
Low Plasma Citrate Levels and Specific Transcriptional Signatures Associated with Quiescence of CD34+ Progenitors Predict Azacitidine Therapy Failure in MDS/AML Patients

 , , , , , ,
, , , , , ,
Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Patients
3. Results
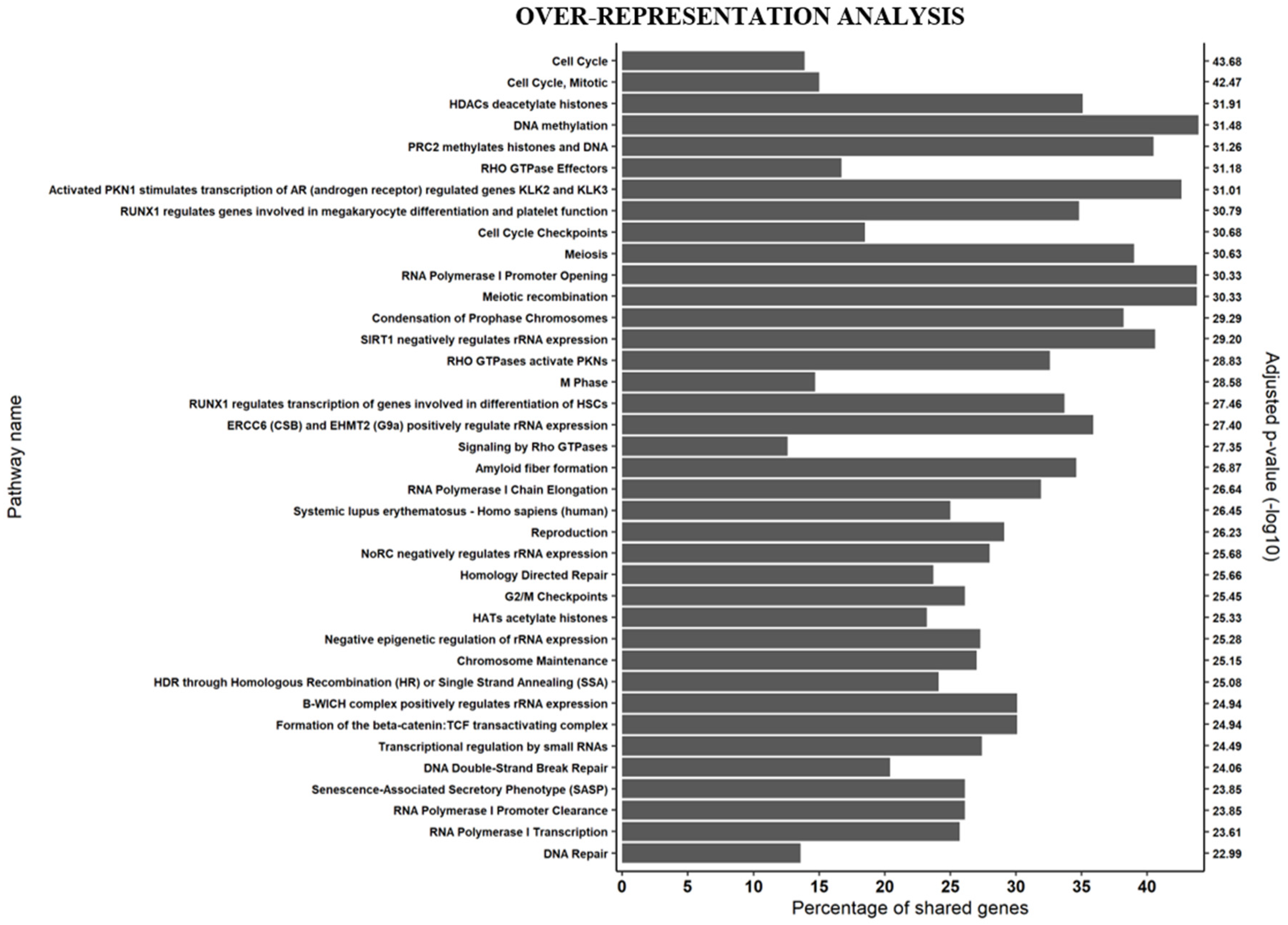
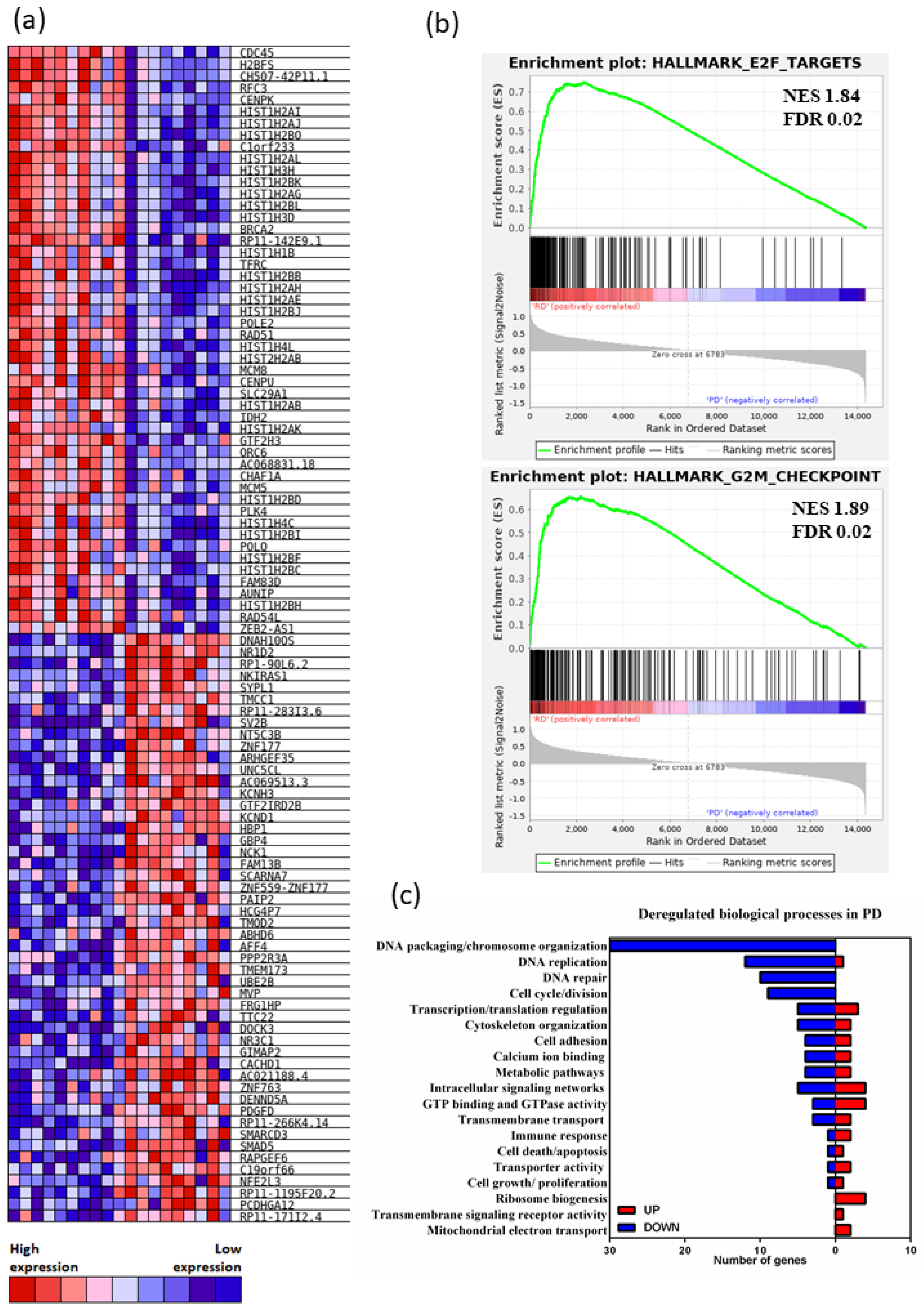
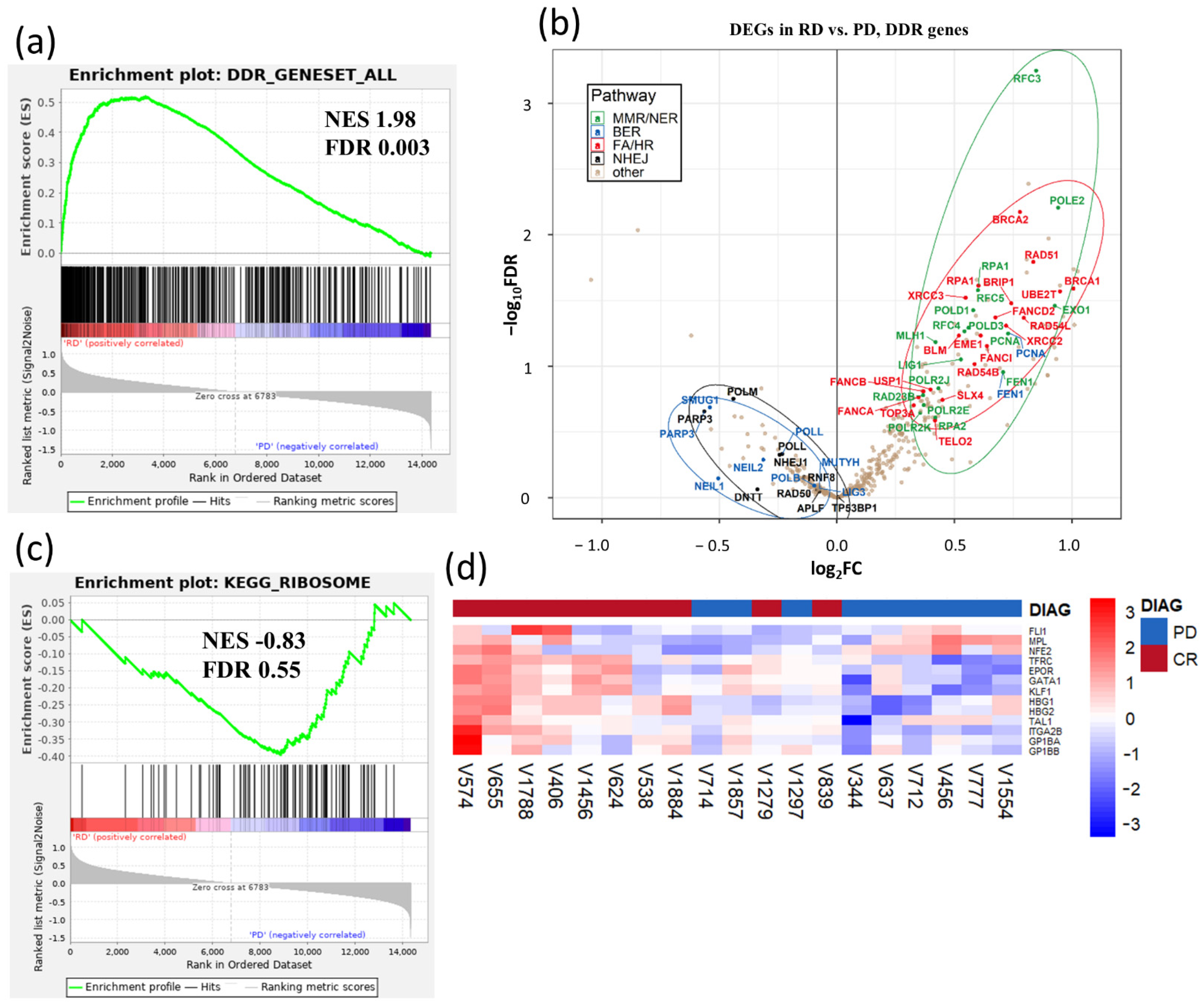
3.1. Gene Expression Signatures Suggest Non-Cycling Status and Diminished Differentiation of CD34+ HSPCs in Patients with AZA Treatment Failure
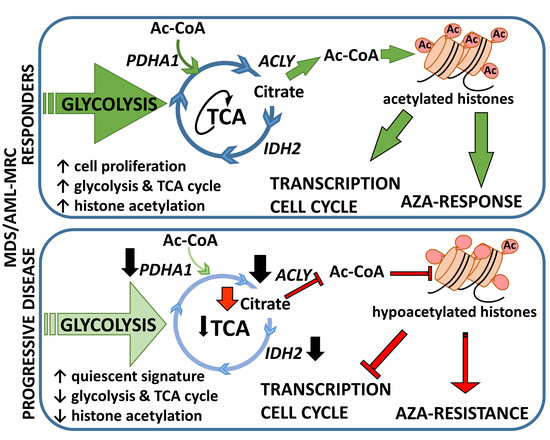
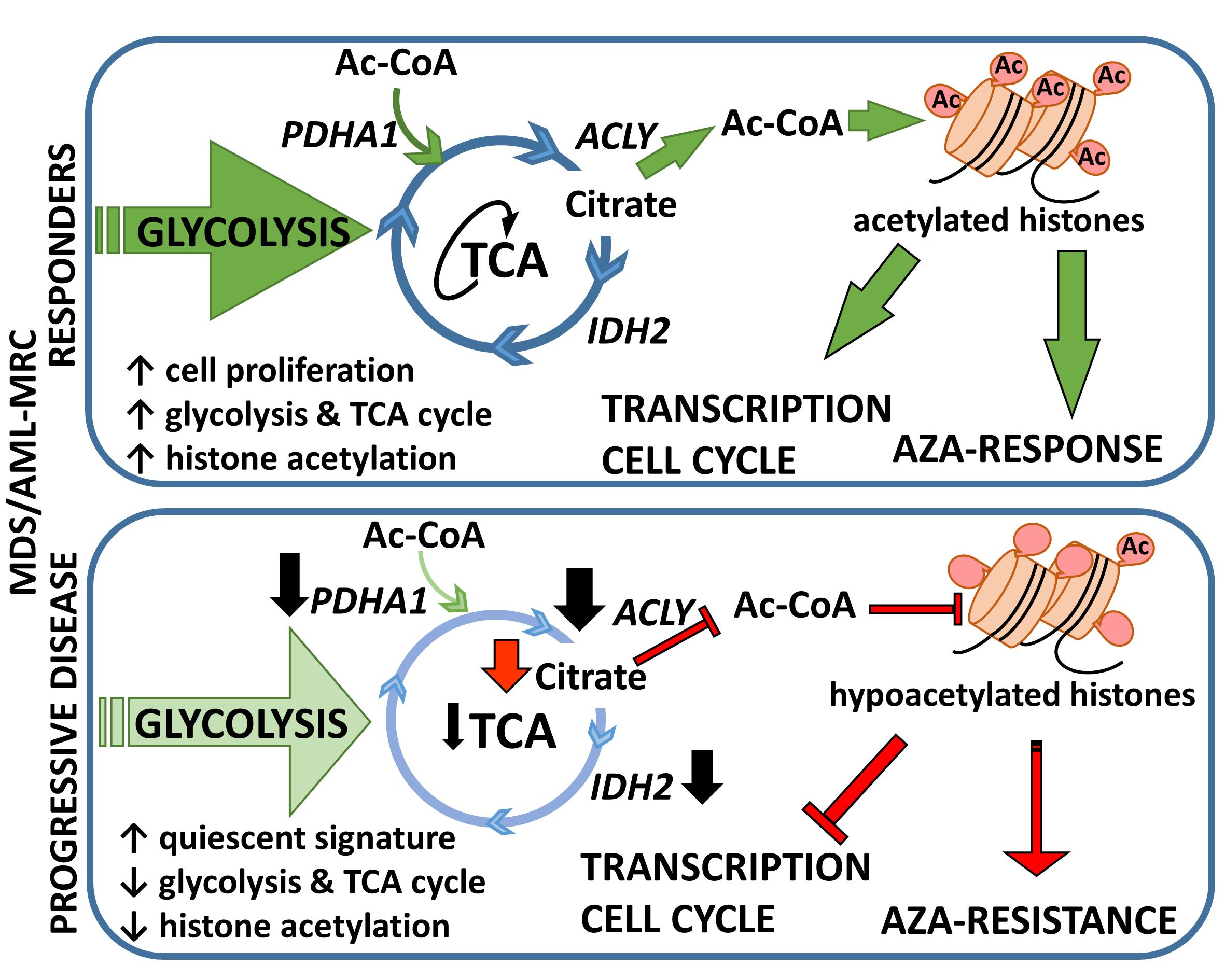
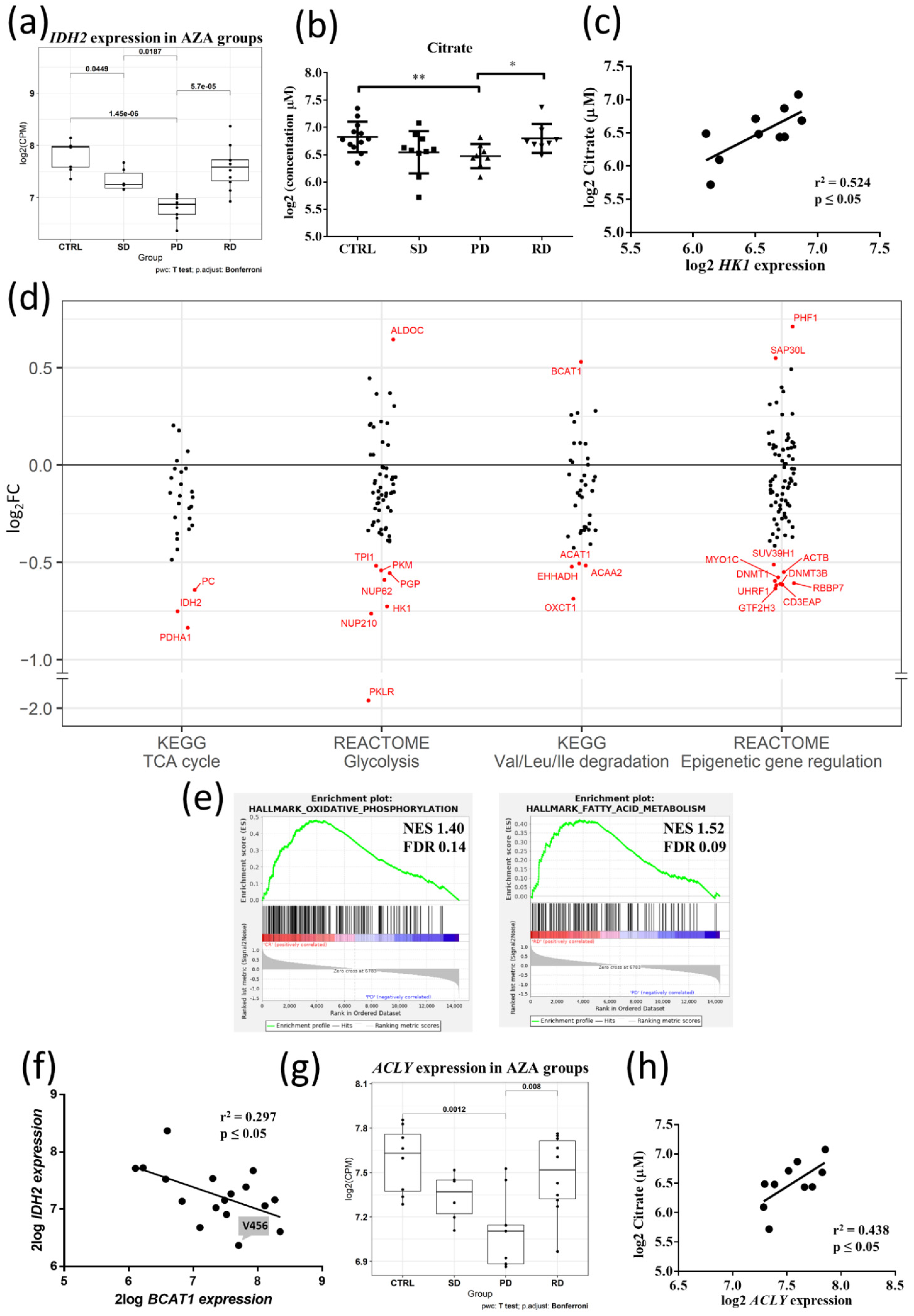
3.2. Metabolic Signature Differences between AZA Pre-Treatment RD and PD Patients
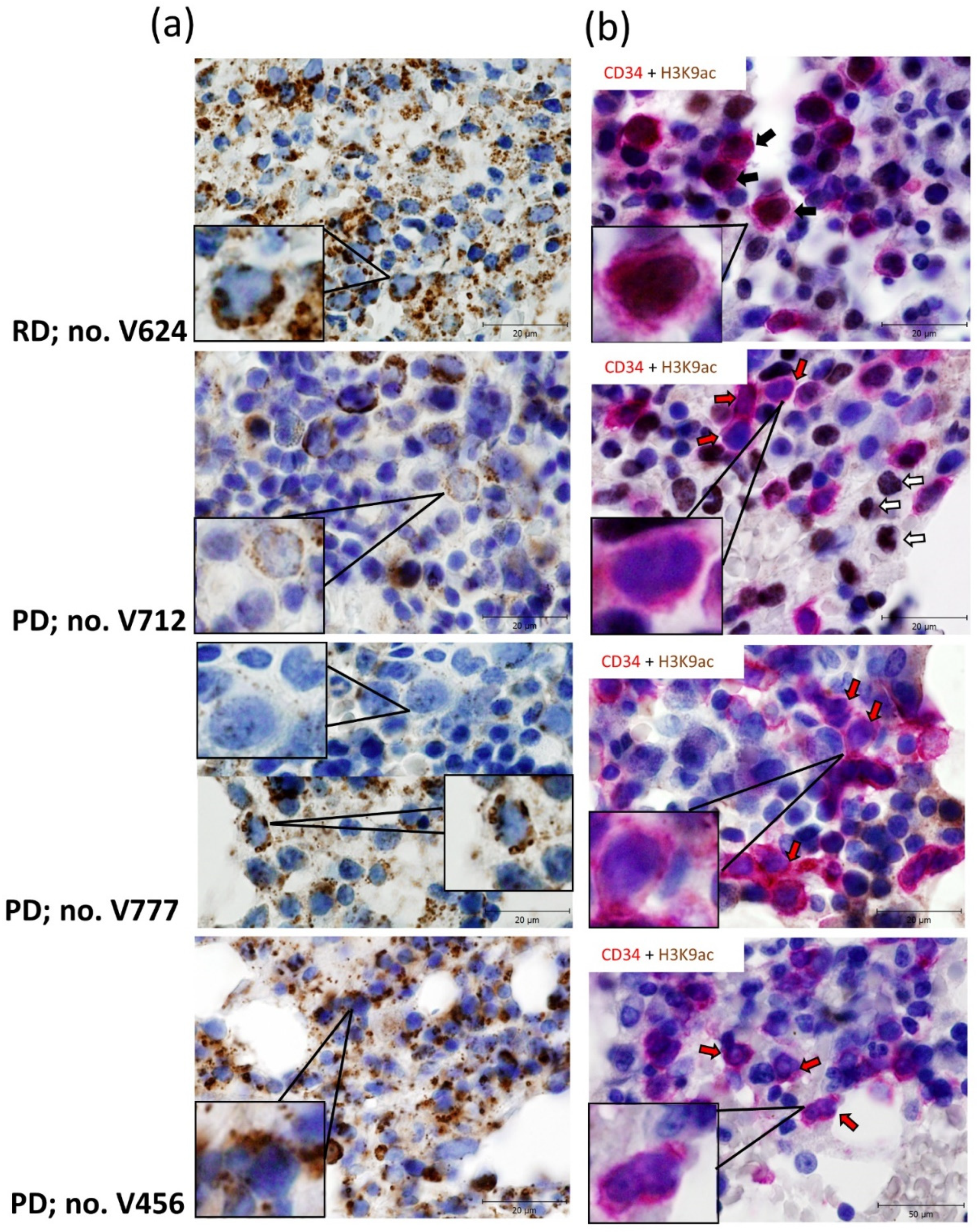
3.3. Relationship between Gene Expression Signatures and Immunohistochemistry (IHC) of IDH2 and Acetylated Histone H3 Lysine 9

4. Discussion
5. Materials and Methods
5.1. CD34+ Cells Isolation and RNA Sequencing
5.2. Mutational Analysis
5.3. Targeted Metabolic Analysis
5.4. IHC of Patients’ Samples
5.5. Bioinformatics and Statistical Analyses
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Appendix B
Appendix C
References
- Silverman, L.R.; Demakos, E.P.; Peterson, B.L.; Kornblith, A.B.; Holland, J.C.; Odchimar-Reissig, R.; Stone, R.M.; Nelson, D.; Powell, B.L.; DeCastro, C.M.; et al. Randomized Controlled Trial of Azacitidine in Patients With the Myelodysplastic Syndrome: A Study of the Cancer and Leukemia Group B. J. Clin. Oncol. 2002, 20, 2429–2440. [Google Scholar] [CrossRef]
- Silverman, L.R.; McKenzie, D.R.; Peterson, B.L.; Holland, J.F.; Backstrom, J.T.; Beach, C.L.; Larson, R.A. Further Analysis of Trials With Azacitidine in Patients With Myelodysplastic Syndrome: Studies 8421, 8921, and 9221 by the Cancer and Leukemia Group B. J. Clin. Oncol. 2006, 24, 3895–3903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fenaux, P.; Mufti, G.J.; Hellstrom-Lindberg, E.; Santini, V.; Finelli, C.; Giagounidis, A.; Schoch, R.; Gattermann, N.; Sanz, G.; List, A.; et al. Efficacy of Azacitidine Compared with That of Conventional Care Regimens in the Treatment of Higher-Risk Myelodysplastic Syndromes: A Randomised, Open-Label, Phase III Study. Lancet Oncol. 2009, 10, 223–232. [Google Scholar] [CrossRef] [Green Version]
- Platzbecker, U. Treatment of MDS. Blood 2019, 133, 1096–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santini, V.; Prebet, T.; Fenaux, P.; Gattermann, N.; Nilsson, L.; Pfeilstöcker, M.; Vyas, P.; List, A.F. Minimizing Risk of Hypomethylating Agent Failure in Patients with Higher-Risk MDS and Practical Management Recommendations. Leuk. Res. 2014, 38, 1381–1391. [Google Scholar] [CrossRef] [Green Version]
- Sekeres, M.A.; Cutler, C. How We Treat Higher-Risk Myelodysplastic Syndromes. Blood 2014, 123, 829–836. [Google Scholar] [CrossRef]
- Santini, V. How I Treat MDS after Hypomethylating Agent Failure. Blood 2019, 133, 521–529. [Google Scholar] [CrossRef] [Green Version]
- Itzykson, R.; Thépot, S.; Quesnel, B.; Dreyfus, F.; Beyne-Rauzy, O.; Turlure, P.; Vey, N.; Recher, C.; Dartigeas, C.; Legros, L.; et al. Prognostic Factors for Response and Overall Survival in 282 Patients with Higher-Risk Myelodysplastic Syndromes Treated with Azacitidine. Blood 2011, 117, 403–411. [Google Scholar] [CrossRef] [Green Version]
- Döhner, H.; Dolnik, A.; Tang, L.; Seymour, J.F.; Minden, M.D.; Stone, R.M.; del Castillo, T.B.; Al-Ali, H.K.; Santini, V.; Vyas, P.; et al. Cytogenetics and Gene Mutations Influence Survival in Older Patients with Acute Myeloid Leukemia Treated with Azacitidine or Conventional Care. Leukemia 2018, 32, 2546–2557. [Google Scholar] [CrossRef]
- Nazha, A.; Sekeres, M.A.; Bejar, R.; Rauh, M.J.; Othus, M.; Komrokji, R.S.; Barnard, J.; Hilton, C.B.; Kerr, C.M.; Steensma, D.P.; et al. Genomic Biomarkers to Predict Resistance to Hypomethylating Agents in Patients With Myelodysplastic Syndromes Using Artificial Intelligence. JCO Precis. Oncol. 2019, 3, 1–11. [Google Scholar] [CrossRef]
- Bejar, R.; Lord, A.; Stevenson, K.; Bar-Natan, M.; Pérez-Ladaga, A.; Zaneveld, J.; Wang, H.; Caughey, B.; Stojanov, P.; Getz, G.; et al. TET2 Mutations Predict Response to Hypomethylating Agents in Myelodysplastic Syndrome Patients. Blood 2014, 124, 2705–2712. [Google Scholar] [CrossRef]
- Traina, F.; Visconte, V.; Elson, P.; Tabarroki, A.; Jankowska, A.M.; Hasrouni, E.; Sugimoto, Y.; Szpurka, H.; Makishima, H.; O’Keefe, C.L.; et al. Impact of Molecular Mutations on Treatment Response to DNMT Inhibitors in Myelodysplasia and Related Neoplasms. Leukemia 2014, 28, 78–87. [Google Scholar] [CrossRef]
- Al-Issa, K.; Mikkael, A.S.; Nielsen, A.D.; Jha, B.; Przychodzen, B.; Aly, M.; Carraway, H.E.; Advani, A.S.; Patel, B.; Clemente, M.J.; et al. TP53 Mutations and Outcome in Patients with Myelodysplastic Syndromes (MDS). Blood 2016, 128, 4336. [Google Scholar] [CrossRef]
- Takahashi, K.; Patel, K.; Bueso-Ramos, C.; Zhang, J.; Gumbs, C.; Jabbour, E.; Kadia, T.; Andreff, M.; Konopleva, M.; DiNardo, C.; et al. Clinical Implications of TP53 Mutations in Myelodysplastic Syndromes Treated with Hypomethylating Agents. Oncotarget 2016, 7, 14172–14187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiNardo, C.D.; Patel, K.P.; Garcia-Manero, G.; Luthra, R.; Pierce, S.; Borthakur, G.; Jabbour, E.; Kadia, T.; Pemmaraju, N.; Konopleva, M.; et al. Lack of Association of IDH1, IDH2 and DNMT3A Mutations with Outcome in Older Patients with Acute Myeloid Leukemia Treated with Hypomethylating Agents. Leuk. Lymphoma 2014, 55, 1925–1929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voso, M.T.; Santini, V.; Fabiani, E.; Fianchi, L.; Criscuolo, M.; Falconi, G.; Guidi, F.; Hohaus, S.; Leone, G. Why Methylation Is Not a Marker Predictive of Response to Hypomethylating Agents. Haematologica 2014, 99, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Treppendahl, M.B.; Kristensen, L.S.; Grønbæk, K. Predicting Response to Epigenetic Therapy. J. Clin. Investig. 2014, 124, 47–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Li, Y.; Lv, N.; Li, Y.; Wang, L.; Yu, L. Predictors of Clinical Responses to Hypomethylating Agents in Acute Myeloid Leukemia or Myelodysplastic Syndromes. Ann. Hematol. 2018, 97, 2025–2038. [Google Scholar] [CrossRef] [PubMed]
- Oellerich, T.; Schneider, C.; Thomas, D.; Knecht, K.M.; Buzovetsky, O.; Kaderali, L.; Schliemann, C.; Bohnenberger, H.; Angenendt, L.; Hartmann, W.; et al. Selective Inactivation of Hypomethylating Agents by SAMHD1 Provides a Rationale for Therapeutic Stratification in AML. Nat. Commun. 2019, 10, 3475. [Google Scholar] [CrossRef]
- Gu, X.; Tohme, R.; Tomlinson, B.; Sakre, N.; Hasipek, M.; Durkin, L.; Schuerger, C.; Grabowski, D.; Zidan, A.M.; Radivoyevitch, T.; et al. Decitabine- and 5-Azacytidine Resistance Emerges from Adaptive Responses of the Pyrimidine Metabolism Network. Leukemia 2020, 35, 1023–1036. [Google Scholar] [CrossRef]
- Diesch, J.; Zwick, A.; Garz, A.-K.; Palau, A.; Buschbeck, M.; Götze, K.S. A Clinical-Molecular Update on Azanucleoside-Based Therapy for the Treatment of Hematologic Cancers. Clin. Epigenetics 2016, 8, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daver, N.; Boddu, P.; Garcia-Manero, G.; Yadav, S.S.; Sharma, P.; Allison, J.; Kantarjian, H. Hypomethylating Agents in Combination with Immune Checkpoint Inhibitors in Acute Myeloid Leukemia and Myelodysplastic Syndromes. Leukemia 2018, 32, 1094–1105. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Pratz, K.; Pullarkat, V.; Jonas, B.A.; Arellano, M.; Becker, P.S.; Frankfurt, O.; Konopleva, M.; Wei, A.H.; Kantarjian, H.M.; et al. Venetoclax Combined with Decitabine or Azacitidine in Treatment-Naive, Elderly Patients with Acute Myeloid Leukemia. Blood 2019, 133, 7–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Manero, G.; Kazmierczak, M.; Fong, C.Y.; Montesinos, P.; Venditti, A.; Mappa, S.; Spezia, R.; Adès, L. A Phase 3 Randomized Study (PRIMULA) of the Epigenetic Combination of Pracinostat, a Pan-Histone Deacetylase (HDAC) Inhibitor, with Azacitidine (AZA) in Patients with Newly Diagnosed Acute Myeloid Leukemia (AML) Unfit for Standard Intensive Chemotherapy (IC). Blood 2019, 134, 2652. [Google Scholar]
- Pericole, F.V.; Lazarini, M.; de Paiva, L.B.; da Silva Santos Duarte, A.; Vieira Ferro, K.P.; Niemann, F.S.; Roversi, F.M.; Olalla Saad, S.T. BRD4 Inhibition Enhances Azacitidine Efficacy in Acute Myeloid Leukemia and Myelodysplastic Syndromes. Front. Oncol. 2019, 9, 16. [Google Scholar] [CrossRef] [Green Version]
- Yalniz, F.F.; Berdeja, J.G.; Maris, M.B.; Lyons, R.M.; Reeves, J.A.; Essell, J.H.; Patel, P.; Sekeres, M.; Hughes, A.; Mappa, S.; et al. A Phase II Study of Addition of Pracinostat to a Hypomethylating Agent in Patients with Myelodysplastic Syndromes Who Have Not Responded to Previous Hypomethylating Agent Therapy. Br. J. Haematol. 2020, 188, 404–412. [Google Scholar] [CrossRef]
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting Epigenetic Regulators for Cancer Therapy: Mechanisms and Advances in Clinical Trials. Signal. Transduct. Target. Ther. 2019, 4, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, D.; Rampal, R.; Mascarenhas, J. Clinical Developments in Epigenetic-Directed Therapies in Acute Myeloid Leukemia. Blood Adv. 2020, 4, 970–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belickova, M.; Merkerova Dostálová, M.D.; Votavova, H.; Valka, J.; Vesela, J.; Pejsova, B.; Hajkova, H.; Klema, J.; Cermak, J.; Jonasova, A. Up-Regulation of Ribosomal Genes Is Associated with a Poor Response to Azacitidine in Myelodysplasia and Related Neoplasms. Int. J. Hematol. 2016, 104, 566–573. [Google Scholar] [CrossRef]
- Lübbert, M.; Ihorst, G.; Sander, P.N.; Bogatyreva, L.; Becker, H.; Wijermans, P.W.; Suciu, S.; Bissé, E.; Claus, R. Elevated Fetal Haemoglobin Is a Predictor of Better Outcome in MDS/AML Patients Receiving 5-Aza-2′-Deoxycytidine (Decitabine). Br. J. Haematol. 2017, 176, 609–617. [Google Scholar] [CrossRef]
- Unnikrishnan, A.; Papaemmanuil, E.; Beck, D.; Deshpande, N.P.; Verma, A.; Kumari, A.; Woll, P.S.; Richards, L.A.; Knezevic, K.; Chandrakanthan, V.; et al. Integrative Genomics Identifies the Molecular Basis of Resistance to Azacitidine Therapy in Myelodysplastic Syndromes. Cell Rep. 2017, 20, 572–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruber, E.; Franich, R.L.; Shortt, J.; Johnstone, R.W.; Kats, L.M. Distinct and Overlapping Mechanisms of Resistance to Azacytidine and Guadecitabine in Acute Myeloid Leukemia. Leukemia 2020, 34, 3388–3392. [Google Scholar] [CrossRef] [PubMed]
- Herwig, R.; Hardt, C.; Lienhard, M.; Kamburov, A. Analyzing and Interpreting Genome Data at the Network Level with ConsensusPathDB. Nat. Protoc. 2016, 11, 1889–1907. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Penneroux, J.; Dal Bello, R.; Massé, A.; Quentin, S.; Unnikrishnan, A.; Hernandez, L.; Raffoux, E.; Ben Abdelali, R.; Renneville, A.; et al. Granulomonocytic Progenitors Are Key Target Cells of Azacytidine in Higher Risk Myelodysplastic Syndromes and Acute Myeloid Leukemia. Leukemia 2018, 32, 1856–1860. [Google Scholar] [CrossRef] [PubMed]
- Beerman, I.; Seita, J.; Inlay, M.A.; Weissman, I.L.; Rossi, D.J. Quiescent Hematopoietic Stem Cells Accumulate DNA Damage during Aging That Is Repaired upon Entry into Cell Cycle. Cell Stem Cell 2014, 15, 37–50. [Google Scholar] [CrossRef] [Green Version]
- Pearl, L.H.; Schierz, A.C.; Ward, S.E.; Al-Lazikani, B.; Pearl, F.M.G. Therapeutic Opportunities within the DNA Damage Response. Nat. Rev. Cancer 2015, 15, 166–180. [Google Scholar] [CrossRef] [Green Version]
- Nabatiyan, A.; Szüts, D.; Krude, T. Induction of CAF-1 Expression in Response to DNA Strand Breaks in Quiescent Human Cells. Mol. Cell. Biol. 2006, 26, 1839–1849. [Google Scholar] [CrossRef] [Green Version]
- Vitale, I.; Manic, G.; De Maria, R.; Kroemer, G.; Galluzzi, L. DNA Damage in Stem Cells. Mol. Cell 2017, 66, 306–319. [Google Scholar] [CrossRef] [Green Version]
- Shin, J.J.; Schröder, M.S.; Caiado, F.; Wyman, S.K.; Bray, N.L.; Bordi, M.; Dewitt, M.A.; Vu, J.T.; Kim, W.-T.; Hockemeyer, D.; et al. Controlled Cycling and Quiescence Enables Efficient HDR in Engraftment-Enriched Adult Hematopoietic Stem and Progenitor Cells. Cell Rep. 2020, 32, 108093. [Google Scholar] [CrossRef]
- Stevens, B.M.; Khan, N.; D’Alessandro, A.; Nemkov, T.; Winters, A.; Jones, C.L.; Zhang, W.; Pollyea, D.A.; Jordan, C.T. Characterization and Targeting of Malignant Stem Cells in Patients with Advanced Myelodysplastic Syndromes. Nat. Commun. 2018, 9, 3694. [Google Scholar] [CrossRef]
- Guo, B.; Zhai, D.; Cabezas, E.; Welsh, K.; Nouraini, S.; Satterthwait, A.C.; Reed, J.C. Humanin Peptide Suppresses Apoptosis by Interfering with Bax Activation. Nature 2003, 423, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Luo, M.; Jeong, M.; Rodriguez, B.; Xia, Z.; Hannah, R.; Wang, H.; Le, T.; Faull, K.F.; Chen, R.; et al. Epigenomic Profiling of Young and Aged HSCs Reveals Concerted Changes during Aging That Reinforce Self-Renewal. Cell Stem Cell 2014, 14, 673–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabezas-Wallscheid, N.; Klimmeck, D.; Hansson, J.; Lipka, D.B.; Reyes, A.; Wang, Q.; Weichenhan, D.; Lier, A.; von Paleske, L.; Renders, S.; et al. Identification of Regulatory Networks in HSCs and Their Immediate Progeny via Integrated Proteome, Transcriptome, and DNA Methylome Analysis. Cell Stem Cell 2014, 15, 507–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poloni, A.; Goteri, G.; Zizzi, A.; Serrani, F.; Trappolini, S.; Costantini, B.; Mariani, M.; Olivieri, A.; Catarini, M.; Centurioni, R.; et al. Prognostic Role of Immunohistochemical Analysis of 5 Mc in Myelodysplastic Syndromes. Eur. J. Haematol. 2013, 91, 219–227. [Google Scholar] [CrossRef]
- Gawlitza, A.L.; Speith, J.; Rinke, J.; Sajzew, R.; Müller, E.K.; Schäfer, V.; Hochhaus, A.; Ernst, T. 5-Azacytidine Modulates CpG Methylation Levels of EZH2 and NOTCH1 in Myelodysplastic Syndromes. J. Cancer Res. Clin. Oncol. 2019, 145, 2835–2843. [Google Scholar] [CrossRef] [PubMed]
- Grövdal, M.; Karimi, M.; Tobiasson, M.; Reinius, L.; Jansson, M.; Ekwall, K.; Ungerstedt, J.; Kere, J.; Greco, D.; Hellström-Lindberg, E. Azacitidine Induces Profound Genome-Wide Hypomethylation in Primary Myelodysplastic Bone Marrow Cultures but May Also Reduce Histone Acetylation. Leukemia 2014, 28, 411–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tobiasson, M.; Abdulkadir, H.; Lennartsson, A.; Katayama, S.; Marabita, F.; Paepe, A.D.; Karimi, M.; Krjutskov, K.; Einarsdottir, E.; Grövdal, M.; et al. Comprehensive Mapping of the Effects of Azacitidine on DNA Methylation, Repressive/Permissive Histone Marks and Gene Expression in Primary Cells from Patients with MDS and MDS-Related Disease. Oncotarget 2017, 8, 28812–28825. [Google Scholar] [CrossRef]
- Kuendgen, A.; Müller-Thomas, C.; Lauseker, M.; Haferlach, T.; Urbaniak, P.; Schroeder, T.; Brings, C.; Wulfert, M.; Meggendorfer, M.; Hildebrandt, B.; et al. Efficacy of Azacitidine Is Independent of Molecular and Clinical Characteristics—An Analysis of 128 Patients with Myelodysplastic Syndromes or Acute Myeloid Leukemia and a Review of the Literature. Oncotarget 2018, 9, 27882–27894. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Li, Y.; Li, T.; Li, Y.; Xing, H.; Sun, H.; Sun, L.; Wan, D.; Liu, Y.; Xie, X.; et al. Gene Mutational Analysis by NGS and Its Clinical Significance in Patients with Myelodysplastic Syndrome and Acute Myeloid Leukemia. Exp. Hematol. Oncol. 2020, 9, 2. [Google Scholar] [CrossRef]
- Reid, M.A.; Dai, Z.; Locasale, J.W. The Impact of Cellular Metabolism on Chromatin Dynamics and Epigenetics. Nat. Cell Biol. 2017, 19, 1298–1306. [Google Scholar] [CrossRef]
- Pollyea, D.A.; Stevens, B.M.; Jones, C.L.; Winters, A.; Pei, S.; Minhajuddin, M.; D’Alessandro, A.; Culp-Hill, R.; Riemondy, K.A.; Gillen, A.E.; et al. Venetoclax with Azacitidine Disrupts Energy Metabolism and Targets Leukemia Stem Cells in Patients with Acute Myeloid Leukemia. Nat. Med. 2018, 24, 1859–1866. [Google Scholar] [CrossRef]
- Coller, H.A. The Paradox of Metabolism in Quiescent Stem Cells. FEBS Lett. 2019, 593, 2817–2839. [Google Scholar] [CrossRef] [PubMed]
- Nakamura-Ishizu, A.; Ito, K.; Suda, T. Hematopoietic Stem Cell Metabolism during Development and Aging. Dev. Cell 2020, 54, 239–255. [Google Scholar] [CrossRef] [PubMed]
- Raffel, S.; Falcone, M.; Kneisel, N.; Hansson, J.; Wang, W.; Lutz, C.; Bullinger, L.; Poschet, G.; Nonnenmacher, Y.; Barnert, A.; et al. BCAT1 Restricts AKG Levels in AML Stem Cells Leading to IDHmut-like DNA Hypermethylation. Nature 2017, 551, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Rashkovan, M.; Ferrando, A. Metabolic Dependencies and Vulnerabilities in Leukemia. Genes Dev. 2019, 33, 1460–1474. [Google Scholar] [CrossRef] [Green Version]
- Kinnaird, A.; Zhao, S.; Wellen, K.E.; Michelakis, E.D. Metabolic Control of Epigenetics in Cancer. Nat. Rev. Cancer 2016, 16, 694–707. [Google Scholar] [CrossRef]
- Etchegaray, J.-P.; Mostoslavsky, R. Interplay between Metabolism and Epigenetics: A Nuclear Adaptation to Environmental Changes. Mol. Cell 2016, 62, 695–711. [Google Scholar] [CrossRef] [Green Version]
- Wellen, K.E.; Hatzivassiliou, G.; Sachdeva, U.M.; Bui, T.V.; Cross, J.R.; Thompson, C.B. ATP-Citrate Lyase Links Cellular Metabolism to Histone Acetylation. Science 2009, 324, 1076–1080. [Google Scholar] [CrossRef] [Green Version]
- Ghoshal, K.; Datta, J.; Majumder, S.; Bai, S.; Kutay, H.; Motiwala, T.; Jacob, S.T. 5-Aza-Deoxycytidine Induces Selective Degradation of DNA Methyltransferase 1 by a Proteasomal Pathway That Requires the KEN Box, Bromo-Adjacent Homology Domain, and Nuclear Localization Signal. Mol. Cell. Biol. 2005, 25, 4727–4741. [Google Scholar] [CrossRef] [Green Version]
- Issa, J.-P.J.; Kantarjian, H.M. Targeting DNA Methylation. Clin. Cancer Res. 2009, 15, 3938–3946. [Google Scholar] [CrossRef] [Green Version]
- Solly, F.; Koering, C.; Mohamed, A.M.; Maucort-Boulch, D.; Robert, G.; Auberger, P.; Flandrin-Gresta, P.; Adès, L.; Fenaux, P.; Kosmider, O.; et al. An MiRNA–DNMT1 Axis Is Involved in Azacitidine Resistance and Predicts Survival in Higher-Risk Myelodysplastic Syndrome and Low Blast Count Acute Myeloid Leukemia. Clin. Cancer Res. 2017, 23, 3025–3034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vispé, S.; Deroide, A.; Davoine, E.; Desjobert, C.; Lestienne, F.; Fournier, L.; Novosad, N.; Bréand, S.; Besse, J.; Busato, F.; et al. Consequences of Combining SiRNA-Mediated DNA Methyltransferase 1 Depletion with 5-Aza-2′-Deoxycytidine in Human Leukemic KG1 Cells. Oncotarget 2015, 6, 15265–15282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, H. Transcription of Mouse DNA Methyltransferase 1 (Dnmt1) Is Regulated by Both E2F-Rb-HDAC-Dependent and -Independent Pathways. Nucleic Acids Res. 2003, 31, 3101–3113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, V.; Sharma, P.; Capalash, N. DNA Methyltransferase-1 Inhibitors as Epigenetic Therapy for Cancer. Curr. Cancer Drug Targets 2013, 13, 379–399. [Google Scholar] [CrossRef]
- Wong, K.K.; Lawrie, C.H.; Green, T.M. Oncogenic Roles and Inhibitors of DNMT1, DNMT3A, and DNMT3B in Acute Myeloid Leukaemia. Biomark Insights 2019, 14, 117727191984645. [Google Scholar] [CrossRef]
- Daugas, E.; Nochy, D.; Ravagnan, L.; Loeffler, M.; Susin, S.A.; Zamzami, N.; Kroemer, G. Apoptosis-Inducing Factor (AIF): A Ubiquitous Mitochondrial Oxidoreductase Involved in Apoptosis. FEBS Lett. 2000, 476, 118–123. [Google Scholar] [CrossRef]
- Vaquero, A.; Scher, M.; Lee, D.; Erdjument-Bromage, H.; Tempst, P.; Reinberg, D. Human SirT1 Interacts with Histone H1 and Promotes Formation of Facultative Heterochromatin. Mol. Cell 2004, 16, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Baur, A.S.; Meugé-Moraw, C.; Schmidt, P.-M.; Parlier, V.; Jotterand, M.; Delacrétaz, F. CD34/QBEND10 Immunostaining in Bone Marrow Biopsies: An Additional Parameterfor the Diagnosis and Classification of Myelodysplastic Syndromes: CD34 in Myelodysplastic Syndromes. Eur. J. Haematol. 2000, 64, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Pollyea, D.A.; Jordan, C.T. Therapeutic Targeting of Acute Myeloid Leukemia Stem Cells. Blood 2017, 129, 1627–1635. [Google Scholar] [CrossRef]
- Thomas, D.; Majeti, R. Biology and Relevance of Human Acute Myeloid Leukemia Stem Cells. Blood 2017, 129, 1577–1585. [Google Scholar] [CrossRef] [PubMed]
- Will, B.; Zhou, L.; Vogler, T.O.; Ben-Neriah, S.; Schinke, C.; Tamari, R.; Yu, Y.; Bhagat, T.D.; Bhattacharyya, S.; Barreyro, L.; et al. Stem and Progenitor Cells in Myelodysplastic Syndromes Show Aberrant Stage-Specific Expansion and Harbor Genetic and Epigenetic Alterations. Blood 2012, 120, 2076–2086. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Kao, Y.-R.; Sun, D.; Todorova, T.I.; Reynolds, D.; Narayanagari, S.-R.; Montagna, C.; Will, B.; Verma, A.; Steidl, U. Myelodysplastic Syndrome Progression to Acute Myeloid Leukemia at the Stem Cell Level. Nat. Med. 2019, 25, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Shastri, A.; Will, B.; Steidl, U.; Verma, A. Stem and Progenitor Cell Alterations in Myelodysplastic Syndromes. Blood 2017, 129, 1586–1594. [Google Scholar] [CrossRef] [PubMed]
- Mejia-Ramirez, E.; Florian, M.C. Understanding Intrinsic Hematopoietic Stem Cell Aging. Haematologica 2020, 105, 22–37. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Xu, C.; Gao, J.; Li, B.; Qin, T.; Xu, Z.; Ren, S.; Zhang, Y.; Jiao, M.; Qu, S.; et al. Severe Ineffective Erythropoiesis Discriminates Prognosis in Myelodysplastic Syndromes: Analysis Based on 776 Patients from a Single Centre. Blood Cancer J. 2020, 10, 83. [Google Scholar] [CrossRef] [PubMed]
- Sancho, M.; Diani, E.; Beato, M.; Jordan, A. Depletion of Human Histone H1 Variants Uncovers Specific Roles in Gene Expression and Cell Growth. PLoS Genet. 2008, 4, e1000227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hakimi, A.A.; Reznik, E.; Lee, C.-H.; Creighton, C.J.; Brannon, A.R.; Luna, A.; Aksoy, B.A.; Liu, E.M.; Shen, R.; Lee, W.; et al. An Integrated Metabolic Atlas of Clear Cell Renal Cell Carcinoma. Cancer Cell 2016, 29, 104–116. [Google Scholar] [CrossRef] [Green Version]
- Rist, M.J.; Roth, A.; Frommherz, L.; Weinert, C.H.; Krüger, R.; Merz, B.; Bunzel, D.; Mack, C.; Egert, B.; Bub, A.; et al. Metabolite Patterns Predicting Sex and Age in Participants of the Karlsruhe Metabolomics and Nutrition (KarMeN) Study. PLoS ONE 2017, 12, e0183228. [Google Scholar] [CrossRef]
- Krumsiek, J.; Mittelstrass, K.; Do, K.T.; Stückler, F.; Ried, J.; Adamski, J.; Peters, A.; Illig, T.; Kronenberg, F.; Friedrich, N.; et al. Gender-Specific Pathway Differences in the Human Serum Metabolome. Metabolomics 2015, 11, 1815–1833. [Google Scholar] [CrossRef] [Green Version]
- Vignoli, A.; Tenori, L.; Luchinat, C.; Saccenti, E. Age and Sex Effects on Plasma Metabolite Association Networks in Healthy Subjects. J. Proteome Res. 2018, 17, 97–107. [Google Scholar] [CrossRef] [Green Version]
- Schvartzman, J.M.; Thompson, C.B.; Finley, L.W.S. Metabolic Regulation of Chromatin Modifications and Gene Expression. J. Cell Biol. 2018, 217, 2247–2259. [Google Scholar] [CrossRef]
- Diehl, K.L.; Muir, T.W. Chromatin as a Key Consumer in the Metabolite Economy. Nat. Chem. Biol. 2020, 16, 620–629. [Google Scholar] [CrossRef]
- Beisel, C.; Paro, R. Silencing Chromatin: Comparing Modes and Mechanisms. Nat. Rev. Genet. 2011, 12, 123–135. [Google Scholar] [CrossRef]
- Toffalorio, F.; Santarpia, M.; Radice, D.; Jaramillo, C.A.; Spitaleri, G.; Manzotti, M.; Catania, C.; Jordheim, L.P.; Pelosi, G.; Peters, G.J.; et al. 5′-Nucleotidase CN-II Emerges as a New Predictive Biomarker of Response to Gemcitabine/Platinum Combination Chemotherapy in Non-Small Cell Lung Cancer. Oncotarget 2018, 9, 16437–16450. [Google Scholar] [CrossRef] [Green Version]
- Quagliano, A.; Gopalakrishnapillai, A.; Barwe, S.P. Understanding the Mechanisms by Which Epigenetic Modifiers Avert Therapy Resistance in Cancer. Front. Oncol. 2020, 10, 992. [Google Scholar] [CrossRef]
- Cameron, E.E.; Bachman, K.E.; Myöhänen, S.; Herman, J.G.; Baylin, S.B. Synergy of Demethylation and Histone Deacetylase Inhibition in the Re-Expression of Genes Silenced in Cancer. Nat. Genet. 1999, 21, 103–107. [Google Scholar] [CrossRef]
- Zhang, B.; Strauss, A.C.; Chu, S.; Li, M.; Ho, Y.; Shiang, K.-D.; Snyder, D.S.; Huettner, C.S.; Shultz, L.; Holyoake, T.; et al. Effective Targeting of Quiescent Chronic Myelogenous Leukemia Stem Cells by Histone Deacetylase Inhibitors in Combination with Imatinib Mesylate. Cancer Cell 2010, 17, 427–442. [Google Scholar] [CrossRef] [Green Version]
- Kovaka, S.; Zimin, A.V.; Pertea, G.M.; Razaghi, R.; Salzberg, S.L.; Pertea, M. Transcriptome Assembly from Long-Read RNA-Seq Alignments with StringTie2. Genome Biol. 2019, 20, 278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babicki, S.; Arndt, D.; Marcu, A.; Liang, Y.; Grant, J.R.; Maciejewski, A.; Wishart, D.S. Heatmapper: Web-Enabled Heat Mapping for All. Nucleic Acids Res. 2016, 44, W147–W153. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 Revision to the World Health Organization Classification of Myeloid Neoplasms and Acute Leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Cheson, B.D. Clinical Application and Proposal for Modification of the International Working Group (IWG) Response Criteria in Myelodysplasia. Blood 2006, 108, 419–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koralkova, P.; Belickova, M.; Kundrat, D.; Dostalova Merkerova, M.; Krejcik, Z.; Szikszai, K.; Kaisrlikova, M.; Vesela, J.; Vyhlidalova, P.; Stetka, J.; et al. Low Plasma Citrate Levels and Specific Transcriptional Signatures Associated with Quiescence of CD34+ Progenitors Predict Azacitidine Therapy Failure in MDS/AML Patients. Cancers 2021, 13, 2161. https://doi.org/10.3390/cancers13092161
Koralkova P, Belickova M, Kundrat D, Dostalova Merkerova M, Krejcik Z, Szikszai K, Kaisrlikova M, Vesela J, Vyhlidalova P, Stetka J, et al. Low Plasma Citrate Levels and Specific Transcriptional Signatures Associated with Quiescence of CD34+ Progenitors Predict Azacitidine Therapy Failure in MDS/AML Patients. Cancers. 2021; 13(9):2161. https://doi.org/10.3390/cancers13092161
Chicago/Turabian StyleKoralkova, Pavla, Monika Belickova, David Kundrat, Michaela Dostalova Merkerova, Zdenek Krejcik, Katarina Szikszai, Monika Kaisrlikova, Jitka Vesela, Pavla Vyhlidalova, Jan Stetka, and et al. 2021. "Low Plasma Citrate Levels and Specific Transcriptional Signatures Associated with Quiescence of CD34+ Progenitors Predict Azacitidine Therapy Failure in MDS/AML Patients" Cancers 13, no. 9: 2161. https://doi.org/10.3390/cancers13092161