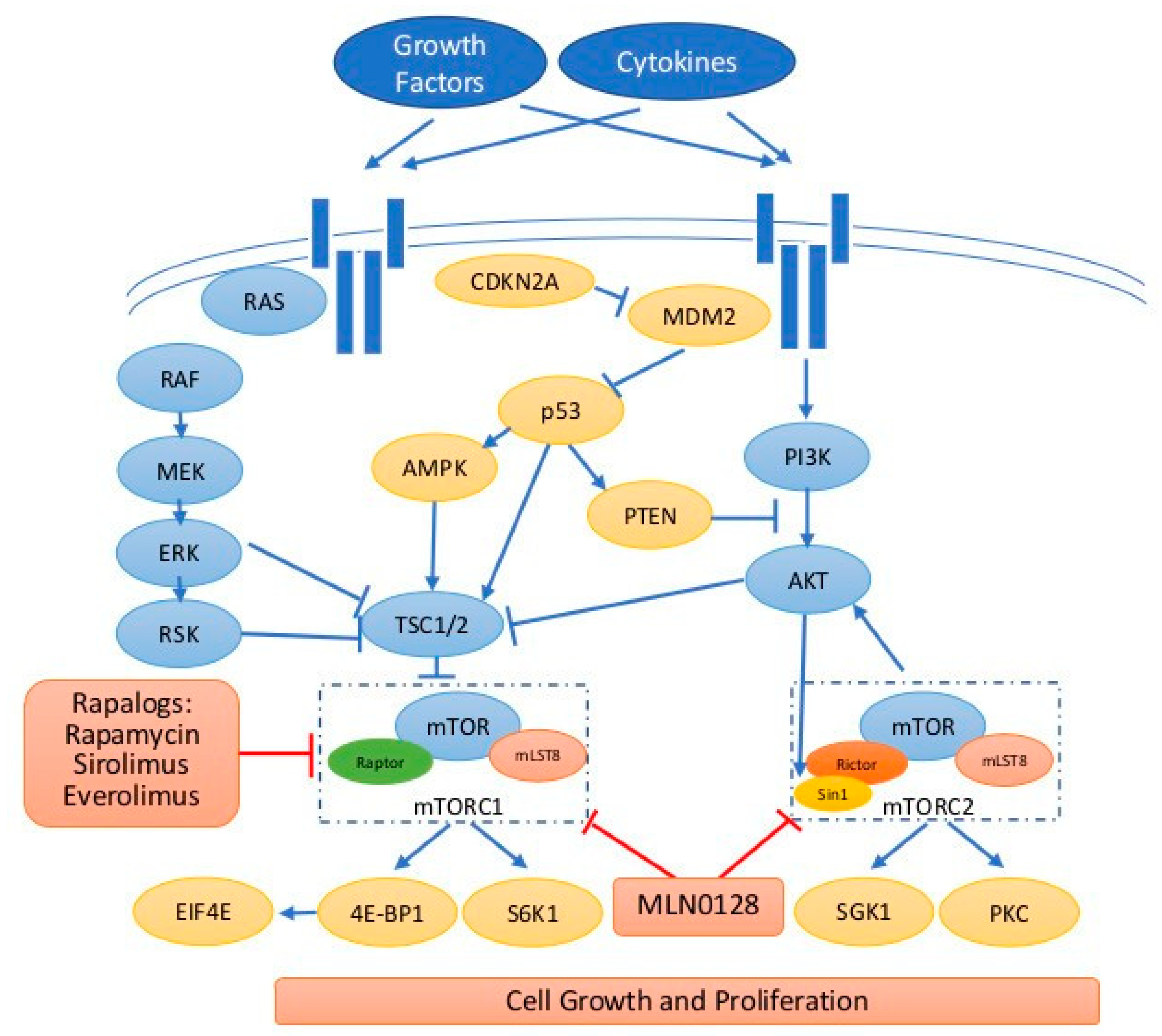
Figure 1.
The signaling transduction of mTOR pathway. Extracellular signals such as growth factors and cytokines binding to the receptors stimulate RAS/RAF/MEK/ERK and PI3K/AKT/mTOR caspases. mTOR exists in two functionally and structurally distinct complexes, mTORC1 and mTORC2. Both mTORC1/2 contain different core components so they phosphorylate a distinct set of substrates and exhibit distinct function. PTEN is a negative regulator for PI3K/AKT. In addition, ERK/RSK, AMPK, and p53 regulate mTORC1 through TSC2 regulation. Rapalogs mainly inhibit mTORC1 and new-generation mTOR inhibitors such as MLN0128 inhibitor both mTORC1/2. Blue t-bar indicates inhibition, blue arrow indicates stimulation/activation, red t-bar indicates inhibition by drugs, and dashed square indicates mTOR1/2 complexes.
Figure 1.
The signaling transduction of mTOR pathway. Extracellular signals such as growth factors and cytokines binding to the receptors stimulate RAS/RAF/MEK/ERK and PI3K/AKT/mTOR caspases. mTOR exists in two functionally and structurally distinct complexes, mTORC1 and mTORC2. Both mTORC1/2 contain different core components so they phosphorylate a distinct set of substrates and exhibit distinct function. PTEN is a negative regulator for PI3K/AKT. In addition, ERK/RSK, AMPK, and p53 regulate mTORC1 through TSC2 regulation. Rapalogs mainly inhibit mTORC1 and new-generation mTOR inhibitors such as MLN0128 inhibitor both mTORC1/2. Blue t-bar indicates inhibition, blue arrow indicates stimulation/activation, red t-bar indicates inhibition by drugs, and dashed square indicates mTOR1/2 complexes.


{kind=link}
Table 1.
Summary of published data regarding mTOR inhibitors in advanced BTC.
| Compound(s) | Phase | Patients | Response | Survival |
|---|---|---|---|---|
| Everolimus, 1 L [106] | Case report | iCCA (n = 1) with PIK3CA mutation | PR | PFS > 6 m |
| Everolimus [107] | Phase I | Advanced BTC (n = 22) | DCR: 50% (11/22) | NA |
| Everolimus (>2 L) [108] | Phase II | Advanced BTC (n = 39) | DCR: 44.7% RR: 5.1% (including 1 CR) | mPFS: 3.2 m (1.8–4.0) mOS: 7.7 m (5.5–13.2) |
| Everolimus [109] | Phase II | CCA (n = 1), PTEN loss | SD | NA |
| Everolimus (1 L) [110] | Phase II | Advanced BTC (n = 27) | DCR at 12 weeks: 48% PR: 12% (3/25) SD: 60% (15/25) | mPFS: 5.5 m (2.2–10.0) mOS: 9.5 m (5.5–16.6) |
| Sirolimus [111] | Phase II | iCCA (n = 9) | SD: 33% (3/9) PD: 67% (6/9) | mOS:7 (2.6–35) |
| Sirolimus [112] | Phase II | hilar CCA (n = 1) with PIK3CA mutation | PD | PFS: 0.9 m |
| Everolimus, gemcitabine, cisplatin (1 L) [113] | Phase I | Cohort III, CCA and GBC (n = 10) | SD: 60% (6/10) PD: 40% (4/10) | NA |
1 L, first line; 2 L, second line; iCCA, intrahepatic cholangiocarcinoma; GBC, gallbladder cancer; BTC, biliary tract cancer; CR, complete response; PR, partial response; SD, stable disease; PD, progressive disease; DCR, disease control rate; mPFS, median progression-free survival; mOS, median overall survival.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Wu, C.-E.; Chen, M.-H.; Yeh, C.-N. mTOR Inhibitors in Advanced Biliary Tract Cancers. Int. J. Mol. Sci. 2019, 20, 500. https://doi.org/10.3390/ijms20030500
AMA Style
Wu C-E, Chen M-H, Yeh C-N. mTOR Inhibitors in Advanced Biliary Tract Cancers. International Journal of Molecular Sciences. 2019; 20(3):500. https://doi.org/10.3390/ijms20030500
Chicago/Turabian StyleWu, Chao-En, Ming-Huang Chen, and Chun-Nan Yeh. 2019. "mTOR Inhibitors in Advanced Biliary Tract Cancers" International Journal of Molecular Sciences 20, no. 3: 500. https://doi.org/10.3390/ijms20030500
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.