Role of Mitochondria-Cytoskeleton Interactions in the Regulation of Mitochondrial Structure and Function in Cancer Stem Cells

1
Brain Korea 21 PLUS Project for Medical Science, Yonsei University College of Medicine, Seoul 03722, Korea
2
Department of Surgery, Yonsei University Health System, Yonsei University College of Medicine, 50 Yonsei-ro, Seodaemun-gu, Seoul 03722, Korea
3
Yonsei Biomedical Research Institute, Yonsei University College of Medicine, Seoul 03722, Korea
4
Department of Biochemistry & Molecular Biology, Yonsei University College of Medicine, Seoul 03722, Korea
5
Department of Biomedical Systems Informatics, Yonsei University College of Medicine, Seoul 03722, Korea
*
Author to whom correspondence should be addressed.
Cells 2020, 9(7), 1691; https://doi.org/10.3390/cells9071691
Submission received: 17 June 2020
/
Revised: 9 July 2020
/
Accepted: 11 July 2020
/
Published: 14 July 2020
(This article belongs to the Special Issue New Aspects of Targeting Cancer Metabolism in Therapeutic Approach)
Abstract
:Despite the promise of cancer medicine, major challenges currently confronting the treatment of cancer patients include chemoresistance and recurrence. The existence of subpopulations of cancer cells, known as cancer stem cells (CSCs), contributes to the failure of cancer therapies and is associated with poor clinical outcomes. Of note, one of the recently characterized features of CSCs is augmented mitochondrial function. The cytoskeleton network is essential in regulating mitochondrial morphology and rearrangement, which are inextricably linked to its functions, such as oxidative phosphorylation (OXPHOS). The interaction between the cytoskeleton and mitochondria can enable CSCs to adapt to challenging conditions, such as a lack of energy sources, and to maintain their stemness. Cytoskeleton-mediated mitochondrial trafficking and relocating to the high energy requirement region are crucial steps in epithelial-to-mesenchymal transition (EMT). In addition, the cytoskeleton itself interplays with and blocks the voltage-dependent anion channel (VDAC) to directly regulate bioenergetics. In this review, we describe the regulation of cellular bioenergetics in CSCs, focusing on the cytoskeleton-mediated dynamic control of mitochondrial structure and function.
1. Introduction
Mitochondria orchestrate diverse fundamental cellular functions, including respiration, calcium homeostasis, reactive oxygen species generation, and programmed cell death [1,2,3,4]. They have been implicated in multiple steps of tumor progression and maintenance of cancer stemness, and heterogeneity in the morphology and spatial distribution of these organelles in cancer is becoming a field of intense investigation [3,5]. The relationship between cancer stem cells (CSCs) and mitochondria has been recently demonstrated to be an important target for cancer treatment [6,7,8,9]. Determining the mitochondrial regulatory mechanisms in order to understand CSC bioenergetics and the role of mitochondria is important in studies of cancer biology. Components of the cytoskeleton play a vital role in the structural and functional organization of the mitochondria, including mitochondrial morphology, dynamics, motility, and intracellular arrangement [5,10]. Additionally, the interaction of mitochondria with cytoskeletal components and their interactions with voltage-dependent anion channels (VDACs) are involved in the coordination of mitochondrial function [11,12,13,14,15]. Moreover, alterations in the cytoskeletal architecture occur to meet the need for cellular energy during proliferation, apoptosis, and adaptation to challenging conditions, such as the lack of an energy source, extrinsic and intrinsic stresses, and mechanical stress [16]. Modulation of mitochondrial dynamics and cytoskeletal features can profoundly impact CSC behavior. Significant efforts in recent years have revealed that mitochondrial function is critical for CSC survival [6,8,9,17]. Although mitochondrial function has now been implicated in multiple CSCs, the underlying mechanisms regulating the mitochondrial architecture and bioenergetics are not well understood. Numerous reports have demonstrated the relationship between the cytoskeleton and CSCs in the context of epithelial-mesenchymal transition (EMT) and metastasis because of the essential role of the cytoskeleton in the maintenance and regulation of cellular architecture [18,19]. Yet, the relationship between the cytoskeleton and CSCs focused on mitochondrial functional augmentation, which can provide CSCs with a survival advantage is unclear. In this review, we summarize previous studies providing evidence for the role of cytoskeletal components in regulating the mitochondrial structure, function, and bioenergetics in a CSC model system. An in-depth understanding of CSC behavior can provide insights into cancer biology that may improve clinical applications aimed at targeting the bioenergetics of CSCs for treating drug-resistant malignant tumors.
2. Unique Features of CSCs
CSCs are more resistant to chemotherapeutic agents compared to bulk tumor cells, and therefore survive conventional therapies, resulting in relapse of the disease. From a bioenergetics perspective, CSCs exhibit mitochondria-centric metabolism and have the capability to adapt to harsh conditions like lack of energy sources, and can thus survive and maintain their stemness features.
2.1. Identification of CSCs
In recent years, CSCs have gained interest as key tumor-initiating cells that may also play an integral role in tumor recurrence and chemoresistance. Identification and isolation of these CSCs using putative surface markers has been a priority in cancer research. The main markers used for isolation and identification of CSCs include surface cell-adhesion molecules (e.g., CD44, CD133, and EpCAM) and cytoprotective enzymes (such as aldehyde dehydrogenase, ALDH). However, the surface markers that are used to isolate or identify CSCs from diverse malignant tissues are expressed not only by CSCs, but also by many normal tissues [20,21,22,23,24]. Fox et al. revealed that normal human tissues express different CD44 isoforms; for instance, normal epithelial tissues and some tumors express a wide range of variants at high levels. The association of putative markers of CSCs such as CD44 and CD133 with drug resistance has been largely based on studies demonstrating reduced sensitivity of CSC marker-positive cells to chemotherapeutic drugs or increased expression of these markers in treatment-resistant tumors [22,25,26]. Elevated ALDH1A1 is observed in CSCs of multiple cancer types [27,28,29,30,31]. Thus, the use of surface marker expression alone is insufficient to identify CSCs. Evaluation of surface markers must be associated with other functional assays, such as sphere-forming assays in a low attachment surface, and measurement of the expression of specific CSC-related genes to provide persuasive evidence for the presence of CSCs. In recent years, mitochondrial and energy metabolism-related indicators of CSCs have been studied by an increasing number of researchers. According to these studies, CSCs are less glycolytic, produce less reactive oxygen species (ROS), and maintain higher ATP levels than bulk tumors. Moreover, the mitochondria in CSCs have increased mass and membrane potential, which is a reflection of augmented mitochondrial function, and enhanced oxygen consumption rates compared with bulk tumors, which generate their energy mainly via glycolysis [32,33,34,35,36,37,38]. Increased mitochondrial mass indicates stem-like phenotypes and is associated with metastatic potential and insensitivity to DNA damage [39]. Taken together, mitochondrial features might be regarded as a useful tool and a novel strategy to identify CSCs, together with the classic CSC markers.
2.2. Cellular Bioenergetics in CSCs
The metabolic characteristics of CSCs have been the subject of intense investigation over the past years. Due to the abnormally controlled proliferation and inadequate blood vessel formation as the tumor progresses, tumor microenvironments are characterized by hypoxic, acidic, and nutrient-poor conditions; therefore, cancer cells must adapt their cellular bioenergetics efficiently to deal with these unfavorable conditions, a process called metabolic reprogramming [40]. Metabolic reprogramming is crucial to sustain proliferation and survival of tumor cells when the oncogenic cue is blocked [6,41]. From a bioenergetics perspective, cancer cells exhibit an aerobic glycolytic metabolism known as the Warburg effect [42], whereas CSCs display distinct metabolic features, known as oxidative phosphorylation (OXPHOS). Metabolic plasticity may allow cancer cell subpopulations to switch between glycolysis and OXPHOS to support their bioenergetic needs and ultimately to survive during and after anticancer treatments [43,44,45,46]. In agreement with the concept of metabolic plasticity of cancer cells, JARID1Bhigh cells [8] revealed a prominent increase in oxygen consumption, resulting in upregulation and downregulation of genes involved in the electron transport chain (ETC) and glycolysis, respectively [1]. In surviving cells after KRAS inactivation, transcriptome analysis revealed that expression of genes involved in mitochondrial function, such as ETC and β-oxidation, is increased, and mitochondria have a larger size and consume more oxygen than bulk pancreatic tumor cells [6]. Furthermore, MYC- and MCL1-positive cells show increased OXPHOS with higher bioenergetic reliance on fatty acid oxidation in triple negative breast cancer (TNBC) [17]. Elevated expression of PGC1-α in melanoma is associated with increased dependency on OXPHOS and decreased glycolytic features [47]. Several studies in diverse cancer types, such as ROSlow quiescent leukemia stem cells, CD133+ cells of glioblastoma, and pancreatic ductal adenocarcinoma, show that they exhibit a preference for OXPHOS rather than glycolysis [33,36,48,49]. OXPHOS is also upregulated as a result of chemotherapy [50]. Increased OXPHOS may support drug resistance by producing sufficient ATP for the removal of drugs via multidrug transporters in a colon cancer model [51]. Oxaliplatin and fluorouracil (5-FU) treatment of colon cancer cells increase OXPHOS through sirtuin 1 (SIRT1) and PGC1-α induction [52]. Furthermore, downregulation of OXPHOS via SIRT1/PGC1-α knockdown resensitizes cancer cells, suggesting that OXPHOS inhibition may represent an attractive addition to adjuvant chemotherapy. Despite mitochondrial function being high in these studies, ROS levels are significantly lower in CSCs [53], which also explains their more powerful antioxidant defense system compared with bulk tumors. A strong quenching process keeps ROS levels low, and helps in the maintenance of the stemness of CSCs, thereby contributing to therapy resistance [54]. According to these observations, understanding the metabolic characteristics of CSCs would be very helpful in designing therapeutic approaches that selectively target CSCs. Furthermore, an effort to investigate the regulatory mechanisms that control cellular bioenergetics of CSCs should provide new therapeutic insights.
3. Mitochondrial Structure and Function
Cellular bioenergetics is the foundation of survival for all organisms, including cancer cells. Mitochondria allow cells to sense cellular stress and to adapt to challenging extrinsic or intrinsic stimuli, conferring a high degree of plasticity to tumor cells for growth and survival.
3.1. Mitochondrial Respiratory Chain
Electrons derived from different metabolic processes are involved in a series of transmembrane protein complexes in the mitochondrial inner membrane (known as the ETC) to fuel the OXPHOS process. Nicotinamide adenine dinucleotide (NADH), flavin adenine dinucleotide (FADH2), and succinate act as electron donors. The ETC is responsible for the transfer of electrons from reduced NADH and FADH2, produced in the tricarboxylic acid (TCA) cycle, to oxygen across complexes I, II, III, and IV. This electron flow provides energy for proton translocation, known as the proton motive force [55,56], thereby generating ATP. Oxygen acts as the terminal electron acceptor (Figure 1). The malate-aspartate shuttle (MAS) is indispensable for the transfer of cytosolic NADH into mitochondria to maintain a high rate of glycolysis and to support tumor cell growth. Moreover, the malate-aspartate shuttle has been reported to oxidize 20–80% of cytosolic NADH in diverse tumor cells [57,58]. The glycerol-3-phosphate shuttle (G3P-DHAP), composed of two distinct enzyme components: (1) NAD+/NADH-linked glycerol 3-phosphate dehydrogenase, which is cytosolic, and (2) flavin adenine dinucleotide (FAD/FADH2)-linked glycerol 3-phosphate dehydrogenase, which is localized to the mitochondrial inner membrane. Unlike the MAS, which transports electrons from cytosolic to mitochondrial NADH, the G3P-DHAP shuttle transfers the electrons directly to ubiquinones that enter the electron transport complex III. Using an intact human platelet study, cell-permeable succinate prodrug (NV118) increased mitochondrial oxygen consumption with energy production in cells intoxicated with complex I inhibitor, metformin [59]. This concept was interpreted as a “bypass” theory. NV118 entered platelets and released succinate; succinate directly donated electrons to complex II and thus reactivated the electron flow to the distal part of the respiratory chain independent of complex I. This result highlights the potential therapeutic application of succinate in conditions accompanied by complex I inhibition. Fatty acids have a higher energy density than glucose, and CSCs attempt to maximize fatty acid utilization under glucose starvation conditions. Several studies that target the mitochondrial complex I might be insensitive in certain cancer cells, which indicates a high degree of redundancy in the survival mechanism of CSCs.
3.2. Mitochondrial Dynamics
Mitochondria continually fuse and divide, and their quality, distribution, size, and motility are finely tuned [60,61]. Mitochondrial fission and fusion are known to play roles in maintaining the integrity of mitochondria, electrical, and biochemical connectivity, and protection of mitochondrial DNA (mtDNA) [62,63,64]. Mitochondrial dynamics are regulated by highly conserved large guanosine triphosphatases (GTPases). Fusion of the outer membrane of mitochondria (OMM) is mediated by mitofusin 1 and mitofusin 2 (Mfn1 and Mfn2) [65,66,67], whereas the inner membrane of mitochondria (IMM) is regulated by optic atrophy 1 (OPA1) [68]. Mitochondrial fission is regulated by dynamin-related protein 1 (DRP1) [69] and mitochondrial fission factor (Mff) [70]. An imbalance between mitochondrial fission and fusion leads to dysfunctional mitochondria and is a key feature of diverse disorders, including cancer [71].
Mitochondrial fusion and fission form an essential axis of mitochondrial quality control. However, quality control may not be the only task carried out by mitochondrial dynamics. Depending on the physiological conditions, the mitochondrial fission–fusion balance may be perturbed and mitochondria may appear fragmented, such as during apoptosis and cell division [2], or elongated, such as during nutrient starvation [32,72] (Figure 2). Mitochondrial fission is increased during the G2/M phase of the cell cycle to guarantee an accurate mitochondrial segregation between the two daughter cells during cell division [73], and mitochondrial fusion prevents the removal of mitochondria by starvation-induced autophagy. Mitochondrial fission is crucially involved in maintenance of healthy mitochondria populations, including mitochondrial inheritance by daughter cells during cellular division. Mitochondrial fragmentation occurs in response to various factors such as intracellular calcium levels and ATP availability [74,75]. For example, the calcium-sensitive phosphatase calcineurin promotes fission by dephosphorylating cytosolic Drp1, which causes translocation to mitochondria [76]. Plasticity bestows the adaptive flexibility needed to adjust and adapt to changing cellular stresses and metabolic demands. In respect to the plasticity of mitochondrial dynamics, we have suggested that constant network-like remodeling establishes a mechanism for quality control of the mitochondrial population with important ramifications for the gain fitness of CSCs. Mitochondrial elongation is necessary to maintain structural and functional homogeneity, to preserve mitochondrial genome integrity, and to ensure the proper balance between energy generation and cellular mass [77,78,79,80,81,82]. Mitochondrial fusion causes an increase in the mitochondrial cristae number, which is associated with dimerization of the ATP synthase and thereby upregulates ATP synthesis activity [72]. Furthermore, this mitochondrial inner membrane rearrangement is involved in cristae remodeling and protection from cytochrome c release [83,84], suggesting that mitochondrial fusion is important for the maintenance of healthy mitochondria. Recent reports demonstrated that mitochondrial fusion during nutrient starvation facilitates fatty acid trafficking and oxidation for survival [85,86]. Upon starvation, cells remodel mitochondria into highly connected networks and also upregulate enzymes required for mitochondrial fatty acid trafficking, import and β-oxidation [56]. Taken together, these results show that mitochondrial morphological dynamics and integrity contribute to changes in mitochondrial function, such as utilization of specific energy sources during nutrient starvation.
3.3. Mitochondrial Structure-Function in CSCs
Although the underlying mechanisms regulating mitochondrial dynamics in CSCs remain unknown, a number of reports have demonstrated that mitochondrial structure is directly related to mitochondrial function. The importance of mitochondrial morphology and function in CSCs is emphasized by the findings of Viale et al. [6]. The authors have shown that oncogene ablation-resistant, surviving cells in pancreatic cancer exhibit elevated mitochondrial bioenergetic capacity along with highly elongated mitochondria. In a similar manner, neuroblastoma cells with increased resistance to apoptosis upon cisplatin treatment show high expression levels of MFN1 following hyperfused mitochondria and increased mitochondrial respiratory capacity [67]. Studies using stress-resistant pancreatic cancer cells demonstrated that suppression of glycolytic capacity dramatically changed mitochondrial morphology; in particular, mitochondrial fusion was more abundant in cells cultured in low-glucose medium [87]. Recent studies have found that mitochondrial fusion and enhanced oxygen consumption capacity are crucial for regulating and maintaining stemness. For example, MIM fusion mediated by OPA1 is critical for regulating tight cristae junctions and the proximity of ETC complexes to each other in memory T cells compared to effector T cells [88].
Although the underlying mechanisms regulating mitochondrial dynamics in CSCs remain unknown, numerous studies have revealed that hyper-activated oncogenic pathways act as potent signals to remodel the mitochondrial structure and metabolism. As specific mitochondrial morphologies are linked to different energetic states of the cells, oncogenic-signaling-mediated mitochondrial structure may induce changes in mitochondrial function to support altered metabolism. For example, KRAS-ablated surviving cells in pancreatic ductal adenocarcinoma (PDAC) displayed fused mitochondria and relied on OXPHOS [6], whereas B-RAFV600E-driven melanoma cells exhibited a fragmented mitochondrial network with upregulated DRP1 expression and increased glycolytic metabolism [89]. Although tumorigenesis can be initiated through oncogenic MAPK or MYC signaling, these pathways result in different mitochondrial phenotypes. Oncogenic MAPK promotes mitochondrial fission, whereas MYC induces mitochondrial fusion. Oncogenic MYC induces the expression of genes involved in cellular metabolism, including diverse genes that regulate mitochondrial mass, and biogenesis results in an increase of metabolic capacity of cancer cells [90]. Knockout of MYC in mouse embryonic fibroblasts (MEFs) exhibited fragmented mitochondria, while re-expression of MYC induces fusion by elevation of OPA1 and MFN2 [91]. In contrast, elevated MAPK signaling amplifies downstream kinase cascades that culminate in increased ERK pathway, immediate activation of DRP1, inactivation of MFN1, and mitochondrial fission [89,92]. In T-cell acute lymphoblastic leukemia cells with acquired drug resistance, ERK activation-induced DRP1 phosphorylation was found to result in pro-glycolytic phenotype switching and mitochondrial fragmentation [93]. Downregulation of mitochondrial fusion proteins reduces OXPHOS and ATP production in diverse cancer models [79,94,95]. In a more complex study, Vaseva et al. found that mutant KRAS regulates MYC via ERK1/2-dependent and independent mechanisms in pancreatic cancer. ERK1/2 inhibition activates a compensatory EGFR-SRC-ERK5 cascade that stabilizes MYC, and combined ERK1/2 and ERK5 inhibition promotes synergistic loss of MYC and suppresses PDAC growth [96]. Studies are needed to test these mechanisms in various model systems to fully understand the role of the oncogenic signaling network in the regulation of mitochondrial structure and function in CSCs. These opposing effects mediated by oncogene activation may be modulated by different capacities of utilization of nutrients from the nutrient-poor tumor microenvironment. As we mentioned above, cancer cells generally rely on glycolysis as the major energy source, whereas CSCs exhibit mitochondrial-centric metabolism, i.e., OXPHOS [97]. Recent reports have suggested that ERK1/2 signaling is necessary to induce glycolysis. The authors’ proposed mechanism for ERK regulation of glycolysis involves the phosphorylation of phosphoglycerate kinase 1 (PGK-1) by ERK1/2 on Ser 203. This phosphorylation induces mitochondrial PGK-1 to act as a protein kinase that phosphorylates and activates PDH kinase 1 (PDHK1) on Thr338. PDHK1, in turn, phosphorylates PDH, inhibiting enzyme activity, and induces glycolysis [98]. In contrast, ATP levels in MYC-positive cells are supported by OXPHOS to a greater extent than in MYC-negative cells [99]. MYC also promotes utilization of TCA cycle intermediates, including fatty acids from acetyl-CoA and NADPH for malic enzymes [100]. In addition, MYC-overexpressing TNBC exhibited elevated fatty acid oxidation and its inhibition suppressed tumorigenesis in vivo [101].
Greer et al. showed that the small-molecule drug ONC201 [102], originally shown to induce the transcription of tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) and to destroy breast cancer cells by activating TRAIL death receptors, induces phosphorylation of AMP-dependent kinase and ATP loss. Cytotoxicity and ATP depletion were significantly enhanced under glucose-limiting conditions, suggesting that ONC201 targets mitochondrial respiration. ONC201 inhibits mitochondrial respiration and induces mitochondrial structural damage (e.g., fragmented mitochondria and matrix-lysed mitochondria). Furthermore, Graves et al. found that caseinolytic protease P (ClpP) is a specific binding protein [103]. ClpP is encoded by a nuclear gene, translated in the cytoplasm and then imported into the mitochondria matrix, which plays a central role in mitochondrial protein degradation within the mitochondria in a process known as mitochondrial proteolysis. In addition, ClpP interacts with mitochondrial respiratory chain proteins and metabolic enzymes. Genetic knockdown of ClpP impairs OXPHOS and complex II [104]. Over the past years we have improved our understanding of how mitochondrial dynamics are regulated, as well as cell-specific contexts in which different mitochondrial morphologies are favored. These studies suggest that CSCs exhibit highly fused mitochondrial networks as means of maintaining OXPHOS, ATP production, and also their stemness.
4. Cytoskeleton
The cytoskeleton is a system of filaments and fibers that are essential for survival and diverse cellular processes in both normal and cancer cells [105,106]. Alterations in cellular structure and intracellular-organelle rearrangement and repositioning lead to changes in cellular metabolism and the acquisition of inappropriate migratory and invasive features, and also accompany the progression of cancer [107]. Actin and the microtubule cytoskeleton are key modulators that underpin these cellular processes. Ultrastructural analysis in diverse model systems have shown that the cytoskeleton is associated with intracellular membranes and organelles [108,109]. It has been postulated that these linkages represent a regulatory process of the distribution, positioning, and trafficking of mitochondria towards energy demanding areas inside cells. An important question arises: how do cells accomplish these processes?
4.1. Role of Actin Microfilaments in the Regulation of Mitochondria
Microfilaments, also called actin filaments, are composed of three isoforms, α-, β-, and γ-actin [110]. Actin exists in two forms: G-actin (globular actin) and F-actin (fibrillar actin). Actin reorganization occurs during cell membrane protrusion and retraction in stress fibers, contractile ring formation during cell division, and mitochondrial fission [111,112,113,114,115]. Several reports have suggested that mitochondria may move along actin filaments in dendrites and axons [116,117,118]. Actin filaments are needed to support microtubules in short-distance trafficking of mitochondria in microtubule-poor regions of the cell [63]. Using live-cell imaging, Moore et al. demonstrated that the association of actin filaments with mitochondria is transient, with rapid disassemble-reassemble cycling throughout the mitochondria within 14 min. In the actin disassemble phase, fragmented mitochondria undergo rapid fusion, which leads to quick local recovery of mitochondrial integrity and maintenance of mitochondrial homeostasis [119]. Moreover, these authors found that F-actin recruitment to mitochondria is dependent on Arp2/3, which has been identified as a component of the mitochondrial outer membrane (MOM). Mitochondrial fission is initiated by marking the division site by the endoplasmic reticulum (ER) tubules, followed by membrane fission via Drp1. The ER-bound inverted formin 2 (INF2), which stimulates polymerization of F-actin at the ER-mitochondria binding sites, leads to mitochondrial fission [115,120,121]. INF2-dependent actin polymerization induces constriction of both mitochondrial membranes during the fission process: IMM constriction is stimulated by increased Ca2+ transfer from ER to mitochondria, and OMM constriction is enhanced by increased recruitment of Drp1. MYO6 is an emerging regulator in cytoskeletal dynamics and cancer progression. MYO6 has been used as a marker of an aggressive phenotype in ovarian cancer. Intriguingly, MYO6 triggers F-actin cage assembly around dysfunctional mitochondria, thereby preventing the spread of damaged mitochondria [122]. These reports suggest that the actin cytoskeleton is essential for spatio-regional trafficking, dynamics, and also quality control of mitochondria.
4.2. Role of Microtubules in the Regulation of Mitochondria
In eukaryotes, microtubules are one of the major components of the cytoskeleton and participate in many vital processes such as mitosis, structural support, intracellular transport of vesicles, and organelles like mitochondria [105,106,123,124,125]. Microtubules are constantly modified in different patterns to enhance their functions. One type of modification is acetylation on lysine 40 of α-tubulin, which results in acetylated α-tubulin (Ac-AT), which recruits motor proteins to facilitate movement of vesicles and mitochondria along its tracks [126,127,128,129,130,131]. Microtubule-associated motor proteins include kinesin and dynein. Dyneins and kinesins transport their cargo toward the minus and plus ends of microtubules (MT), respectively. In neuronal studies, kinesins and dyneins transport mitochondria along Ac-AT throughout axonal and dendritic cells towards the high energy consumption area (e.g., groups of ion pumps or cell protrusion zones) to generate and distribute ATP [132,133,134]. Dynein has been reported to be associated with mitochondria, possibly via interaction with the outer membrane protein VDAC [135]. It has been noted that, in vitro, loss of acetylated residues reduces the interaction of kinesin with MT, with a subsequent decrease in motility of the motor protein [126,127]. Trafficking of mitochondria along MT tracks is regulated by second messengers generated from signaling events. Several studies have demonstrated that intracellular Ca2+, when elevated, halts MT-based mitochondrial movement in many cell types [63,134,136,137]; however, the rapid axonal transport of other cargo persists in the presence of Ca2+ [138]. Ca2+ influx occurs in areas of high metabolic demand, such as nerve terminals, protrusion zones, and the leading edge of pro-metastatic regions, where energetically valuable mitochondria are clustered [5,139]. Furthermore, sustained elevated cytosolic Ca2+ may be a symptom of insufficient local ATP to pump Ca2+ out across the plasma membrane, or a defective buffering capacity of mitochondria, owing to decreased activity of the ETC. Intracellular Ca2+ level-dependent inhibition of mitochondrial motility occurs in response to spatial and temporal requirements and in order to enhance local Ca2+ buffering. In a previous report, we showed that CSCs express sarco/endoplasmic reticulum Ca2+-ATPase to avoid apoptosis, which occurs after Ca2+ overload under glucose deprived conditions [140]. How Ca2+-dependent mitochondrial movement in CSCs is accomplished is presently not well understood. Taken together, the microtubule cytoskeleton is an essential component of mitochondrial trafficking and anchoring through Ca2+ regulation.
5. Cytoskeleton-Mitochondria Interaction
In addition to regulating mitochondrial dynamics, several pieces of evidence demonstrate that the cytoskeletal network interacts with mitochondria to control mitochondrial function [15,108,109].
5.1. Interaction and Regulation of Mitochondrial Function by Actin
Among the most abundant proteins in the cell, actin can self-assemble into polymers (F-actin). As a component of the cytoskeleton, it has the property of interacting with diverse proteins such as the voltage-dependent anion-selective channel (VDAC) and MtCK. VDAC is the most abundant protein of the MOM. It has an important role in the energy management of the cell by regulating the metabolic fluxes and is involved in diverse cellular processes. VDAC acts as a channel or as an anchoring protein and interacts with actin filaments. G-actin, not F-actin, induces the closure of VDAC in vitro [141]. Overexpression of mitochondrial creatine kinase (MtCK) has been reported in several tumors with poor prognosis [142]. Furthermore, increased levels of MtCK in malignant cells may be part of a metabolic adaptation of cancer cells to increase growth rates under oxygen- and glucose-restricted conditions. Upregulated MtCK levels could help to sustain a high energy turnover, but would also be protective against stress conditions such as hypoxia and eventually apoptotic death [143]. Guzun et al. reported that the regulation of energy fluxes is dependent on the interaction of mitochondria with cytoskeletal proteins. The results show that MtCK, which localizes with myosin surrounded by actin filaments in cells, limit VDAC permeability, mostly decreasing ATP and ADP levels. Strongly decreased permeability of MOM for adenine nucleotides significantly enhances the functional coupling between MtCK and ANT (adenine nucleotide translocator), increasing the rate of ADP and ATP recycling in the mitochondrial matrix inner membrane space [144]. Actin, on its own, is suggested to be a regulator of mitochondrial function via interactions with VDAC and MtCK.
5.2. Interaction and Regulation of Mitochondrial Function by Microtubules
Microtubules have long been known to interact strongly with mitochondria in many cell types [145]. A growing body of evidence suggests that the microtubule network may interact with mitochondria to control mitochondrial respiration [15,108,109]. Co-localization of β-tubulin with mitochondria and its association with the MOM was demonstrated first by Saetersdal et al. [146]. Additionally, the presence of tubulin in mitochondria has been shown by Carre et al. in different cell types, where tubulin is localized at the MOM. Several studies suggested that ‘mitochondrial’ tubulin can be organized in α/β-dimers, and, using immunoprecipitation, the authors found that this tubulin is associated with the mitochondrial VDAC [15,146,147,148,149,150]. The VDAC controls diverse cellular processes such as cellular metabolism and apoptosis. The VDAC forms channels on the MOM, where the opening/closure of the channel is critical for controlling the permeability of the mitochondrial membrane and the flux of metabolites into the organelle [150]. Mitochondrial tubulin interacts with the VDAC, and nano-molar concentrations of α β-heterodimers can lead to closure of the VDAC [15,148]. In vitro measurements showed the insertion of tubulin’s polyanionic C terminus into the lumen of the VDAC β-barrel as the mechanism modulating the flow of ATP and ADP through the VDAC and regulating metabolite fluxes across the MOM [127,128]. Several studies have shown that the rate of mitochondrial respiration in isolated mitochondria in vitro is strictly regulated by the availability of ADP for ANT in the mitochondrial inner membrane (MIM) [151]. In isolated mitochondria, the MOM is permeable to metabolites with molecular mass lower than 7 kDa, owing to the open state of the VDAC in MOM [133], and the efficiency of mitochondrial function regulation in vitro by extra ADP depends only upon the affinity of ANT for ADP, which is very high. In cancer cells, the closure of the VDAC by tubulin leads to decreased mitochondrial membrane potential, which could alter mitochondrial metabolism [11,152]. Collectively, microtubules regulate mitochondrial function via interaction with mitochondrial outer membrane components. However, many aspects related to the mitochondria–tubulin interplay in CSCs have yet to be elucidated. In particular, revealing the precise nature and possible functional roles of various α-tubulin isoforms and their post-translational modifications will be required.
5.3. Cytoskeleton Rearrangement in the Regulation of Cancer Metabolism
CSCs exhibit elevated mitochondrial fusion with a metabolic rewiring to OXPHOS [145,146] and a rearranged cytoskeleton network. Cytoskeletal rearrangement is essential for promoting cell movement, invasion, and proliferation. Emerging evidence is beginning to highlight that cytoskeleton rearrangement somehow changes cellular bioenergetics. In yeast, decreased actin turnover, leading to accumulation of aggregates of F-actin, triggers an increase in the production of ROS by mitochondria [153,154]. Some evidence implies a role for actin in ROS production and mitochondrial clustering following cell death signaling [155]. Recently, Hu et al. revealed a mechanism by which PI3K signaling promotes glycolysis. Aldolase functions not only as a key metabolic enzyme for glycolysis but also interacts with cytoskeletal components that control actin polymerization [156]. Activation of the PI3K pathway allows for the physical dissociation of aldolase from F-actin into the cytoplasm, where it is active. These data show an AKT-independent role of PI3K in altering glycolysis through spatial redistribution of aldolase via cytoskeletal rearrangement. These data suggest a mechanism of activating a metabolic enzyme through rearrangement of the cytoskeleton to increase the metabolic flux. Amelia et al. demonstrated that βIII-tubulin promotes the reduction of the reliance of lung cancer cells on glycolysis when glucose becomes limiting. When glucose supply is abundant, βIII-tubulin reduces the reliance of tumor cells on glucose metabolism and promotes responsive AKT signaling to prime cells to adapt when glucose becomes lacking. BIII-tubulin protects tumor cells from ER stress in nutrient-starved conditions via modulation of glycolytic dependency [120]. Recently, the cytoskeletal protein syntaphilin (SNPH), known for arresting mitochondrial movement at sites of high energy demands [157], was found to be expressed in cancer [10,157]. In addition, a recent study revealed that a novel isoform of SNPH maintains the activity of ETC II and local tumor growth. Thus, SNPH downregulation increases mitochondrial superoxide production and reduces cell proliferation [158]. Therefore, the regulatory mechanism between cytoskeletal rearrangement or cytoskeletal regulators and cellular bioenergetics is crucial for understanding tumor cells in response to diverse stimuli.
5.4. Cytoskeleton-Mitochondria Interplay Regulates EMT
A growing body of evidence points to the functional interaction between the cytoskeleton and mitochondria as a crucial regulatory center. Interestingly, mitochondria, which are affected by autophagy-related processes, define the energy supply that cancer cells use to reorganize the cytoskeleton and sustain cell movement during EMT [5,159,160,161]. EMT is firstly determined by morphological reprogramming of the cellular architecture, which is guided by changes in the interaction properties of cells with the surrounding microenvironment and is supported by a reorganized cytoskeleton [162,163]. Accumulation of mitochondria near the cell membrane is critical to promote the formation of filopodia, lamellipodia, and other cellular structures for motility during EMT [160,161]. The involvement of ROS in regulating EMT via actin reorganization has been suggested in recent years. Upon exposure to temporary ROS, oxidized actin filaments could promote the contractility of actomyosin by inducing the disassembly of actin and myosin, leading to the formation of stress fibers and the promotion of cell spreading [164]. At the leading edge, mitochondria support enhanced cell motility and invasion through providing a local source of energy. The importance of regional ATP production for mitochondrial repositioning at the leading edge was demonstrated by using tumor cells that contain OXPHOS-deficient mitochondria (called ρ0 cells). ρ0 cells failed to reposition mitochondria to focal adhesion complexes [5,165]. Furthermore, Max-Hinderk Schuler et al. demonstrated that Miro-1-mediated mitochondrial repositioning at the leading edge provides localized energy demands to promote cell migration by supporting membrane protrusion and focal adhesion stability [165]. Another independent study that relied on misfolding of the ETC II subunit SDHB by treatment with the mitochondrial HSP90 inhibitor Gamitrinib prevented accumulation of mitochondria to focal adhesion. In addition, pharmacological inhibition of ETC I, III, and V (rotenone, antimycin A, and oligomycin) inhibited mitochondrial repositioning to the cortical cytoskeleton [166]. The same group showed that under nutrient deprivation conditions, cancer cells protect cytoskeletal dynamics and motility through the chaperone function of mitochondria-associated HSP90. Increased expression of HSP90 preserves residual OXPHOS and ATP production, preventing mitochondrial disruption [167]. Mutations in mtDNA content are also associated with EMT in cancer cells. Studies using NSCLC cells showed that EMT induced by TGF-β leads to an increase in mtDNA copy number [168]. Increased mtDNA is associated with a higher energy requirement for EMT and with metastasis. Indeed, mitochondrial localization through cytoskeleton provides a means to concentrated energy where is most needed.
6. Therapeutic Strategies Targeting CSCs
Improvement of anticancer therapeutic strategies is often limited by incomplete knowledge of the molecular mechanisms underlying cancer biology and cellular metabolism in response to treatment. CSCs exhibit elevated mitochondrial fusion with a metabolic rewiring to OXPHOS [169,170] and a rearranged cytoskeleton network. Increased mitochondrial fusion promotes ATP production via OXPHOS, allowing the cancer cells to survive without energy constraints. A list of inhibitors under study in vivo and in the clinic is shown in Table 1 and Figure 1.
6.1. Targeting Mitochondrial Dynamics
Inhibition of mitochondrial fusion is a promising strategy because it plays extensive roles in cancer biology. Changes in levels of mitochondrial fusion proteins MFN 1 and 2 affect mitochondrial morphology and integrity. MFN 1- and 2-deficient cells are characterized by elevated mitochondrial fragmentation with a loss of mitochondrial membrane potential and defects in mitochondrial respiration [79]. Moreover, MFN2 depletion downregulates genes that participate in OXPHOS complex, such as ETC I (p39), II (p70), III (p49,) and V (α subunit), leading to a decrease in their enzymatic activity [94,172]. A selective inhibitor of MFN1, Beta II protein kinase C (βIIPKC) accumulates on the mitochondrial outer membrane and phosphorylates MFN1 at serine 86. Phosphorylation of MFN1 induces a partial loss of its GTPases activity and results in buildup of fragmented and dysfunctional mitochondria [173]. Thus, examining other MFN- and OPA1-targeting molecules could further improve our understanding of the role of mitochondrial dynamics in CSC maintenance from a cellular bioenergetics perspective.
6.2. Targeting OXPHOS
OXPHOS inhibitors are promising in targeting tumors with increased dependency on OXPHOS. One of the most promising OXPHOS inhibitors is metformin, a Food and Drug Administration (FDA)-approved diabetic drug repurposed for cancer treatment. By inhibiting ETC I, metformin reduces OXPHOS and tumor growth in multiple cancer models [174,175]. Phenformin, an alternative to metformin, is another biguanide that is readily transported into cancer cells and shows a higher affinity to the mitochondrial membrane than metformin [175]. In non-small cell lung cancer (NSCLC), ALDH-positive cells exhibit CSC phenotypes such as self-renewal and multidrug resistance. An ALDH inhibitor, gossypol reduces ATP production with a decrease in NADH levels in NSCLC. A new metformin derivative, IM156, targets slow-cycling tumor cells that tend to escape from treatment with conventional chemotherapies that have been designed to target fast-cycling cells. In addition, IM156-mediated inhibition of mitochondrial function prevents the growth of oral cancer and glioblastoma [176,177]. Although the above-mentioned therapeutics have been demonstrated to reduce mitochondrial function and survival of CSCs, it should be considered that inhibition of a single specific component of OXPHOS and ATP production may induce alternative pathways, precluding the efficacy of regimens to ultimately eradicate CSCs.
6.3. Targeting the Cytoskeleton
The coupling between mitochondria and the cytoskeleton in the regulation of mitochondrial dynamics and respiration can be a novel target for CSCs. Nowadays, microtubule-targeted agents (MTAs) constitute a class of anticancer drugs largely used in the clinic. Among them, taxanes and vinca alkaloids are powerful inhibitors of microtubule dynamics and are used to treat multiple malignancies. Several studies show that cytochalasin D stimulates actin depolymerization and downregulates mitochondrial function via VDAC closure. Koyama et al. demonstrated that inhibiting Rho-kinase with Y-27632, diminishes ATP production [178]. Paclitaxel and colchicine, which target β-tubulin, increase the mitochondrial membrane potential by decreasing free cytosolic tubulin, which leads to opening of VDAC. This process may explain chemoresistance against tubulin-targeted agents. Because cytoskeletal rearrangement contributes mostly to maintaining quality control and stress resistance of cancers, investigating possible combinations with mitochondria-targeted drugs or synthetic lethal targets may be a promising strategy for overcoming drug resistance in CSCs.
7. Conclusions
In this review, we summarized the mechanisms regulating mitochondrial dynamics and functions that are modulated by the interaction between mitochondria and the cytoskeleton (Figure 3). All these interactions play important roles in the regulation of mitochondrial dynamics, tempo-regional repositioning, re-arrangement, and energy fluxes in the cell, demonstrating system-level properties of coordinating cellular bioenergetics and cellular behavior. In particular, fatty acid oxidation, one of the major characteristics of CSCs, is also regulated by mitochondrial dynamics in response to cytoskeletal remodeling. This metabolic preference is a promising target for certain types of cancer with elevated fatty acid utilization. Many recent studies have demonstrated that mitochondria-centric metabolism, OXPHOS, is upregulated in a variety of cancers, potentially rendering them sensitive to mitochondria-targeted drugs. Cancer’s intrinsic sensitivity to mitochondria-targeted drugs should continue to be characterized, environmental drivers of cancer cell susceptibility to these drugs must be recognized, and combinations with cytoskeleton-targeted drugs should be evaluated. Tumor heterogeneity may also affect cytoskeletal protein expression and organization to utilize cellular dynamics and coordination. Thus, understanding the mechanisms of mitochondrial fitness, including the impact of the cytoskeletal contribution to extrinsic and intrinsic stress responses, can facilitate the development of novel therapeutic strategies that directly eradicate CSCs in patients with drug-resistant tumors.
Author Contributions
Conceptualization, J.K. and J.-H.C.; writing—original draft preparation, J.K. and J.-H.C.; writing—review and editing, J.K. and J.-H.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Research Foundation of Korea (NRF), the Korean Government (MSIT) (grant NRF-2018R1A5A2025079).
Acknowledgments
This work was supported by the Brain Korea 21 PLUS Project for Medical Science, Yonsei University.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Chan, D.C. Mitochondria: Dynamic organelles in disease, aging, and development. Cell 2006, 125, 1241–1252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suen, D.F.; Norris, K.L.; Youle, R.J. Mitochondrial dynamics and apoptosis. Genes Dev. 2008, 22, 1577–1590. [Google Scholar] [CrossRef] [Green Version]
- Trotta, A.P.; Chipuk, J.E. Mitochondrial dynamics as regulators of cancer biology. Cell Mol. Life Sci. 2017, 74, 1999–2017. [Google Scholar] [CrossRef] [PubMed]
- Lewis, M.R.; Lewis, W.H. Mitochondria in Tissue Culture. Science 1914, 39, 330–333. [Google Scholar] [CrossRef]
- Desai, S.P.; Bhatia, S.N.; Toner, M.; Irimia, D. Mitochondrial localization and the persistent migration of epithelial cancer cells. Biophys. J. 2013, 104, 2077–2088. [Google Scholar] [CrossRef] [Green Version]
- Viale, A.; Pettazzoni, P.; Lyssiotis, C.A.; Ying, H.; Sanchez, N.; Marchesini, M.; Carugo, A.; Green, T.; Seth, S.; Giuliani, V.; et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 2014, 514, 628–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Lim, S.K.; Liang, Q.; Iyer, S.V.; Wang, H.Y.; Wang, Z.; Xie, X.; Sun, D.; Chen, Y.J.; Tabar, V.; et al. Gboxin is an oxidative phosphorylation inhibitor that targets glioblastoma. Nature 2019, 567, 341–346. [Google Scholar] [CrossRef]
- Roesch, A.; Vultur, A.; Bogeski, I.; Wang, H.; Zimmermann, K.M.; Speicher, D.; Korbel, C.; Laschke, M.W.; Gimotty, P.A.; Philipp, S.E.; et al. Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1B(high) cells. Cancer Cell 2013, 23, 811–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeBleu, V.S.; O’Connell, J.T.; Gonzalez Herrera, K.N.; Wikman, H.; Pantel, K.; Haigis, M.C.; de Carvalho, F.M.; Damascena, A.; Domingos Chinen, L.T.; Rocha, R.M.; et al. PGC-1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell Biol. 2014, 16, 992–1003, 1001–1015. [Google Scholar] [CrossRef] [Green Version]
- Caino, M.C.; Seo, J.H.; Aguinaldo, A.; Wait, E.; Bryant, K.G.; Kossenkov, A.V.; Hayden, J.E.; Vaira, V.; Morotti, A.; Ferrero, S.; et al. A neuronal network of mitochondrial dynamics regulates metastasis. Nat. Commun. 2016, 7, 13730. [Google Scholar] [CrossRef]
- Maldonado, E.N.; Patnaik, J.; Mullins, M.R.; Lemasters, J.J. Free tubulin modulates mitochondrial membrane potential in cancer cells. Cancer Res. 2010, 70, 10192–10201. [Google Scholar] [CrossRef] [Green Version]
- Monge, C.; Beraud, N.; Kuznetsov, A.V.; Rostovtseva, T.; Sackett, D.; Schlattner, U.; Vendelin, M.; Saks, V.A. Regulation of respiration in brain mitochondria and synaptosomes: Restrictions of ADP diffusion in situ, roles of tubulin, and mitochondrial creatine kinase. Mol. Cell Biochem. 2008, 318, 147–165. [Google Scholar] [CrossRef]
- Noskov, S.Y.; Rostovtseva, T.K.; Bezrukov, S.M. ATP transport through VDAC and the VDAC-tubulin complex probed by equilibrium and nonequilibrium MD simulations. Biochemistry 2013, 52, 9246–9256. [Google Scholar] [CrossRef]
- Rostovtseva, T.K.; Bezrukov, S.M. VDAC regulation: Role of cytosolic proteins and mitochondrial lipids. J. Bioenerg. Biomembr. 2008, 40, 163–170. [Google Scholar] [CrossRef] [Green Version]
- Rostovtseva, T.K.; Sheldon, K.L.; Hassanzadeh, E.; Monge, C.; Saks, V.; Bezrukov, S.M.; Sackett, D.L. Tubulin binding blocks mitochondrial voltage-dependent anion channel and regulates respiration. Proc. Natl. Acad. Sci. USA 2008, 105, 18746–18751. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Schaedel, L.; Portran, D.; Aguilar, A.; Gaillard, J.; Marinkovich, M.P.; Thery, M.; Nachury, M.V. Microtubules acquire resistance from mechanical breakage through intralumenal acetylation. Science 2017, 356, 328–332. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.M.; Giltnane, J.M.; Balko, J.M.; Schwarz, L.J.; Guerrero-Zotano, A.L.; Hutchinson, K.E.; Nixon, M.J.; Estrada, M.V.; Sanchez, V.; Sanders, M.E.; et al. MYC and MCL1 Cooperatively Promote Chemotherapy-Resistant Breast Cancer Stem Cells via Regulation of Mitochondrial Oxidative Phosphorylation. Cell Metab. 2017, 26, 633–647.e7. [Google Scholar] [CrossRef] [Green Version]
- Samardzija, C.; Greening, D.W.; Escalona, R.; Chen, M.; Bilandzic, M.; Luwor, R.; Kannourakis, G.; Findlay, J.K.; Ahmed, N. Knockdown of stem cell regulator Oct4A in ovarian cancer reveals cellular reprogramming associated with key regulators of cytoskeleton-extracellular matrix remodelling. Sci. Rep. 2017, 7, 46312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashida, T.; Jinno, H.; Kitagawa, Y.; Kitajima, M. Cooperation of cancer stem cell properties and epithelial-mesenchymal transition in the establishment of breast cancer metastasis. J. Oncol. 2011, 2011, 591427. [Google Scholar] [CrossRef] [Green Version]
- Pasonen-Seppanen, S.; Hyttinen, J.M.; Rilla, K.; Jokela, T.; Noble, P.W.; Tammi, M.; Tammi, R. Role of CD44 in the organization of keratinocyte pericellular hyaluronan. Histochem Cell Biol. 2012, 137, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Eberlein, C.; Rooney, C.; Ross, S.J.; Farren, M.; Weir, H.M.; Barry, S.T. E-Cadherin and EpCAM expression by NSCLC tumour cells associate with normal fibroblast activation through a pathway initiated by integrin alphavbeta6 and maintained through TGFbeta signalling. Oncogene 2015, 34, 704–716. [Google Scholar] [CrossRef]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keller, L.; Werner, S.; Pantel, K. Biology and clinical relevance of EpCAM. Cell Stress 2019, 3, 165–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nam-Cha, S.H.; Serrano-Vargas, R.; Escario, E.; Azana, J.M.; Calero-Oliver, R.; Martin, A.G.; Poblet, E. CD133 expression in normal skin and in epithelial cutaneous tumors. Biomed. Res. Int. 2013, 2013, 385604. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Shi, S.; Yen, Y.; Brown, J.; Ta, J.Q.; Le, A.D. A subpopulation of CD133(+) cancer stem-like cells characterized in human oral squamous cell carcinoma confer resistance to chemotherapy. Cancer Lett. 2010, 289, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Auvinen, P.; Tammi, R.; Tammi, M.; Johansson, R.; Kosma, V.M. Expression of CD 44 s, CD 44 v 3 and CD 44 v 6 in benign and malignant breast lesions: Correlation and colocalization with hyaluronan. Histopathology 2005, 47, 420–428. [Google Scholar] [CrossRef]
- Marcato, P.; Dean, C.A.; Giacomantonio, C.A.; Lee, P.W. Aldehyde dehydrogenase: Its role as a cancer stem cell marker comes down to the specific isoform. Cell Cycle 2011, 10, 1378–1384. [Google Scholar] [CrossRef]
- Tanei, T.; Morimoto, K.; Shimazu, K.; Kim, S.J.; Tanji, Y.; Taguchi, T.; Tamaki, Y.; Noguchi, S. Association of breast cancer stem cells identified by aldehyde dehydrogenase 1 expression with resistance to sequential Paclitaxel and epirubicin-based chemotherapy for breast cancers. Clin. Cancer Res. 2009, 15, 4234–4241. [Google Scholar] [CrossRef] [Green Version]
- Tomita, H.; Tanaka, K.; Tanaka, T.; Hara, A. Aldehyde dehydrogenase 1A1 in stem cells and cancer. Oncotarget 2016, 7, 11018–11032. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.H.; Lee, S.H.; Hong, D.; Lee, J.S.; Ahn, H.S.; Ahn, J.H.; Seong, T.W.; Lee, C.H.; Jang, H.; Hong, K.M.; et al. Aldehyde dehydrogenase is used by cancer cells for energy metabolism. Exp. Mol. Med. 2016, 48, e272. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Shim, J.K.; Kang, J.H.; Choi, J.; Chang, J.H.; Kim, S.Y.; Kang, S.G. Regulation of bioenergetics through dual inhibition of aldehyde dehydrogenase and mitochondrial complex I suppresses glioblastoma tumorspheres. Neuro. Oncol. 2018, 20, 954–965. [Google Scholar] [CrossRef] [Green Version]
- Rambold, A.S.; Kostelecky, B.; Elia, N.; Lippincott-Schwartz, J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc. Natl. Acad. Sci. USA 2011, 108, 10190–10195. [Google Scholar] [CrossRef] [Green Version]
- Janiszewska, M.; Suva, M.L.; Riggi, N.; Houtkooper, R.H.; Auwerx, J.; Clement-Schatlo, V.; Radovanovic, I.; Rheinbay, E.; Provero, P.; Stamenkovic, I. Imp2 controls oxidative phosphorylation and is crucial for preserving glioblastoma cancer stem cells. Genes Dev. 2012, 26, 1926–1944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasto, A.; Bellio, C.; Pilotto, G.; Ciminale, V.; Silic-Benussi, M.; Guzzo, G.; Rasola, A.; Frasson, C.; Nardo, G.; Zulato, E.; et al. Cancer stem cells from epithelial ovarian cancer patients privilege oxidative phosphorylation, and resist glucose deprivation. Oncotarget 2014, 5, 4305–4319. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.A.; Lin, C.H.; Chi, W.H.; Wang, C.Y.; Hsieh, Y.T.; Wei, Y.H.; Chen, Y.J. Resveratrol Impedes the Stemness, Epithelial-Mesenchymal Transition, and Metabolic Reprogramming of Cancer Stem Cells in Nasopharyngeal Carcinoma through p53 Activation. Evid. Based Complement. Alternat. Med. 2013, 2013, 590393. [Google Scholar] [CrossRef]
- Lagadinou, E.D.; Sach, A.; Callahan, K.; Rossi, R.M.; Neering, S.J.; Minhajuddin, M.; Ashton, J.M.; Pei, S.; Grose, V.; O’Dwyer, K.M.; et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 2013, 12, 329–341. [Google Scholar] [CrossRef] [Green Version]
- De Luca, A.; Fiorillo, M.; Peiris-Pages, M.; Ozsvari, B.; Smith, D.L.; Sanchez-Alvarez, R.; Martinez-Outschoorn, U.E.; Cappello, A.R.; Pezzi, V.; Lisanti, M.P.; et al. Mitochondrial biogenesis is required for the anchorage-independent survival and propagation of stem-like cancer cells. Oncotarget 2015, 6, 14777–14795. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.Q.; Li, Q.; Wang, G.H.; Sun, F.F.; Huang, G.J.; Bian, X.W.; Yu, S.C.; Qian, G.S. Mitochondrial and energy metabolism-related properties as novel indicators of lung cancer stem cells. Int. J. Cancer 2011, 129, 820–831. [Google Scholar] [CrossRef]
- Farnie, G.; Sotgia, F.; Lisanti, M.P. High mitochondrial mass identifies a sub-population of stem-like cancer cells that are chemo-resistant. Oncotarget 2015, 6, 30472–30486. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.; Yang, J.; Ku, M.; Kim, N.H.; Park, Y.; Park, C.B.; Suh, J.S.; Park, E.S.; Yook, J.I.; Mills, G.B.; et al. Metabolic stress induces a Wnt-dependent cancer stem cell-like state transition. Cell Death Dis. 2015, 6, e1805. [Google Scholar] [CrossRef] [Green Version]
- Lue, H.W.; Podolak, J.; Kolahi, K.; Cheng, L.; Rao, S.; Garg, D.; Xue, C.H.; Rantala, J.K.; Tyner, J.W.; Thornburg, K.L.; et al. Metabolic reprogramming ensures cancer cell survival despite oncogenic signaling blockade. Genes Dev. 2017, 31, 2067–2084. [Google Scholar] [CrossRef]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef]
- Araujo, E.P.; Carvalheira, J.B.; Velloso, L.A. Disruption of metabolic pathways--perspectives for the treatment of cancer. Curr. Cancer Drug Targets 2006, 6, 77–87. [Google Scholar] [CrossRef]
- Kroemer, G.; Pouyssegur, J. Tumor cell metabolism: Cancer’s Achilles’ heel. Cancer Cell 2008, 13, 472–482. [Google Scholar] [CrossRef]
- Obre, E.; Rossignol, R. Emerging concepts in bioenergetics and cancer research: Metabolic flexibility, coupling, symbiosis, switch, oxidative tumors, metabolic remodeling, signaling and bioenergetic therapy. Int. J. Biochem. Cell Biol. 2015, 59, 167–181. [Google Scholar] [CrossRef]
- Kim, S.Y. Targeting cancer energy metabolism: A potential systemic cure for cancer. Arch. Pharm. Res. 2019, 42, 140–149. [Google Scholar] [CrossRef]
- Vazquez, F.; Lim, J.H.; Chim, H.; Bhalla, K.; Girnun, G.; Pierce, K.; Clish, C.B.; Granter, S.R.; Widlund, H.R.; Spiegelman, B.M.; et al. PGC1alpha expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell 2013, 23, 287–301. [Google Scholar] [CrossRef] [Green Version]
- Sancho, P.; Burgos-Ramos, E.; Tavera, A.; Bou Kheir, T.; Jagust, P.; Schoenhals, M.; Barneda, D.; Sellers, K.; Campos-Olivas, R.; Grana, O.; et al. MYC/PGC-1alpha Balance Determines the Metabolic Phenotype and Plasticity of Pancreatic Cancer Stem Cells. Cell Metab. 2015, 22, 590–605. [Google Scholar] [CrossRef] [Green Version]
- Vlashi, E.; Lagadec, C.; Vergnes, L.; Matsutani, T.; Masui, K.; Poulou, M.; Popescu, R.; Della Donna, L.; Evers, P.; Dekmezian, C.; et al. Metabolic state of glioma stem cells and nontumorigenic cells. Proc. Natl. Acad. Sci. USA 2011, 108, 16062–16067. [Google Scholar] [CrossRef] [Green Version]
- Morandi, A.; Indraccolo, S. Linking metabolic reprogramming to therapy resistance in cancer. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 1–6. [Google Scholar] [CrossRef]
- Zhou, Y.; Tozzi, F.; Chen, J.; Fan, F.; Xia, L.; Wang, J.; Gao, G.; Zhang, A.; Xia, X.; Brasher, H.; et al. Intracellular ATP levels are a pivotal determinant of chemoresistance in colon cancer cells. Cancer Res. 2012, 72, 304–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vellinga, T.T.; Borovski, T.; de Boer, V.C.; Fatrai, S.; van Schelven, S.; Trumpi, K.; Verheem, A.; Snoeren, N.; Emmink, B.L.; Koster, J.; et al. SIRT1/PGC1alpha-Dependent Increase in Oxidative Phosphorylation Supports Chemotherapy Resistance of Colon Cancer. Clin. Cancer Res. 2015, 21, 2870–2879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, H.J.; Jhe, Y.L.; Kim, J.; Lim, J.Y.; Lee, J.E.; Shin, M.K.; Cheong, J.H. FoxM1-dependent and fatty acid oxidation-mediated ROS modulation is a cell-intrinsic drug resistance mechanism in cancer stem-like cells. Redox Biol. 2020, 36, 101589. [Google Scholar] [CrossRef] [PubMed]
- Diehn, M.; Cho, R.W.; Lobo, N.A.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.; Lam, J.S.; Ailles, L.E.; Wong, M.; et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009, 458, 780–783. [Google Scholar] [CrossRef]
- Reid, R.A.; Moyle, J.; Mitchell, P. Synthesis of adenosine triphosphate by a protonmotive force in rat liver mitochondria. Nature 1966, 212, 257–258. [Google Scholar] [CrossRef]
- Mitchell, P. Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature 1961, 191, 144–148. [Google Scholar] [CrossRef]
- Greenhouse, W.V.; Lehninger, A.L. Magnitude of malate-aspartate reduced nicotinamide adenine dinucleotide shuttle activity in intact respiring tumor cells. Cancer Res. 1977, 37, 4173–4181. [Google Scholar]
- Dionisi, O.; Longhi, G.; Eboli, M.L.; Galeotti, T.; Terranova, T. Oxidation of reduced nicotinamide-adenine dinucleotide by the malate-aspartate shuttle in Ehrlich ascites tumour cells. Biochim. Biophys. Acta 1974, 333, 577–580. [Google Scholar] [CrossRef]
- Piel, S.; Ehinger, J.K.; Chamkha, I.; Frostner, E.A.; Sjovall, F.; Elmer, E.; Hansson, M.J. Bioenergetic bypass using cell-permeable succinate, but not methylene blue, attenuates metformin-induced lactate production. Intensive Care Med. Exp. 2018, 6, 22. [Google Scholar] [CrossRef]
- Frederick, R.L.; Shaw, J.M. Moving mitochondria: Establishing distribution of an essential organelle. Traffic 2007, 8, 1668–1675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, F.; Hamann, A.; Osiewacz, H.D. Mitochondrial quality control: An integrated network of pathways. Trends Biochem. Sci. 2012, 37, 284–292. [Google Scholar] [CrossRef]
- Westermann, B. Merging mitochondria matters: Cellular role and molecular machinery of mitochondrial fusion. EMBO Rep. 2002, 3, 527–531. [Google Scholar] [CrossRef] [Green Version]
- Hollenbeck, P.J.; Saxton, W.M. The axonal transport of mitochondria. J. Cell Sci. 2005, 118, 5411–5419. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, K.; Shaw, J.M. Mitochondrial morphology and dynamics in yeast and multicellular eukaryotes. Annu. Rev. Genet. 2005, 39, 503–536. [Google Scholar] [CrossRef]
- Eura, Y.; Ishihara, N.; Yokota, S.; Mihara, K. Two mitofusin proteins, mammalian homologues of FZO, with distinct functions are both required for mitochondrial fusion. J. Biochem. 2003, 134, 333–344. [Google Scholar] [CrossRef]
- Qi, Y.; Yan, L.; Yu, C.; Guo, X.; Zhou, X.; Hu, X.; Huang, X.; Rao, Z.; Lou, Z.; Hu, J. Structures of human mitofusin 1 provide insight into mitochondrial tethering. J. Cell Biol. 2016, 215, 621–629. [Google Scholar] [CrossRef] [Green Version]
- Santin, G.; Piccolini, V.M.; Barni, S.; Veneroni, P.; Giansanti, V.; Dal Bo, V.; Bernocchi, G.; Bottone, M.G. Mitochondrial fusion: A mechanism of cisplatin-induced resistance in neuroblastoma cells? Neurotoxicology 2013, 34, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Cipolat, S.; Martins de Brito, O.; Dal Zilio, B.; Scorrano, L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc. Natl. Acad. Sci. USA 2004, 101, 15927–15932. [Google Scholar] [CrossRef] [Green Version]
- Otera, H.; Ishihara, N.; Mihara, K. New insights into the function and regulation of mitochondrial fission. Biochim. Biophys. Acta 2013, 1833, 1256–1268. [Google Scholar] [CrossRef] [Green Version]
- Otera, H.; Wang, C.; Cleland, M.M.; Setoguchi, K.; Yokota, S.; Youle, R.J.; Mihara, K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J. Cell Biol. 2010, 191, 1141–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Archer, S.L. Mitochondrial dynamics--mitochondrial fission and fusion in human diseases. N. Engl. J. Med. 2013, 369, 2236–2251. [Google Scholar] [CrossRef] [Green Version]
- Gomes, L.C.; Di Benedetto, G.; Scorrano, L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 2011, 13, 589–598. [Google Scholar] [CrossRef] [Green Version]
- Smirnova, E.; Griparic, L.; Shurland, D.L.; van der Bliek, A.M. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell 2001, 12, 2245–2256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, Y.; Krueger, E.W.; Oswald, B.J.; McNiven, M.A. The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol. Cell Biol. 2003, 23, 5409–5420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, D.; Xu, L.; Yu, Y.; Zhu, W.; Andrews, D.W.; Yoon, Y.; Kuo, T.H. Regulation of Ca2+-induced permeability transition by Bcl-2 is antagonized by Drpl and hFis1. Mol. Cell Biochem. 2005, 272, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Cereghetti, G.M.; Costa, V.; Scorrano, L. Inhibition of Drp1-dependent mitochondrial fragmentation and apoptosis by a polypeptide antagonist of calcineurin. Cell Death Differ. 2010, 17, 1785–1794. [Google Scholar] [CrossRef] [Green Version]
- Olichon, A.; Baricault, L.; Gas, N.; Guillou, E.; Valette, A.; Belenguer, P.; Lenaers, G. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J. Biol. Chem. 2003, 278, 7743–7746. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Chan, D.C. Emerging functions of mammalian mitochondrial fusion and fission. Hum. Mol. Genet. 2005, 14, R283–R289. [Google Scholar] [CrossRef]
- Chen, H.; Chomyn, A.; Chan, D.C. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J. Biol. Chem. 2005, 280, 26185–26192. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Chan, D.C. Mitochondrial dynamics--fusion, fission, movement, and mitophagy--in neurodegenerative diseases. Hum. Mol. Genet. 2009, 18, R169–R176. [Google Scholar] [CrossRef]
- Zorzano, A.; Liesa, M.; Sebastian, D.; Segales, J.; Palacin, M. Mitochondrial fusion proteins: Dual regulators of morphology and metabolism. Semin. Cell Dev. Biol. 2010, 21, 566–574. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, T.P.; Bjorklund, M. Mitochondrial Function and Cell Size: An Allometric Relationship. Trends Cell Biol. 2017, 27, 393–402. [Google Scholar] [CrossRef]
- Scorrano, L.; Ashiya, M.; Buttle, K.; Weiler, S.; Oakes, S.A.; Mannella, C.A.; Korsmeyer, S.J. A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev. Cell 2002, 2, 55–67. [Google Scholar] [CrossRef] [Green Version]
- Roucou, X.; Montessuit, S.; Antonsson, B.; Martinou, J.C. Bax oligomerization in mitochondrial membranes requires tBid (caspase-8-cleaved Bid) and a mitochondrial protein. Biochem. J. 2002, 368, 915–921. [Google Scholar] [CrossRef] [PubMed]
- Boutant, M.; Kulkarni, S.S.; Joffraud, M.; Ratajczak, J.; Valera-Alberni, M.; Combe, R.; Zorzano, A.; Canto, C. Mfn2 is critical for brown adipose tissue thermogenic function. EMBO J. 2017, 36, 1543–1558. [Google Scholar] [CrossRef] [Green Version]
- Rambold, A.S.; Cohen, S.; Lippincott-Schwartz, J. Fatty acid trafficking in starved cells: Regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev. Cell 2015, 32, 678–692. [Google Scholar] [CrossRef] [Green Version]
- Shiratori, R.; Furuichi, K.; Yamaguchi, M.; Miyazaki, N.; Aoki, H.; Chibana, H.; Ito, K.; Aoki, S. Glycolytic suppression dramatically changes the intracellular metabolic profile of multiple cancer cell lines in a mitochondrial metabolism-dependent manner. Sci. Rep. 2019, 9, 18699. [Google Scholar] [CrossRef] [Green Version]
- Buck, M.D.; O’Sullivan, D.; Klein Geltink, R.I.; Curtis, J.D.; Chang, C.H.; Sanin, D.E.; Qiu, J.; Kretz, O.; Braas, D.; van der Windt, G.J.; et al. Mitochondrial Dynamics Controls T Cell Fate through Metabolic Programming. Cell 2016, 166, 63–76. [Google Scholar] [CrossRef] [Green Version]
- Serasinghe, M.N.; Wieder, S.Y.; Renault, T.T.; Elkholi, R.; Asciolla, J.J.; Yao, J.L.; Jabado, O.; Hoehn, K.; Kageyama, Y.; Sesaki, H.; et al. Mitochondrial division is requisite to RAS-induced transformation and targeted by oncogenic MAPK pathway inhibitors. Mol. Cell 2015, 57, 521–536. [Google Scholar] [CrossRef] [Green Version]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and Cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef]
- Graves, J.A.; Wang, Y.; Sims-Lucas, S.; Cherok, E.; Rothermund, K.; Branca, M.F.; Elster, J.; Beer-Stolz, D.; Van Houten, B.; Vockley, J.; et al. Mitochondrial structure, function and dynamics are temporally controlled by c-Myc. PLoS ONE 2012, 7, e37699. [Google Scholar] [CrossRef] [Green Version]
- Kashatus, J.A.; Nascimento, A.; Myers, L.J.; Sher, A.; Byrne, F.L.; Hoehn, K.L.; Counter, C.M.; Kashatus, D.F. Erk2 phosphorylation of Drp1 promotes mitochondrial fission and MAPK-driven tumor growth. Mol. Cell 2015, 57, 537–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, J.; Wang, J.; Huang, Y.; Wu, H.; Xia, T.; Xiao, J.; Chen, X.; Li, H.; Qiu, Y.; Wang, Y.; et al. ERK/Drp1-dependent mitochondrial fission is involved in the MSC-induced drug resistance of T-cell acute lymphoblastic leukemia cells. Cell Death Dis. 2016, 7, e2459. [Google Scholar] [CrossRef] [PubMed]
- Loiseau, D.; Chevrollier, A.; Verny, C.; Guillet, V.; Gueguen, N.; Pou de Crescenzo, M.A.; Ferre, M.; Malinge, M.C.; Guichet, A.; Nicolas, G.; et al. Mitochondrial coupling defect in Charcot-Marie-Tooth type 2A disease. Ann. Neurol. 2007, 61, 315–323. [Google Scholar] [CrossRef]
- Picard, M.; Shirihai, O.S.; Gentil, B.J.; Burelle, Y. Mitochondrial morphology transitions and functions: Implications for retrograde signaling? Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 304, R393–R406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaseva, A.V.; Blake, D.R.; Gilbert, T.S.K.; Ng, S.; Hostetter, G.; Azam, S.H.; Ozkan-Dagliyan, I.; Gautam, P.; Bryant, K.L.; Pearce, K.H.; et al. KRAS Suppression-Induced Degradation of MYC Is Antagonized by a MEK5-ERK5 Compensatory Mechanism. Cancer Cell 2018, 34, 807–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, M.K.; Cheong, J.H. Mitochondria-centric bioenergetic characteristics in cancer stem-like cells. Arch. Pharm. Res. 2019, 42, 113–127. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Jiang, Y.; Meisenhelder, J.; Yang, W.; Hawke, D.H.; Zheng, Y.; Xia, Y.; Aldape, K.; He, J.; Hunter, T.; et al. Mitochondria-Translocated PGK1 Functions as a Protein Kinase to Coordinate Glycolysis and the TCA Cycle in Tumorigenesis. Mol. Cell 2016, 61, 705–719. [Google Scholar] [CrossRef] [Green Version]
- Murphy, T.A.; Dang, C.V.; Young, J.D. Isotopically nonstationary 13C flux analysis of Myc-induced metabolic reprogramming in B-cells. Metab. Eng. 2013, 15, 206–217. [Google Scholar] [CrossRef] [Green Version]
- Morrish, F.; Noonan, J.; Perez-Olsen, C.; Gafken, P.R.; Fitzgibbon, M.; Kelleher, J.; VanGilst, M.; Hockenbery, D. Myc-dependent mitochondrial generation of acetyl-CoA contributes to fatty acid biosynthesis and histone acetylation during cell cycle entry. J. Biol. Chem. 2010, 285, 36267–36274. [Google Scholar] [CrossRef] [Green Version]
- Camarda, R.; Williams, J.; Goga, A. In vivo Reprogramming of Cancer Metabolism by MYC. Front Cell Dev. Biol. 2017, 5, 35. [Google Scholar] [CrossRef] [Green Version]
- Greer, Y.E.; Porat-Shliom, N.; Nagashima, K.; Stuelten, C.; Crooks, D.; Koparde, V.N.; Gilbert, S.F.; Islam, C.; Ubaldini, A.; Ji, Y.; et al. ONC201 kills breast cancer cells in vitro by targeting mitochondria. Oncotarget 2018, 9, 18454–18479. [Google Scholar] [CrossRef] [Green Version]
- Graves, P.R.; Aponte-Collazo, L.J.; Fennell, E.M.J.; Graves, A.C.; Hale, A.E.; Dicheva, N.; Herring, L.E.; Gilbert, T.S.K.; East, M.P.; McDonald, I.M.; et al. Mitochondrial Protease ClpP is a Target for the Anticancer Compounds ONC201 and Related Analogues. ACS Chem. Biol. 2019, 14, 1020–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole, A.; Wang, Z.; Coyaud, E.; Voisin, V.; Gronda, M.; Jitkova, Y.; Mattson, R.; Hurren, R.; Babovic, S.; Maclean, N.; et al. Inhibition of the Mitochondrial Protease ClpP as a Therapeutic Strategy for Human Acute Myeloid Leukemia. Cancer Cell 2015, 27, 864–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nogales, E. Structural insight into microtubule function. Annu. Rev. Biophys. Biomol. Struct. 2001, 30, 397–420. [Google Scholar] [CrossRef]
- Heald, R.; Nogales, E. Microtubule dynamics. J. Cell Sci. 2002, 115, 3–4. [Google Scholar]
- Fife, C.M.; McCarroll, J.A.; Kavallaris, M. Movers and shakers: Cell cytoskeleton in cancer metastasis. Br. J. Pharm. 2014, 171, 5507–5523. [Google Scholar] [CrossRef] [Green Version]
- Appaix, F.; Kuznetsov, A.V.; Usson, Y.; Kay, L.; Andrienko, T.; Olivares, J.; Kaambre, T.; Sikk, P.; Margreiter, R.; Saks, V. Possible role of cytoskeleton in intracellular arrangement and regulation of mitochondria. Exp. Physiol. 2003, 88, 175–190. [Google Scholar] [CrossRef] [Green Version]
- Kuznetsov, A.V.; Tiivel, T.; Sikk, P.; Kaambre, T.; Kay, L.; Daneshrad, Z.; Rossi, A.; Kadaja, L.; Peet, N.; Seppet, E.; et al. Striking differences between the kinetics of regulation of respiration by ADP in slow-twitch and fast-twitch muscles in vivo. Eur. J. Biochem. 1996, 241, 909–915. [Google Scholar] [CrossRef]
- Rao, J.; Li, N. Microfilament actin remodeling as a potential target for cancer drug development. Curr. Cancer Drug Targets 2004, 4, 345–354. [Google Scholar] [CrossRef]
- Pollard, T.D.; Blanchoin, L.; Mullins, R.D. Actin dynamics. J. Cell Sci. 2001, 114, 3–4. [Google Scholar] [PubMed]
- Pollard, T.D.; Blanchoin, L.; Mullins, R.D. Molecular mechanisms controlling actin filament dynamics in nonmuscle cells. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 545–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korobova, F.; Ramabhadran, V.; Higgs, H.N. An actin-dependent step in mitochondrial fission mediated by the ER-associated formin INF2. Science 2013, 339, 464–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korobova, F.; Gauvin, T.J.; Higgs, H.N. A role for myosin II in mammalian mitochondrial fission. Curr. Biol. 2014, 24, 409–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manor, U.; Bartholomew, S.; Golani, G.; Christenson, E.; Kozlov, M.; Higgs, H.; Spudich, J.; Lippincott-Schwartz, J. A mitochondria-anchored isoform of the actin-nucleating spire protein regulates mitochondrial division. Elife 2015, 4. [Google Scholar] [CrossRef]
- Langford, G.M. Actin- and microtubule-dependent organelle motors: Interrelationships between the two motility systems. Curr. Opin. Cell Biol. 1995, 7, 82–88. [Google Scholar] [CrossRef]
- Morris, R.L.; Hollenbeck, P.J. Axonal transport of mitochondria along microtubules and F-actin in living vertebrate neurons. J. Cell Biol. 1995, 131, 1315–1326. [Google Scholar] [CrossRef] [Green Version]
- Sheetz, M.P.; Steuer, E.R.; Schroer, T.A. The mechanism and regulation of fast axonal transport. Trends Neurosci. 1989, 12, 474–478. [Google Scholar] [CrossRef]
- Moore, A.S.; Wong, Y.C.; Simpson, C.L.; Holzbaur, E.L. Dynamic actin cycling through mitochondrial subpopulations locally regulates the fission-fusion balance within mitochondrial networks. Nat. Commun. 2016, 7, 12886. [Google Scholar] [CrossRef]
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER tubules mark sites of mitochondrial division. Science 2011, 334, 358–362. [Google Scholar] [CrossRef] [Green Version]
- Ji, W.K.; Hatch, A.L.; Merrill, R.A.; Strack, S.; Higgs, H.N. Actin filaments target the oligomeric maturation of the dynamin GTPase Drp1 to mitochondrial fission sites. Elife 2015, 4, e11553. [Google Scholar] [CrossRef]
- Kruppa, A.J.; Kishi-Itakura, C.; Masters, T.A.; Rorbach, J.E.; Grice, G.L.; Kendrick-Jones, J.; Nathan, J.A.; Minczuk, M.; Buss, F. Myosin VI-Dependent Actin Cages Encapsulate Parkin-Positive Damaged Mitochondria. Dev. Cell 2018, 44, 484–499.e6. [Google Scholar] [CrossRef]
- Desai, A.; Mitchison, T.J. Microtubule polymerization dynamics. Annu. Rev. Cell Dev. Biol. 1997, 13, 83–117. [Google Scholar] [CrossRef] [Green Version]
- Downing, K.H. Structural basis for the interaction of tubulin with proteins and drugs that affect microtubule dynamics. Annu. Rev. Cell Dev. Biol. 2000, 16, 89–111. [Google Scholar] [CrossRef] [PubMed]
- Straight, A.F.; Field, C.M. Microtubules, membranes and cytokinesis. Curr. Biol. 2000, 10, R760–R770. [Google Scholar] [CrossRef] [Green Version]
- Dompierre, J.P.; Godin, J.D.; Charrin, B.C.; Cordelieres, F.P.; King, S.J.; Humbert, S.; Saudou, F. Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington’s disease by increasing tubulin acetylation. J. Neurosci. 2007, 27, 3571–3583. [Google Scholar] [CrossRef] [PubMed]
- Reed, N.A.; Cai, D.; Blasius, T.L.; Jih, G.T.; Meyhofer, E.; Gaertig, J.; Verhey, K.J. Microtubule acetylation promotes kinesin-1 binding and transport. Curr. Biol. 2006, 16, 2166–2172. [Google Scholar] [CrossRef] [Green Version]
- Bulinski, J.C. Microtubule modification: Acetylation speeds anterograde traffic flow. Curr. Biol. 2007, 17, R18–R20. [Google Scholar] [CrossRef] [Green Version]
- Cagalinec, M.; Safiulina, D.; Liiv, M.; Liiv, J.; Choubey, V.; Wareski, P.; Veksler, V.; Kaasik, A. Principles of the mitochondrial fusion and fission cycle in neurons. J. Cell Sci. 2013, 126, 2187–2197. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Weaver, D.; Shirihai, O.; Hajnoczky, G. Mitochondrial ‘kiss-and-run’: Interplay between mitochondrial motility and fusion-fission dynamics. EMBO J. 2009, 28, 3074–3089. [Google Scholar] [CrossRef] [Green Version]
- Martz, D.; Lasek, R.J.; Brady, S.T.; Allen, R.D. Mitochondrial motility in axons: Membranous organelles may interact with the force generating system through multiple surface binding sites. Cell Motil. 1984, 4, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Vallee, R.B.; Bloom, G.S. Mechanisms of fast and slow axonal transport. Annu. Rev. Neurosci. 1991, 14, 59–92. [Google Scholar] [CrossRef] [PubMed]
- Ligon, L.A.; Steward, O. Role of microtubules and actin filaments in the movement of mitochondria in the axons and dendrites of cultured hippocampal neurons. J. Comp. Neurol. 2000, 427, 351–361. [Google Scholar] [CrossRef]
- Saxton, W.M.; Hollenbeck, P.J. The axonal transport of mitochondria. J. Cell Sci. 2012, 125, 2095–2104. [Google Scholar] [CrossRef] [Green Version]
- Schwarzer, C.; Barnikol-Watanabe, S.; Thinnes, F.P.; Hilschmann, N. Voltage-dependent anion-selective channel (VDAC) interacts with the dynein light chain Tctex1 and the heat-shock protein PBP74. Int. J. Biochem. Cell Biol. 2002, 34, 1059–1070. [Google Scholar] [CrossRef]
- Chang, D.T.; Honick, A.S.; Reynolds, I.J. Mitochondrial trafficking to synapses in cultured primary cortical neurons. J. Neurosci. 2006, 26, 7035–7045. [Google Scholar] [CrossRef]
- Rintoul, G.L.; Filiano, A.J.; Brocard, J.B.; Kress, G.J.; Reynolds, I.J. Glutamate decreases mitochondrial size and movement in primary forebrain neurons. J. Neurosci. 2003, 23, 7881–7888. [Google Scholar] [CrossRef] [Green Version]
- Brady, S.T.; Lasek, R.J.; Allen, R.D.; Yin, H.L.; Stossel, T.P. Gelsolin inhibition of fast axonal transport indicates a requirement for actin microfilaments. Nature 1984, 310, 56–58. [Google Scholar] [CrossRef] [PubMed]
- Infante, A.S.; Stein, M.S.; Zhai, Y.; Borisy, G.G.; Gundersen, G.G. Detyrosinated (Glu) microtubules are stabilized by an ATP-sensitive plus-end cap. J. Cell Sci. 2000, 113 (Pt 22), 3907–3919. [Google Scholar]
- Park, K.C.; Kim, S.W.; Jeon, J.Y.; Jo, A.R.; Choi, H.J.; Kim, J.; Lee, H.G.; Kim, Y.; Mills, G.B.; Noh, S.H.; et al. Survival of Cancer Stem-Like Cells Under Metabolic Stress via CaMK2alpha-mediated Upregulation of Sarco/Endoplasmic Reticulum Calcium ATPase Expression. Clin. Cancer Res. 2018, 24, 1677–1690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roman, I.; Figys, J.; Steurs, G.; Zizi, M. Direct measurement of VDAC-actin interaction by surface plasmon resonance. Biochim. Biophys. Acta 2006, 1758, 479–486. [Google Scholar] [CrossRef] [Green Version]
- DeLuca, M.; Hall, N.; Rice, R.; Kaplan, N.O. Creatine kinase isozymes in human tumors. Biochem. Biophys. Res. Commun. 1981, 99, 189–195. [Google Scholar] [CrossRef]
- Dang, C.V.; Semenza, G.L. Oncogenic alterations of metabolism. Trends Biochem. Sci. 1999, 24, 68–72. [Google Scholar] [CrossRef]
- Guzun, R.; Timohhina, N.; Tepp, K.; Monge, C.; Kaambre, T.; Sikk, P.; Kuznetsov, A.V.; Pison, C.; Saks, V. Regulation of respiration controlled by mitochondrial creatine kinase in permeabilized cardiac cells in situ. Importance of system level properties. Biochim. Biophys. Acta 2009, 1787, 1089–1105. [Google Scholar] [CrossRef]
- Heggeness, M.H.; Simon, M.; Singer, S.J. Association of mitochondria with microtubules in cultured cells. Proc. Natl. Acad. Sci. USA 1978, 75, 3863–3866. [Google Scholar] [CrossRef] [Green Version]
- Saetersdal, T.; Greve, G.; Dalen, H. Associations between beta-tubulin and mitochondria in adult isolated heart myocytes as shown by immunofluorescence and immunoelectron microscopy. Histochemistry 1990, 95, 1–10. [Google Scholar] [CrossRef]
- Bernier-Valentin, F.; Aunis, D.; Rousset, B. Evidence for tubulin-binding sites on cellular membranes: Plasma membranes, mitochondrial membranes, and secretory granule membranes. J. Cell Biol. 1983, 97, 209–216. [Google Scholar] [CrossRef]
- Carre, M.; Andre, N.; Carles, G.; Borghi, H.; Brichese, L.; Briand, C.; Braguer, D. Tubulin is an inherent component of mitochondrial membranes that interacts with the voltage-dependent anion channel. J. Biol. Chem. 2002, 277, 33664–33669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cicchillitti, L.; Penci, R.; Di Michele, M.; Filippetti, F.; Rotilio, D.; Donati, M.B.; Scambia, G.; Ferlini, C. Proteomic characterization of cytoskeletal and mitochondrial class III beta-tubulin. Mol. Cancer 2008, 7, 2070–2079. [Google Scholar] [CrossRef] [Green Version]
- Magri, A.; Reina, S.; De Pinto, V. VDAC1 as Pharmacological Target in Cancer and Neurodegeneration: Focus on Its Role in Apoptosis. Front. Chem. 2018, 6, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chae, Y.C.; Angelin, A.; Lisanti, S.; Kossenkov, A.V.; Speicher, K.D.; Wang, H.; Powers, J.F.; Tischler, A.S.; Pacak, K.; Fliedner, S.; et al. Landscape of the mitochondrial Hsp90 metabolome in tumours. Nat. Commun. 2013, 4, 2139. [Google Scholar] [CrossRef] [Green Version]
- Maldonado, E.N.; Sheldon, K.L.; DeHart, D.N.; Patnaik, J.; Manevich, Y.; Townsend, D.M.; Bezrukov, S.M.; Rostovtseva, T.K.; Lemasters, J.J. Voltage-dependent anion channels modulate mitochondrial metabolism in cancer cells: Regulation by free tubulin and erastin. J. Biol. Chem. 2013, 288, 11920–11929. [Google Scholar] [CrossRef] [Green Version]
- Gourlay, C.W.; Carpp, L.N.; Timpson, P.; Winder, S.J.; Ayscough, K.R. A role for the actin cytoskeleton in cell death and aging in yeast. J. Cell Biol. 2004, 164, 803–809. [Google Scholar] [CrossRef] [Green Version]
- Drubin, D.G.; Jones, H.D.; Wertman, K.F. Actin structure and function: Roles in mitochondrial organization and morphogenesis in budding yeast and identification of the phalloidin-binding site. Mol. Biol. Cell 1993, 4, 1277–1294. [Google Scholar] [CrossRef]
- Li, J.; Li, Q.; Xie, C.; Zhou, H.; Wang, Y.; Zhang, N.; Shao, H.; Chan, S.C.; Peng, X.; Lin, S.C.; et al. Beta-actin is required for mitochondria clustering and ROS generation in TNF-induced, caspase-independent cell death. J. Cell Sci. 2004, 117, 4673–4680. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Juvekar, A.; Lyssiotis, C.A.; Lien, E.C.; Albeck, J.G.; Oh, D.; Varma, G.; Hung, Y.P.; Ullas, S.; Lauring, J.; et al. Phosphoinositide 3-Kinase Regulates Glycolysis through Mobilization of Aldolase from the Actin Cytoskeleton. Cell 2016, 164, 433–446. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.S.; Tian, J.H.; Pan, P.Y.; Zald, P.; Li, C.; Deng, C.; Sheng, Z.H. Docking of axonal mitochondria by syntaphilin controls their mobility and affects short-term facilitation. Cell 2008, 132, 137–148. [Google Scholar] [CrossRef] [Green Version]
- Caino, M.C.; Seo, J.H.; Wang, Y.; Rivadeneira, D.B.; Gabrilovich, D.I.; Kim, E.T.; Weeraratna, A.T.; Languino, L.R.; Altieri, D.C. Syntaphilin controls a mitochondrial rheostat for proliferation-motility decisions in cancer. J. Clin. Invest. 2017, 127, 3755–3769. [Google Scholar] [CrossRef] [Green Version]
- Pongrakhananon, V.; Wattanathamsan, O.; Takeichi, M.; Chetprayoon, P.; Chanvorachote, P. Loss of CAMSAP3 promotes EMT via the modification of microtubule-Akt machinery. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [Green Version]
- Ketschek, A.; Gallo, G. Nerve growth factor induces axonal filopodia through localized microdomains of phosphoinositide 3-kinase activity that drive the formation of cytoskeletal precursors to filopodia. J. Neurosci. 2010, 30, 12185–12197. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, J.; Yu, M.; Xie, Y.; Huang, Y.; Wolff, D.W.; Abel, P.W.; Tu, Y. Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene 2013, 32, 4814–4824. [Google Scholar] [CrossRef] [PubMed]
- Shankar, J.; Nabi, I.R. Actin cytoskeleton regulation of epithelial mesenchymal transition in metastatic cancer cells. PLoS ONE 2015, 10, e0119954. [Google Scholar] [CrossRef] [Green Version]
- Tojkander, S.; Gateva, G.; Lappalainen, P. Actin stress fibers--assembly, dynamics and biological roles. J. Cell Sci. 2012, 125, 1855–1864. [Google Scholar] [CrossRef] [Green Version]
- Taulet, N.; Delorme-Walker, V.D.; DerMardirossian, C. Reactive oxygen species regulate protrusion efficiency by controlling actin dynamics. PLoS ONE 2012, 7, e41342. [Google Scholar] [CrossRef]
- Schuler, M.H.; Lewandowska, A.; Caprio, G.D.; Skillern, W.; Upadhyayula, S.; Kirchhausen, T.; Shaw, J.M.; Cunniff, B. Miro1-mediated mitochondrial positioning shapes intracellular energy gradients required for cell migration. Mol. Biol. Cell 2017, 28, 2159–2169. [Google Scholar] [CrossRef]
- Caino, M.C.; Ghosh, J.C.; Chae, Y.C.; Vaira, V.; Rivadeneira, D.B.; Faversani, A.; Rampini, P.; Kossenkov, A.V.; Aird, K.M.; Zhang, R.; et al. PI3K therapy reprograms mitochondrial trafficking to fuel tumor cell invasion. Proc. Natl. Acad. Sci. USA 2015, 112, 8638–8643. [Google Scholar] [CrossRef] [Green Version]
- Caino, M.C.; Chae, Y.C.; Vaira, V.; Ferrero, S.; Nosotti, M.; Martin, N.M.; Weeraratna, A.; O’Connell, M.; Jernigan, D.; Fatatis, A.; et al. Metabolic stress regulates cytoskeletal dynamics and metastasis of cancer cells. J. Clin. Invest. 2013, 123, 2907–2920. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Lu, S. Transforming growth factor-beta1-induced epithelial to mesenchymal transition increases mitochondrial content in the A549 non-small cell lung cancer cell line. Mol. Med. Rep. 2015, 11, 417–421. [Google Scholar] [CrossRef]
- Kim, S.Y. Cancer Energy Metabolism: Shutting Power off Cancer Factory. Biomol. Ther. 2018, 26, 39–44. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.Y. Cancer Metabolism: A Hope for Curing Cancer. Biomol. Ther. 2018, 26, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Ralff, M.D.; Lulla, A.R.; Wagner, J.; El-Deiry, W.S. ONC201: A new treatment option being tested clinically for recurrent glioblastoma. Transl Cancer Res. 2017, 6, S1239–S1243. [Google Scholar] [CrossRef] [PubMed]
- Pich, S.; Bach, D.; Briones, P.; Liesa, M.; Camps, M.; Testar, X.; Palacin, M.; Zorzano, A. The Charcot-Marie-Tooth type 2A gene product, Mfn2, up-regulates fuel oxidation through expression of OXPHOS system. Hum. Mol. Genet. 2005, 14, 1405–1415. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, J.C.B.; Campos, J.C.; Qvit, N.; Qi, X.; Bozi, L.H.M.; Bechara, L.R.G.; Lima, V.M.; Queliconi, B.B.; Disatnik, M.H.; Dourado, P.M.M.; et al. A selective inhibitor of mitofusin 1-betaIIPKC association improves heart failure outcome in rats. Nat. Commun. 2019, 10, 329. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, E.; Cioffi, M.; Sancho, P.; Sanchez-Ripoll, Y.; Trabulo, S.M.; Dorado, J.; Balic, A.; Hidalgo, M.; Heeschen, C. Metformin targets the metabolic achilles heel of human pancreatic cancer stem cells. PLoS ONE 2013, 8, e76518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. Elife 2014, 3, e02242. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Lee, J.H.; Koh, I.; Shim, J.K.; Park, J.; Jeon, J.Y.; Yun, M.; Kim, S.H.; Yook, J.I.; Kim, E.H.; et al. Inhibiting stemness and invasive properties of glioblastoma tumorsphere by combined treatment with temozolomide and a newly designed biguanide (HL156A). Oncotarget 2016, 7, 65643–65659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, T.G.; Jeong, Y.S.; Kim, S.A.; Ahn, S.G. New metformin derivative HL156A prevents oral cancer progression by inhibiting the insulin-like growth factor/AKT/mammalian target of rapamycin pathways. Cancer Sci. 2018, 109, 699–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koyama, T.; Oike, M.; Ito, Y. Involvement of Rho-kinase and tyrosine kinase in hypotonic stress-induced ATP release in bovine aortic endothelial cells. J. Physiol. 2001, 532, 759–769. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Inhibitors of oxidative phosphorylation (OXPHOS). The OXPHOS metabolic pathway generates ATP by transporting electrons through a series of transmembrane complexes in the mitochondrial inner membrane, known as the electron transport chain (ETC). Electrons flow through complex I, complex II, coenzyme Q (Q), complex III, cytochrome c (C), and complex IV, with O2 acting as the terminal electron acceptor. Therapeutic agents that are OXPHOS inhibitors are shown in red boxes. 3-NPA: 3-nitropropionic acid; TTFA: 2-thenoyltrifluoroacetone.
Figure 1.
Inhibitors of oxidative phosphorylation (OXPHOS). The OXPHOS metabolic pathway generates ATP by transporting electrons through a series of transmembrane complexes in the mitochondrial inner membrane, known as the electron transport chain (ETC). Electrons flow through complex I, complex II, coenzyme Q (Q), complex III, cytochrome c (C), and complex IV, with O2 acting as the terminal electron acceptor. Therapeutic agents that are OXPHOS inhibitors are shown in red boxes. 3-NPA: 3-nitropropionic acid; TTFA: 2-thenoyltrifluoroacetone.

Figure 2.
A simplified scheme of mitochondrial dynamics in cancer cells and cancer stem cells (CSCs). In cancer cells, mitochondria are usually localized in the nuclear periphery and characterized by sphere-like, fragmented, and punctate morphology. Correspondingly, mitochondrial fission is high, whereas mitochondrial biogenesis is low. Cancer cells generally rely on glycolysis as the major energy source and have low levels of OXPHOS, ATP production, and fatty acid oxidation levels. In CSCs, mitochondria exhibit more elongated and enlarged tubular morphology. Correspondingly, mitochondrial fusion and biogenesis increase with the densely packed mitochondria. Comparably, CSCs have higher OXPHOS, ATP production, and fatty acid oxidation levels. Blue: nucleus; red lines: mitochondria.
Figure 2.
A simplified scheme of mitochondrial dynamics in cancer cells and cancer stem cells (CSCs). In cancer cells, mitochondria are usually localized in the nuclear periphery and characterized by sphere-like, fragmented, and punctate morphology. Correspondingly, mitochondrial fission is high, whereas mitochondrial biogenesis is low. Cancer cells generally rely on glycolysis as the major energy source and have low levels of OXPHOS, ATP production, and fatty acid oxidation levels. In CSCs, mitochondria exhibit more elongated and enlarged tubular morphology. Correspondingly, mitochondrial fusion and biogenesis increase with the densely packed mitochondria. Comparably, CSCs have higher OXPHOS, ATP production, and fatty acid oxidation levels. Blue: nucleus; red lines: mitochondria.

Figure 3.
Schematic overview of therapeutic targeting of cancer stem cells (CSCs). Therapeutic approaches (indicated in red font) against CSCs may include reduction of metabolic plasticity, pharmacological targeting of mitochondrial dynamics and morphology, and cytoskeleton-mitochondria interaction-targeted drugs. For detailed mechanisms see text.
Figure 3.
Schematic overview of therapeutic targeting of cancer stem cells (CSCs). Therapeutic approaches (indicated in red font) against CSCs may include reduction of metabolic plasticity, pharmacological targeting of mitochondrial dynamics and morphology, and cytoskeleton-mitochondria interaction-targeted drugs. For detailed mechanisms see text.


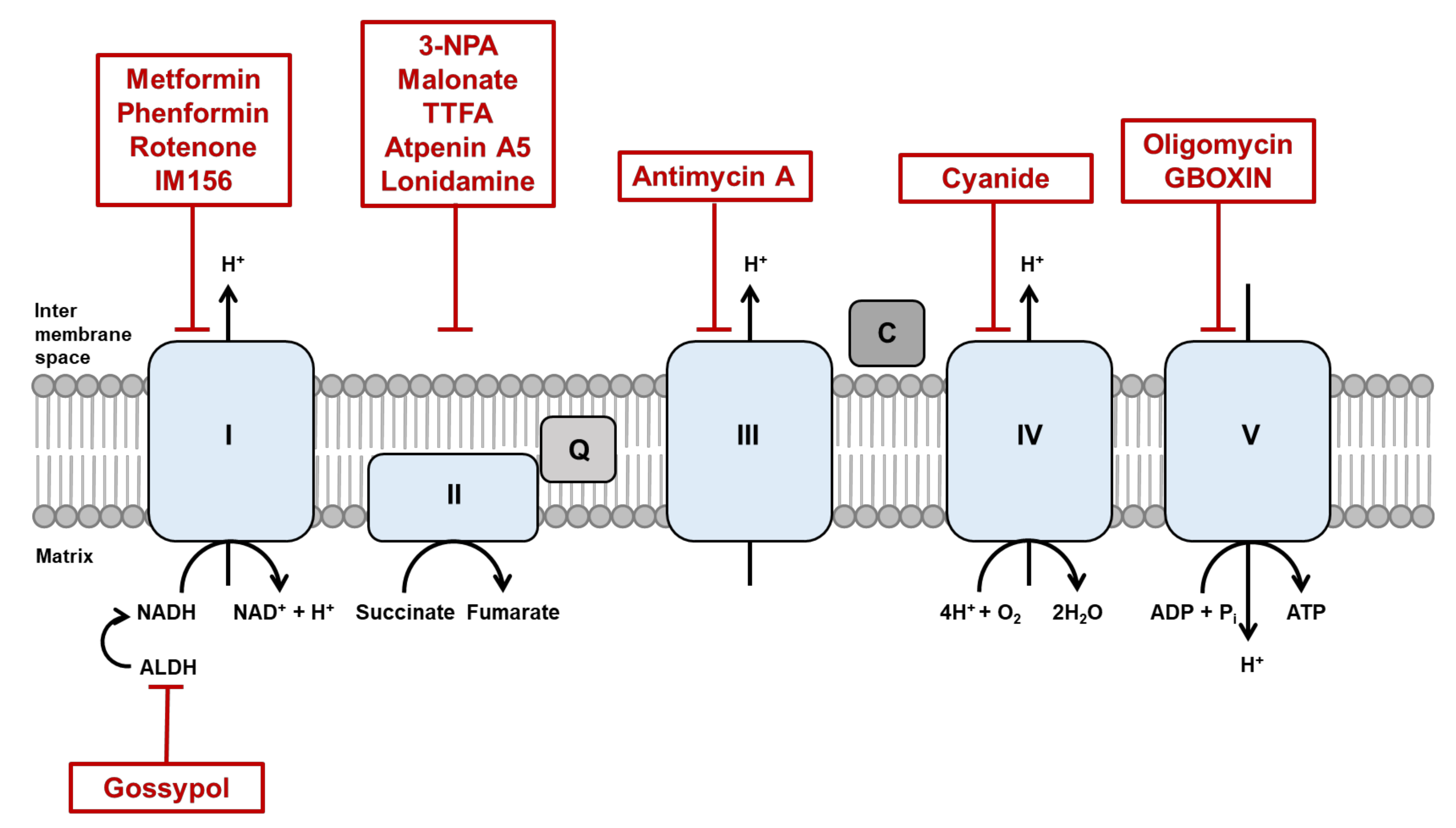{kind=link}
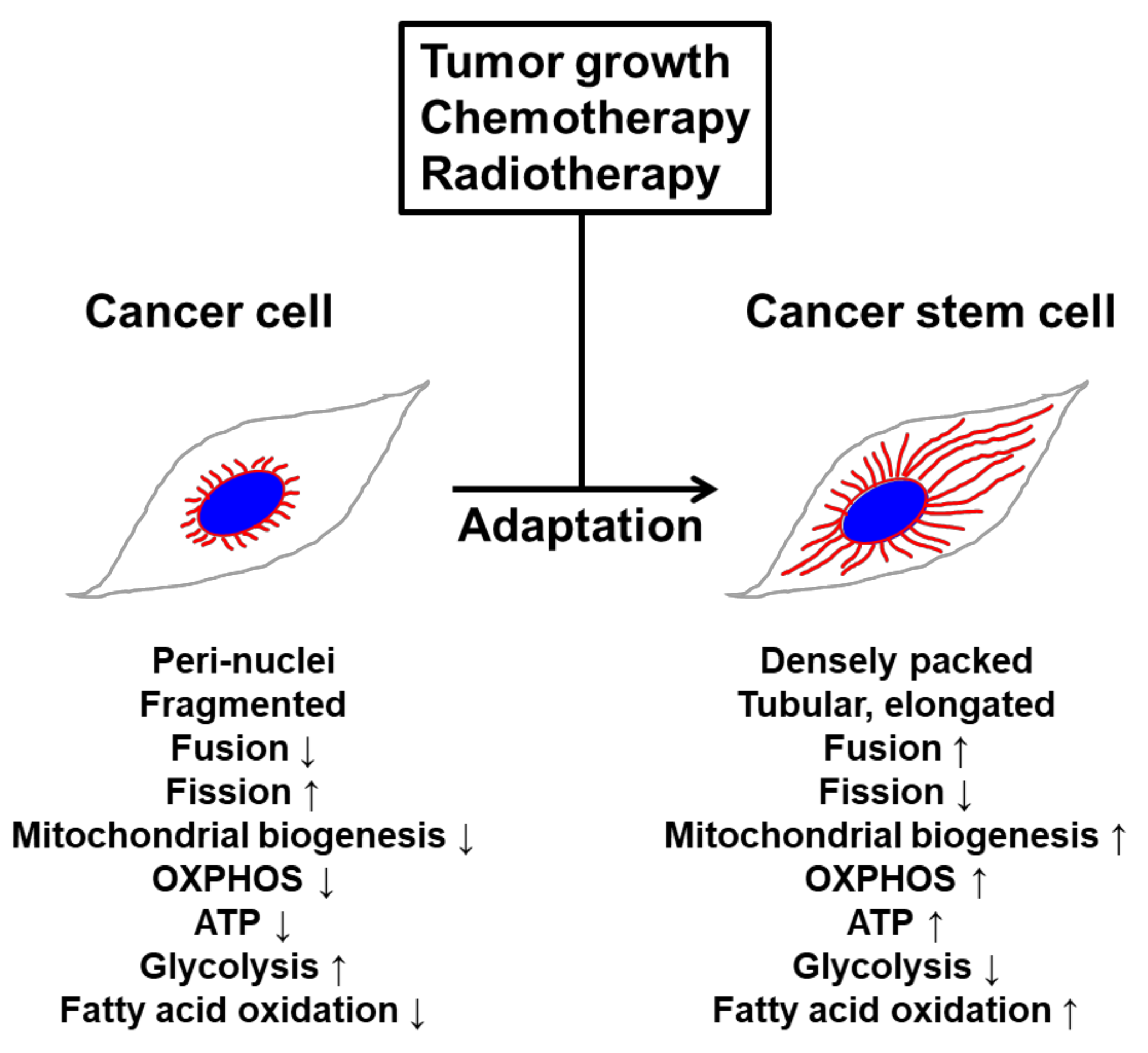{kind=link}
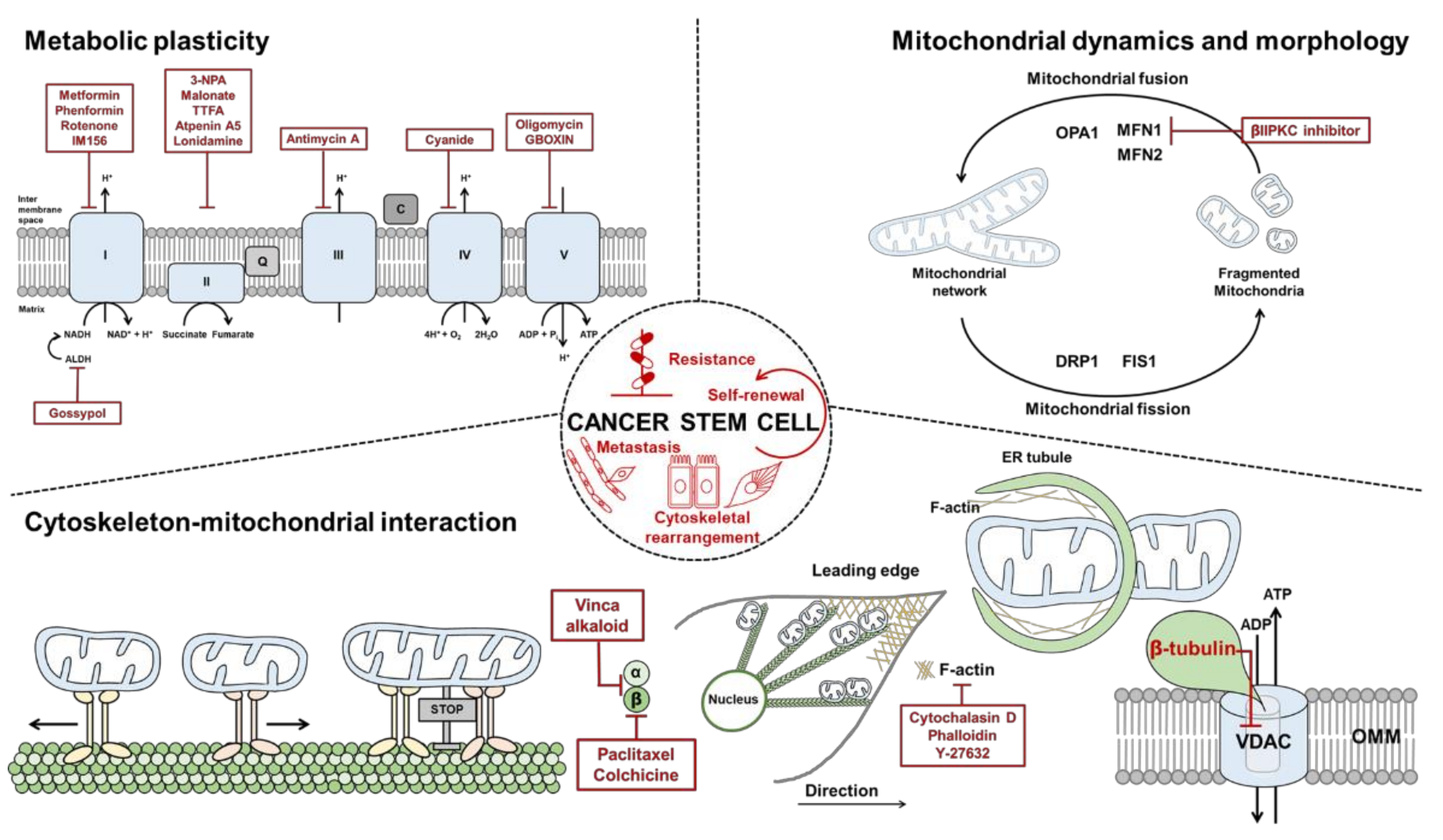{kind=link}
Table 1.
List of Inhibitors under study in vivo or in the clinic as anticancer therapeutics.
| Categories | Compound | Target | Conditions | Clinical Trials | Clinical Trials Identifier/Ref. |
|---|---|---|---|---|---|
| Mitochondrial dynamics and function | βIIPKC inhibitor | MFN1 | Heart failure | Experimental | [156,157] |
| ONC201 | Mitochondrial respiration | Multiple cancers | Several trials in process | [171] | |
| NADH production | Gossypol | Aldehyde dehydrogenase | Multiple cancers | Several trials in process | [158] |
| Microtubule | Paclitaxel | β-Tubulin subunit | Multiple cancers | Several trials in process | [159,160] |
| Colchicine | β-Tubulin subunit | Multiple cancers | Several trials in process | [161,162] | |
| Vinca alkaloid | Tubulin dimer | Multiple cancers | Several trials in process | [163,164] | |
| Actin | Cytochalasin D | Actin monomer | Infertility | Preclinical | NCT03677492 |
| Phalloidin | F-actin | Depression | Preclinical recruiting | NCT04137458 | |
| Y-27632 | Rho-associated kinase | Hippocampal neurons | Preclinical | [165] | |
| ETC | Metformin | Complex I | Multiple cancers | Several trials in progress | NCT03477162 |
| Phenformin | Complex I | Diabetes mellitus, type 2 | Preclinical completed | NCT02475499 | |
| Rotenone | Complex I | Parkinson’s disease | Preclinical enrolling | NCT04287543 | |
| IM156 | Complex I | Advanced solid tumor and lymphoma | Phase I active | NCT03272256 | |
| 3-NPA | Complex II | Parkinson’s disease | Preclinical | [166] | |
| Malonate | Complex II | Parkinson’s disease | Phase I terminated | NCT01476085 | |
| TTFA | Complex II | Cancer | Experimental | [167] | |
| Atpenin A5 | Complex II | Cardiac ischemia-reperfusion injury | Preclinical | [168] | |
| Lonidamine | Complex II | Symptomatic benign prostatic hyperplasia, enlarged prostate | Phase 2, 3 terminated | NCT00237536, NCT00435448 | |
| Antimycin A | Complex III | Lung cancer | Experimental | [169] | |
| Cyanide | Complex IV | Malignant glioma | Phase 2 recruiting | NCT00075387 | |
| Oligomycin | Complex V | Cancer cachexia, transthyretin amyloidosis | Preclinical recruiting | NCT03144128, NCT03328338 | |
| GBOXIN | Complex V | Glioblastoma | Preclinical | [170] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kim, J.; Cheong, J.-H. Role of Mitochondria-Cytoskeleton Interactions in the Regulation of Mitochondrial Structure and Function in Cancer Stem Cells. Cells 2020, 9, 1691. https://doi.org/10.3390/cells9071691
AMA Style
Kim J, Cheong J-H. Role of Mitochondria-Cytoskeleton Interactions in the Regulation of Mitochondrial Structure and Function in Cancer Stem Cells. Cells. 2020; 9(7):1691. https://doi.org/10.3390/cells9071691
Chicago/Turabian StyleKim, Jungmin, and Jae-Ho Cheong. 2020. "Role of Mitochondria-Cytoskeleton Interactions in the Regulation of Mitochondrial Structure and Function in Cancer Stem Cells" Cells 9, no. 7: 1691. https://doi.org/10.3390/cells9071691
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.