Comprehensive Assessment of Copy Number Alterations Uncovers Recurrent AIFM3 and DLK1 Copy Gain in Medullary Thyroid Carcinoma


 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
2.1. Identification of CNAs in MTC Genome
2.2. Recurrent Gains in Medullary Thyroid Carcinoma
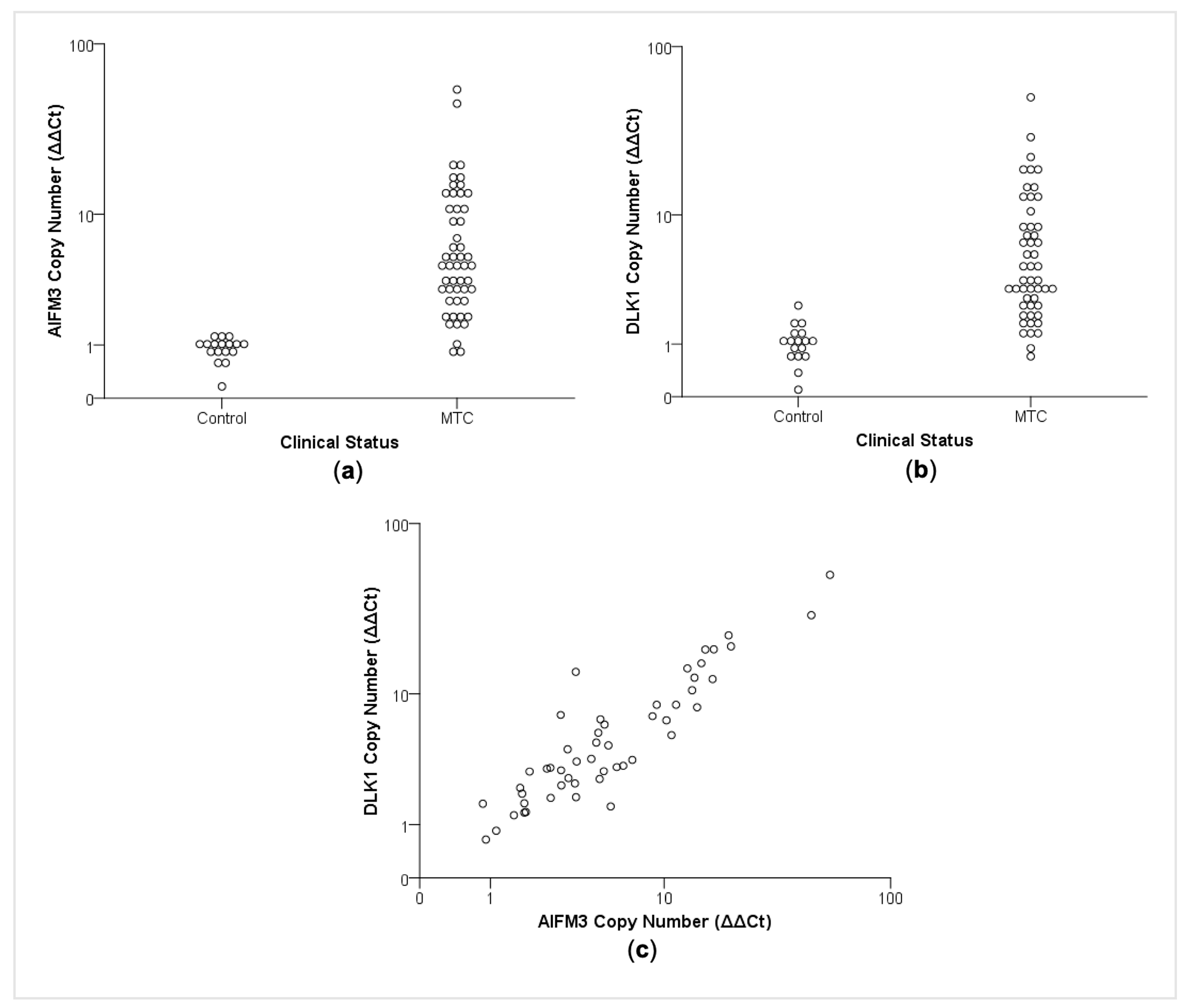
2.3. CNA Affects AIFM3 and DLK1 Expression in Medullary Thyroid Carcinomas
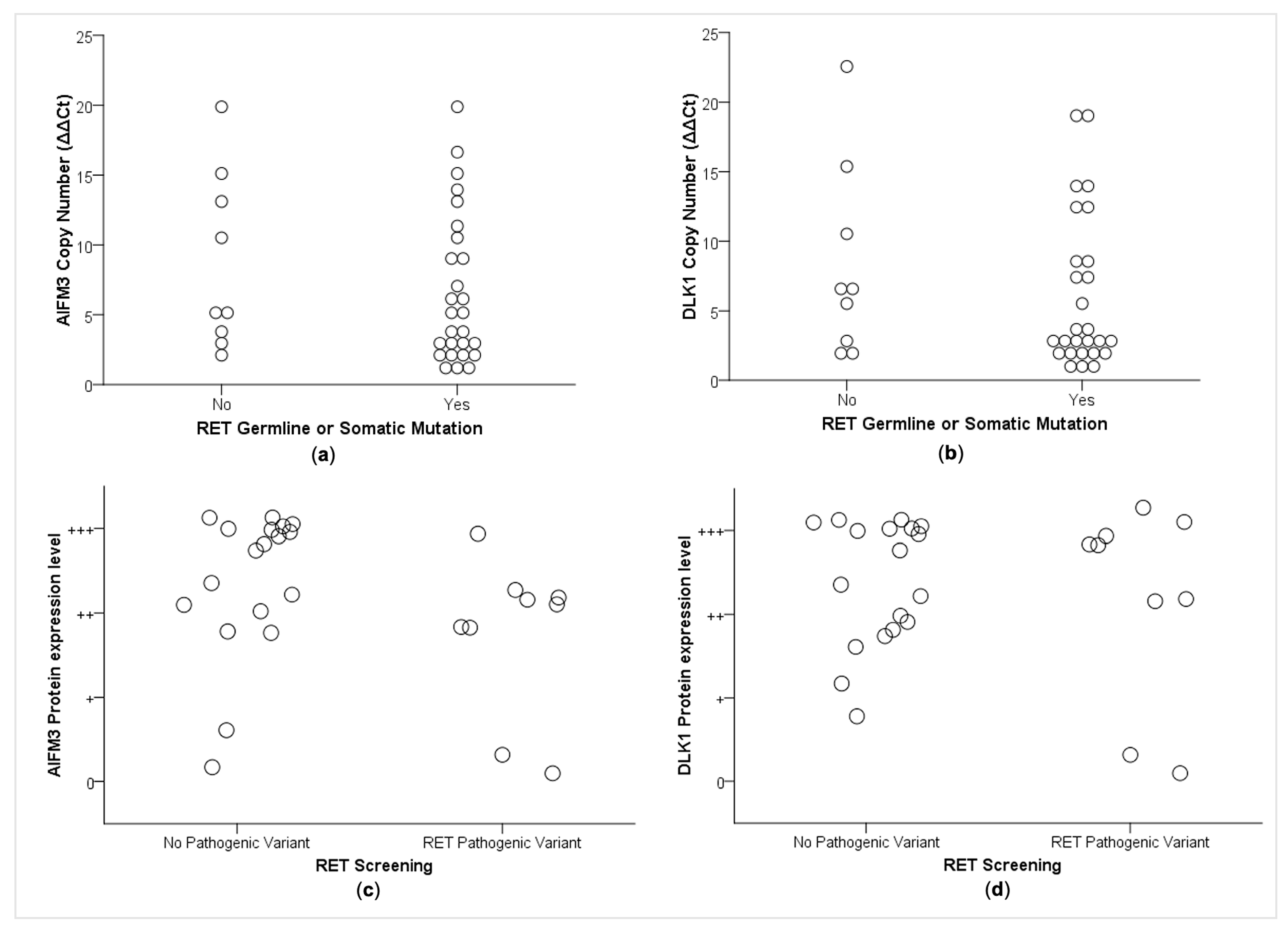
2.4. Correlations between CNA and RET Mutational Status
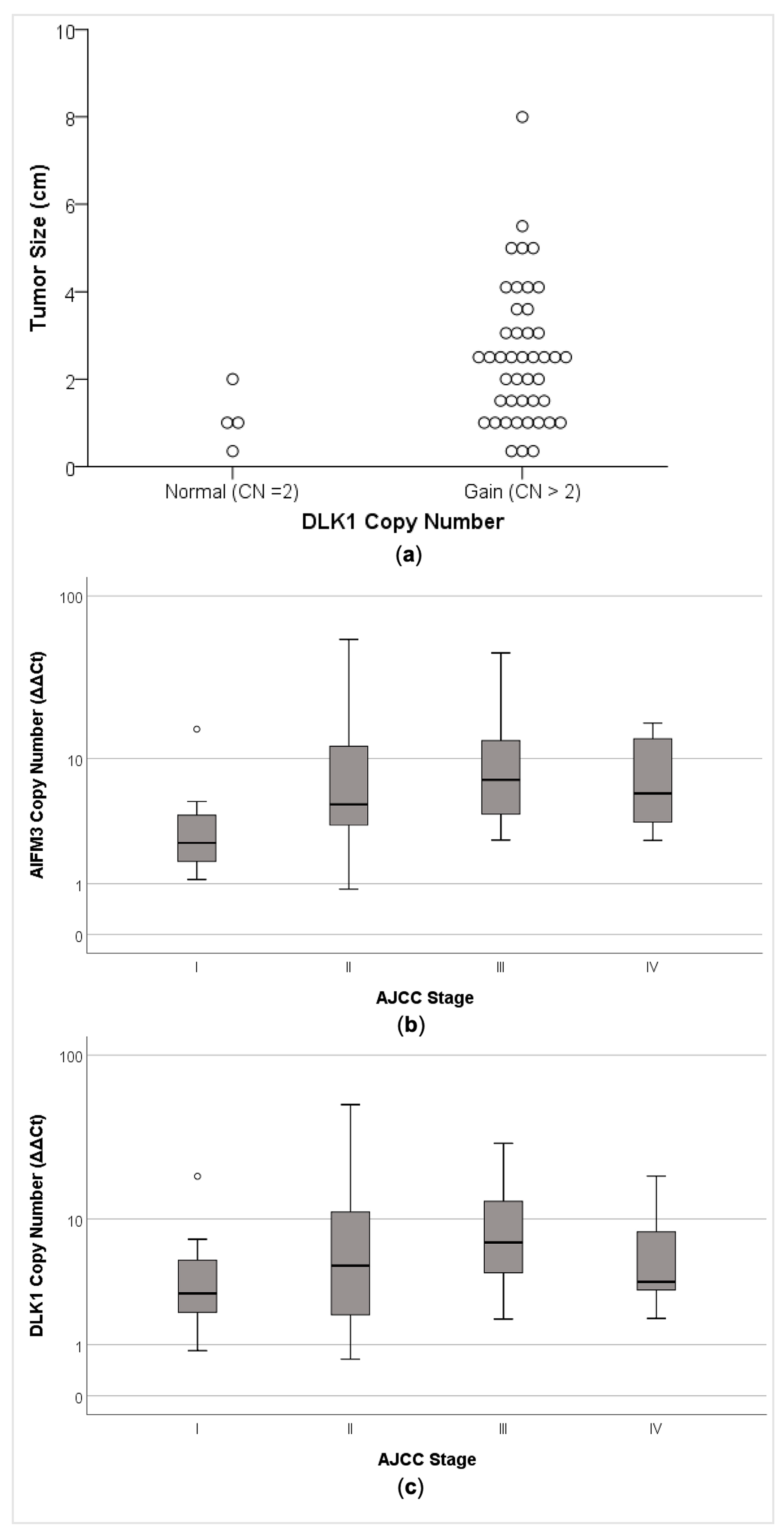
2.5. AIFM3 and DLK1 Copy Number Alteration was Associated with Clinical–Pathological Parameters
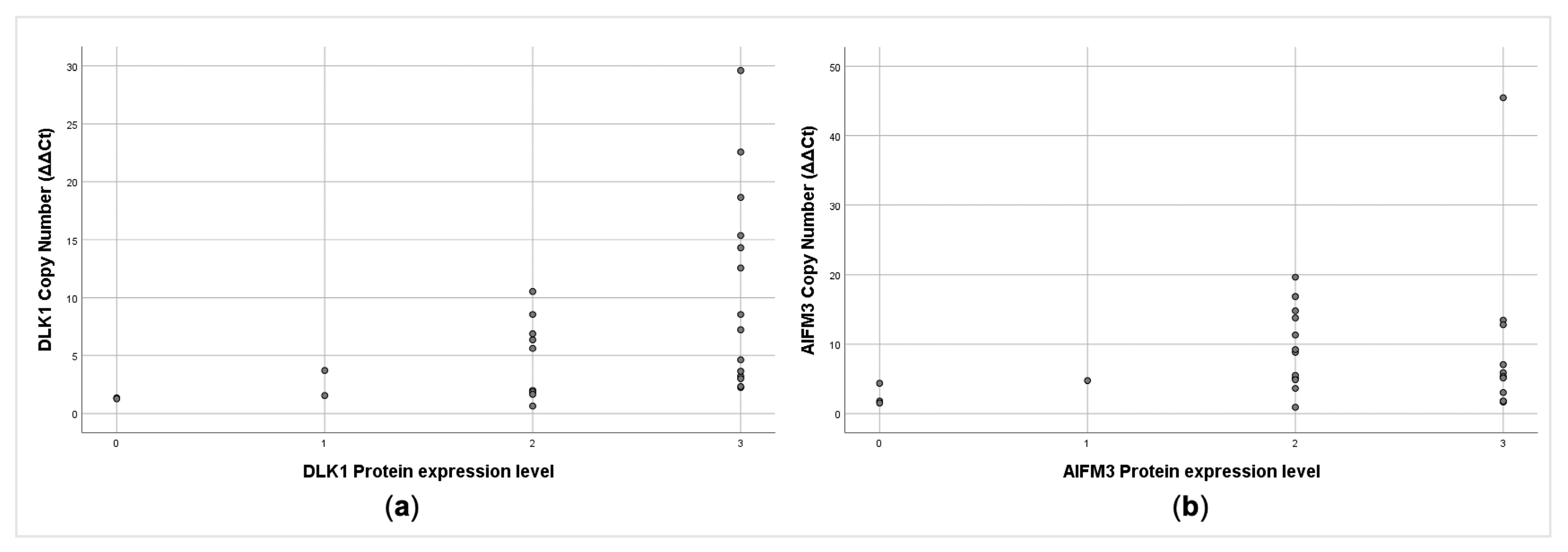
2.6. AIFM3 and DLK1 Protein Expression and Clinical–Pathological Parameters
3. Discussion
4. Materials and Methods
4.1. Subjects
4.2. DNA Isolation
4.3. RET Mutation Analysis
4.4. Copy Number Analysis
4.5. Recurrent CNAs were Experimentally Validated by Quantitative PCR
4.6. Association of CNA with Protein Expression
4.7. Correlations of CNA Alteration and Protein Expression with RET Status, Demographic and Clinical–Pathological Parameters
4.8. Functional Annotation of Identified CNAs and Enrichment Analysis
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wells, S.A., Jr.; Asa, S.L.; Dralle, H.; Elisei, R.; Evans, D.B.; Gagel, R.F.; Lee, N.; Machens, A.; Moley, J.F.; Pacini, F.; et al. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid 2015, 25, 567–610. [Google Scholar] [CrossRef]
- Ciampi, R.; Mian, C.; Fugazzola, L.; Cosci, B.; Romei, C.; Barollo, S.; Cirello, V.; Bottici, V.; Marconcini, G.; Rosa, P.M.; et al. Evidence of a low prevalence of RAS mutations in a large medullary thyroid cancer series. Thyroid 2013, 23, 50–57. [Google Scholar] [CrossRef]
- Moura, M.M.; Cavaco, B.M.; Leite, V. RAS proto-oncogene in medullary thyroid carcinoma. Endocr. Relat. Cancer 2015, 22, 235–252. [Google Scholar] [CrossRef]
- Mulligan, L.M.; Kwok, J.B.; Healey, C.S.; Elsdon, M.J.; Eng, C.; Gardner, E.; Love, D.R.; Mole, S.E.; Moore, J.K.; Papi, L.; et al. Germ-line mutations of the RET proto-oncogene in multiple endocrine neoplasia type 2A. Nature 1993, 363, 458–460. [Google Scholar] [CrossRef]
- Michiels, F.M.; Chappuis, S.; Caillou, B.; Pasini, A.; Talbot, M.; Monier, R.; Lenoir, G.M.; Feunteun, J.; Billaud, M. Development of medullary thyroid carcinoma in transgenic mice expressing the RET protooncogene altered by a multiple endocrine neoplasia type 2A mutation. Proc. Natl. Acad. Sci. USA 1997, 94, 3330–3335. [Google Scholar] [CrossRef] [Green Version]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef]
- Redon, R.; Ishikawa, S.; Fitch, K.R.; Feuk, L.; Perry, G.H.; Andrews, T.D.; Fiegler, H.; Shapero, M.H.; Carson, A.R.; Chen, W.; et al. Global variation in copy number in the human genome. Nature 2006, 444, 444–454. [Google Scholar] [CrossRef] [Green Version]
- Bignell, G.R.; Greenman, C.D.; Davies, H.; Butler, A.P.; Edkins, S.; Andrews, J.M.; Buck, G.; Chen, L.; Beare, D.; Latimer, C.; et al. Signatures of mutation and selection in the cancer genome. Nature 2010, 463, 893–898. [Google Scholar] [CrossRef] [PubMed]
- Miyaguchi, K.; Fukuoka, Y.; Mizushima, H.; Yasen, M.; Nemoto, S.; Ishikawa, T.; Uetake, H.; Tanaka, S.; Sugihara, K.; Arii, S.; et al. Genome-wide integrative analysis revealed a correlation between lengths of copy number segments and corresponding gene expression profile. Bioinformation 2011, 7, 280–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stranger, B.E.; Forrest, M.S.; Dunning, M.; Ingle, C.E.; Beazley, C.; Thorne, N.; Redon, R.; Bird, C.P.; de Grassi, A.; Lee, C.; et al. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science 2007, 315, 848–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulligan, L.M.; Gardner, E.; Smith, B.A.; Mathew, C.G.; Ponder, B.A. Genetic events in tumour initiation and progression in multiple endocrine neoplasia type 2. Genes Chromosomes Cancer 1993, 6, 166–177. [Google Scholar] [CrossRef] [PubMed]
- Hemmer, S.; Wasenius, V.M.; Knuutila, S.; Franssila, K.; Joensuu, H. DNA copy number changes in thyroid carcinoma. Am. J. Pathol. 1999, 154, 1539–1547. [Google Scholar] [CrossRef] [Green Version]
- Ye, L.; Santarpia, L.; Cote, G.J.; El-Naggar, A.K.; Gagel, R.F. High resolution array-comparative genomic hybridization profiling reveals deoxyribonucleic acid copy number alterations associated with medullary thyroid carcinoma. J. Clin. Endocrinol. Metab. 2008, 93, 4367–4372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsh, D.J.; Theodosopoulos, G.; Martin-Schulte, K.; Richardson, A.L.; Philips, J.; Roher, H.D.; Delbridge, L.; Robinson, B.G. Genome-wide copy number imbalances identified in familial and sporadic medullary thyroid carcinoma. J. Clin. Endocrinol. Metab. 2003, 88, 1866–1872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flicker, K.; Ulz, P.; Hoger, H.; Zeitlhofer, P.; Haas, O.A.; Behmel, A.; Buchinger, W.; Scheuba, C.; Niederle, B.; Pfragner, R.; et al. High-resolution analysis of alterations in medullary thyroid carcinoma genomes. Int. J. Cancer 2012, 131, E66–E73. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Li, L.; Ye, L.; Jiang, X.; Shen, L.; Gao, Z.; Fang, W.; Huang, F.; Su, T.; Zhou, Y.; et al. Exome sequencing reveals mutant genes with low penetrance involved in MEN2A-associated tumorigenesis. Endocr. Relat. Cancer 2015, 22, 23–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grubbs, E.G.; Williams, M.D.; Scheet, P.; Vattathil, S.; Perrier, N.D.; Lee, J.E.; Gagel, R.F.; Hai, T.; Feng, L.; Cabanillas, M.E.; et al. Role of CDKN2C Copy Number in Sporadic Medullary Thyroid Carcinoma. Thyroid 2016, 26, 1553–1562. [Google Scholar] [CrossRef]
- Wang, N.; Kjellin, H.; Sofiadis, A.; Fotouhi, O.; Juhlin, C.C.; Backdahl, M.; Zedenius, J.; Xu, D.; Lehtio, J.; Larsson, C. Genetic and epigenetic background and protein expression profiles in relation to telomerase activation in medullary thyroid carcinoma. Oncotarget 2016, 7, 21332–21346. [Google Scholar] [CrossRef] [Green Version]
- Shlien, A.; Malkin, D. Copy number variations and cancer. Genome Med. 2009, 1, 62. [Google Scholar] [CrossRef] [Green Version]
- Dvorakova, S.; Vaclavikova, E.; Sykorova, V.; Vcelak, J.; Novak, Z.; Duskova, J.; Ryska, A.; Laco, J.; Cap, J.; Kodetova, D.; et al. Somatic mutations in the RET proto-oncogene in sporadic medullary thyroid carcinomas. Mol. Cell Endocrinol. 2008, 284, 21–27. [Google Scholar] [CrossRef]
- Bim, L.V.; Navarro, F.C.P.; Valente, F.O.F.; Lima-Junior, J.V.; Delcelo, R.; Dias-da-Silva, M.R.; Maciel, R.M.B.; Galante, P.A.F.; Cerutti, J.M. Retroposed copies of RET gene: A somatically acquired event in medullary thyroid carcinoma. BMC Med. Genom. 2019, 12, 104. [Google Scholar] [CrossRef] [PubMed]
- Alaei-Mahabadi, B.; Bhadury, J.; Karlsson, J.W.; Nilsson, J.A.; Larsson, E. Global analysis of somatic structural genomic alterations and their impact on gene expression in diverse human cancers. Proc. Natl. Acad. Sci. USA 2016, 113, 13768–13773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, L.; Li, Q.; Du, Y.; Yun, J.; Xie, Y.; deBerardinis, R.J.; Xiao, G. Genomic regression analysis of coordinated expression. Nat. Commun. 2017, 19, 2187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Q.; Lin, T.; Zhang, Y.; Zheng, J.; Bonanno, J.A. Molecular cloning and characterization of a human AIF-like gene with ability to induce apoptosis. J. Biol. Chem. 2005, 280, 19673–19681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Sun, T.; Cao, J.; Liu, F.; Tian, Y.; Zhu, W. Downregulation of miR-210 expression inhibits proliferation, induces apoptosis and enhances radiosensitivity in hypoxic human hepatoma cells in vitro. Exp. Cell Res. 2012, 318, 944–954. [Google Scholar] [CrossRef]
- Mutharasan, R.K.; Nagpal, V.; Ichikawa, Y.; Ardehali, H. microRNA-210 is upregulated in hypoxic cardiomyocytes through Akt- and p53-dependent pathways and exerts cytoprotective effects. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1519–H1530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chua-On, D.; Proungvitaya, T.; Techasen, A.; Limpaiboon, T.; Roytrakul, S.; Wongkham, S.; Wongkham, C.; Somintara, O.; Sungkhamanon, S.; Proungvitaya, S. High expression of apoptosis-inducing factor, mitochondrion-associated 3 (AIFM3) in human cholangiocarcinoma. Tumour. Biol. 2016, 37, 13659–13667. [Google Scholar] [CrossRef]
- Zheng, A.; Zhang, L.; Song, X.; Wang, Y.; Wei, M.; Jin, F. Clinical implications of a novel prognostic factor AIFM3 in breast cancer patients. BMC Cancer 2019, 19, 451. [Google Scholar] [CrossRef] [Green Version]
- Traustadottir, G.A.; Lagoni, L.V.; Ankerstjerne, L.B.S.; Bisgaard, H.C.; Jensen, C.H.; Andersen, D.C. The imprinted gene Delta like non-canonical Notch ligand 1 (Dlk1) is conserved in mammals, and serves a growth modulatory role during tissue development and regeneration through Notch dependent and independent mechanisms. Cytokine Growth Factor Rev. 2019, 46, 17–27. [Google Scholar] [CrossRef]
- Garcia-Lopez, S.; Albo-Castellanos, C.; Urdinguio, R.G.; Canon, S.; Sanchez-Cabo, F.; Martinez-Serrano, A.; Fraga, M.F.; Bernad, A. Deregulation of the imprinted DLK1-DIO3 locus ncRNAs is associated with replicative senescence of human adipose-derived stem cells. PLoS ONE 2018, 13, e0206534. [Google Scholar] [CrossRef]
- Aw, D.K.; Sinha, R.A.; Tan, H.C.; Loh, L.M.; Salvatore, D.; Yen, P.M. Studies of molecular mechanisms associated with increased deiodinase 3 expression in a case of consumptive hypothyroidism. J. Clin. Endocrinol. Metab. 2014, 99, 3965–3971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romitti, M.; Wajner, S.M.; Ceolin, L.; Ferreira, C.V.; Ribeiro, R.V.; Rohenkohl, H.C.; Weber, S. de S.; Lopez, P.L.; Fuziwara, C.S.; Kimura, E.T.; et al. MAPK and SHH pathways modulate type 3 deiodinase expression in papillary thyroid carcinoma. Endocr. Relat. Cancer 2016, 23, 135–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrault, L.; Gide, J.; Qing, T.; Lesueur, L.; Tost, J.; Denis, J.A.; Cailleret, M.; Aubry, L.; Peschanski, M.; Martinat, C.; et al. Expression of miRNAs from the Imprinted DLK1/DIO3Locus Signals the Osteogenic Potential of Human Pluripotent Stem Cells. Cells 2019, 8, 1523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laborda, J.; Sausville, E.A.; Hoffman, T.; Notario, V. Dlk, a putative mammalian homeotic gene differentially expressed in small cell lung carcinoma and neuroendocrine tumor cell line. J. Biol. Chem. 1993, 268, 3817–3820. [Google Scholar] [CrossRef]
- Turanyi, E.; Dezso, K.; Paku, S.; Nagy, P. DLK is a novel immunohistochemical marker for adrenal gland tumors. Virchows Arch. 2009, 455, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.C.; Cheng, S.H.; Wu, C.H.; Li, W.Y.; Wang, J.S.; Kung, M.L.; Chu, T.H.; Huang, S.T.; Feng, C.T.; Huang, S.C.; et al. Delta-like 1 homologue promotes tumorigenesis and epithelial-mesenchymal transition of ovarian high-grade serous carcinoma through activation of Notch signaling. Oncogene 2019, 38, 3201–3215. [Google Scholar] [CrossRef]
- Da Silva, A.M.; Maciel, R.M.; Da Silva, M.R.; Toledo, S.R.; De Carvalho, M.B.; Cerutti, J.M. A novel germ-line point mutation in RET exon 8 (Gly(533)Cys) in a large kindred with familial medullary thyroid carcinoma. J. Clin. Endocrinol. Metab. 2003, 88, 5438–5443. [Google Scholar] [CrossRef] [PubMed]
- Maciel, R.M.B.; Camacho, C.P.; Assumpcao, L.V.M.; Bufalo, N.E.; Carvalho, A.L.; de Carvalho, G.A.; Castroneves, L.A.; de Castro, F.M.; Ceolin, L.; Cerutti, J.M.; et al. Genotype and phenotype landscape of MEN2 in 554 medullary thyroid cancer patients: The BrasMEN study. Endocr. Connect 2019, 8, 289–298. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Li, M.; Hadley, D.; Liu, R.; Glessner, J.; Grant, S.F.; Hakonarson, H.; Bucan, M. PennCNV: An integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007, 17, 1665–1674. [Google Scholar] [CrossRef] [Green Version]
- Cerutti, J.M.; Delcelo, R.; Amadei, M.J.; Nakabashi, C.; Maciel, R.M.; Peterson, B.; Shoemaker, J.; Riggins, G.J. A preoperative diagnostic test that distinguishes benign from malignant thyroid carcinoma based on gene expression. J. Clin. Investig. 2004, 113, 1234–1242. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Software | MTC * Sample | Chr | Start Position ** | End Position | Markers | Length (kb) | CN | CN | Genes |
|---|---|---|---|---|---|---|---|---|---|
| PennCNV | 2, 3 | 15q11.2 | 22849641 | 22885568 | 37 | 36 | 4 | gain | SNORD116-2, SNORD116-3, SNORD116-4, SNORD116-5, SNORD116-6, SNORD116-7, SNORD116-8, SNORD116-9, SNORD116-10, SNORD116-11, SNORD116-12, SNORD116-13, SNORD116-14, SNORD116-15, SNORD116-16, SNORD116-17, SNORD116-18, SNORD116-19, SNORD116-20, SNORD116-21. |
| Genotyping Console | 1, 2 | 4q28.3 | 137428273 | 137528519 | 67 | 100 | 1 | loss | none |
| 15q11.1 | 20130129 | 20291301 | 30 | 161 | 3 | gain | none *** | ||
| 1, 3 | 9p12 | 41475094 | 44244868 | 84 | 2770 | 3 | gain | SPATA31A5, SPATA31A7, FAM74A1, FAM74A6, ZNF658B, GLIDR, FGF7P3, ANKRD20A2, ANKRD20A3, GXYLT1P3, FAM95B1, FOXD4L4, AQP7P3, FAM74A7, SPATA31A6, CNTNAP3B, CNTNAP3P2. | |
| 2, 3 | 10q26.3 | 135076596 | 135184421 | 36 | 108 | 3 | gain | ADAM8, TUBGCP2, ZNF511, CALY, PRAP1, FUOM, ECHS1. | |
| 14q32.2 | 101178067 | 101300567 | 75 | 123 | 3 | gain | DLK1, MIR2392, MEG3. | ||
| 22q11.21 | 21298060 | 21407086 | 66 | 109 | 3 | gain | CRKL, LINC01637, AIFM3, LZTR1, THAP7, TUBA3FP, P2RX6, SLC7A4, MIR649, P2RX6P, LRRC74B. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Araujo, A.N.; Camacho, C.P.; Mendes, T.B.; Lindsey, S.C.; Moraes, L.; Miyazawa, M.; Delcelo, R.; Pellegrino, R.; Mazzotti, D.R.; Maciel, R.M.d.B.; et al. Comprehensive Assessment of Copy Number Alterations Uncovers Recurrent AIFM3 and DLK1 Copy Gain in Medullary Thyroid Carcinoma. Cancers 2021, 13, 218. https://doi.org/10.3390/cancers13020218
Araujo AN, Camacho CP, Mendes TB, Lindsey SC, Moraes L, Miyazawa M, Delcelo R, Pellegrino R, Mazzotti DR, Maciel RMdB, et al. Comprehensive Assessment of Copy Number Alterations Uncovers Recurrent AIFM3 and DLK1 Copy Gain in Medullary Thyroid Carcinoma. Cancers. 2021; 13(2):218. https://doi.org/10.3390/cancers13020218
Chicago/Turabian StyleAraujo, Aline Neves, Cléber Pinto Camacho, Thais Biude Mendes, Susan Chow Lindsey, Lais Moraes, Marta Miyazawa, Rosana Delcelo, Renata Pellegrino, Diego Robles Mazzotti, Rui Monteiro de Barros Maciel, and et al. 2021. "Comprehensive Assessment of Copy Number Alterations Uncovers Recurrent AIFM3 and DLK1 Copy Gain in Medullary Thyroid Carcinoma" Cancers 13, no. 2: 218. https://doi.org/10.3390/cancers13020218