Role of Fibroblast Growth Factors Receptors (FGFRs) in Brain Tumors, Focus on Astrocytoma and Glioblastoma

 , , , , , and
, , , , , and
Abstract
:Simple Summary
Abstract
1. Introduction
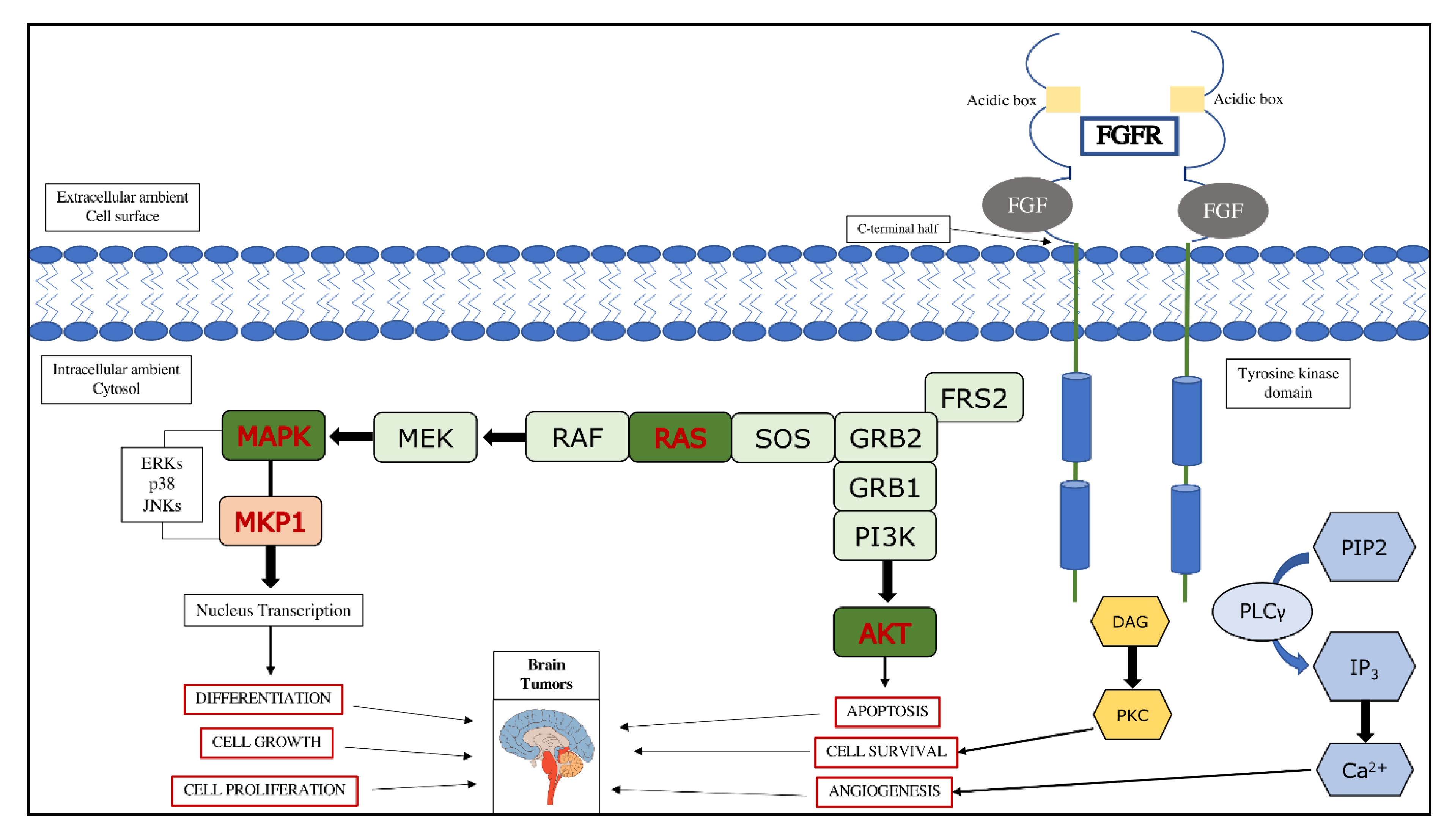
2. Fibroblast Growth Factors (FGFs) and Fibroblast Growth Factors Receptors (FGFRs)
2.1. FGFRs and Cell Adhesion Molecules (CAMs)
2.2. Involvement of FGFRs Subtypes in Cancer
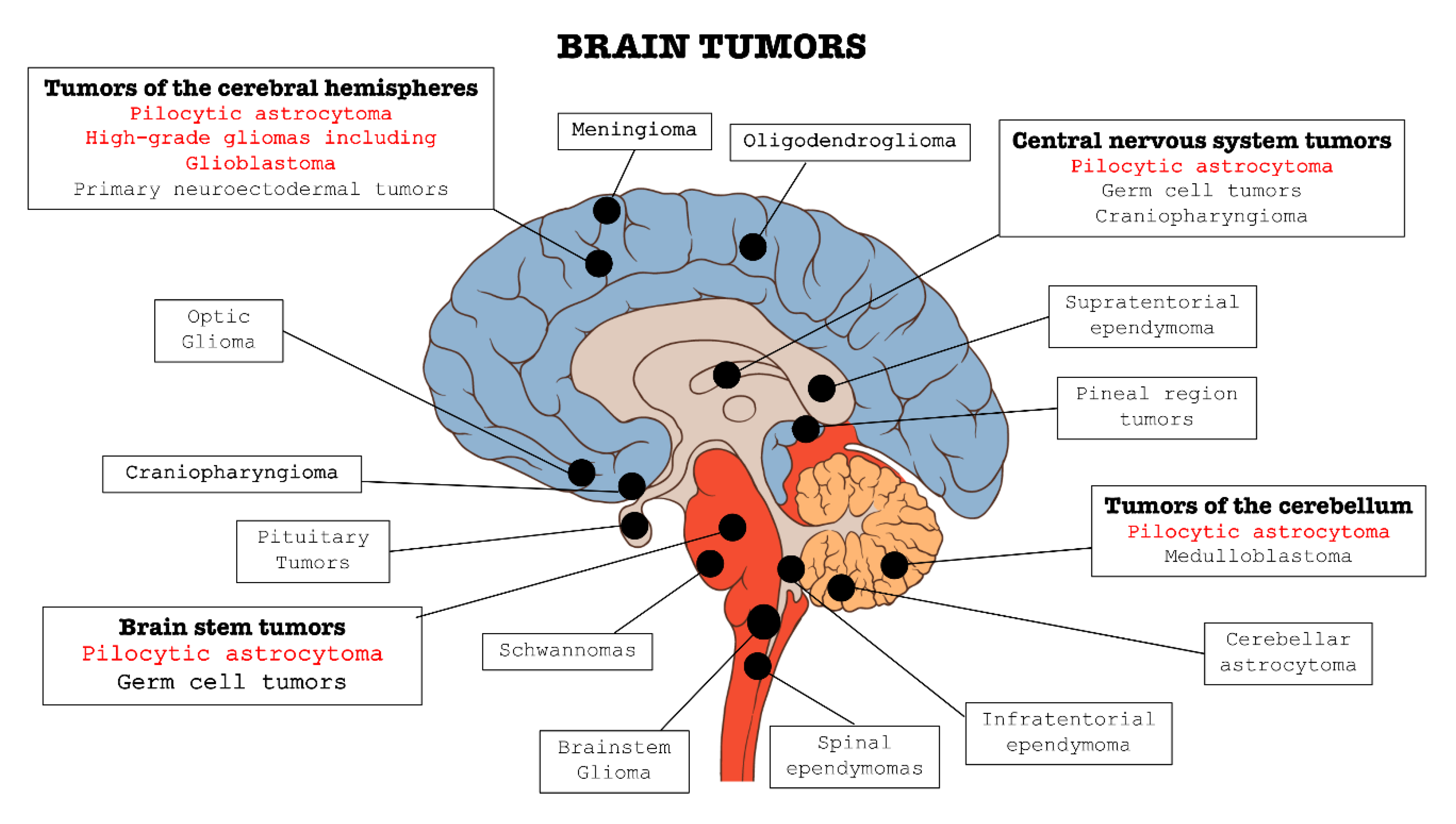
3. Role of FGFRs in Brain Tumors
3.1. Role of FGFRs in Astrocytoma
3.2. Role of FGFRs in Glioblastoma
4. FGFRs Inhibitor
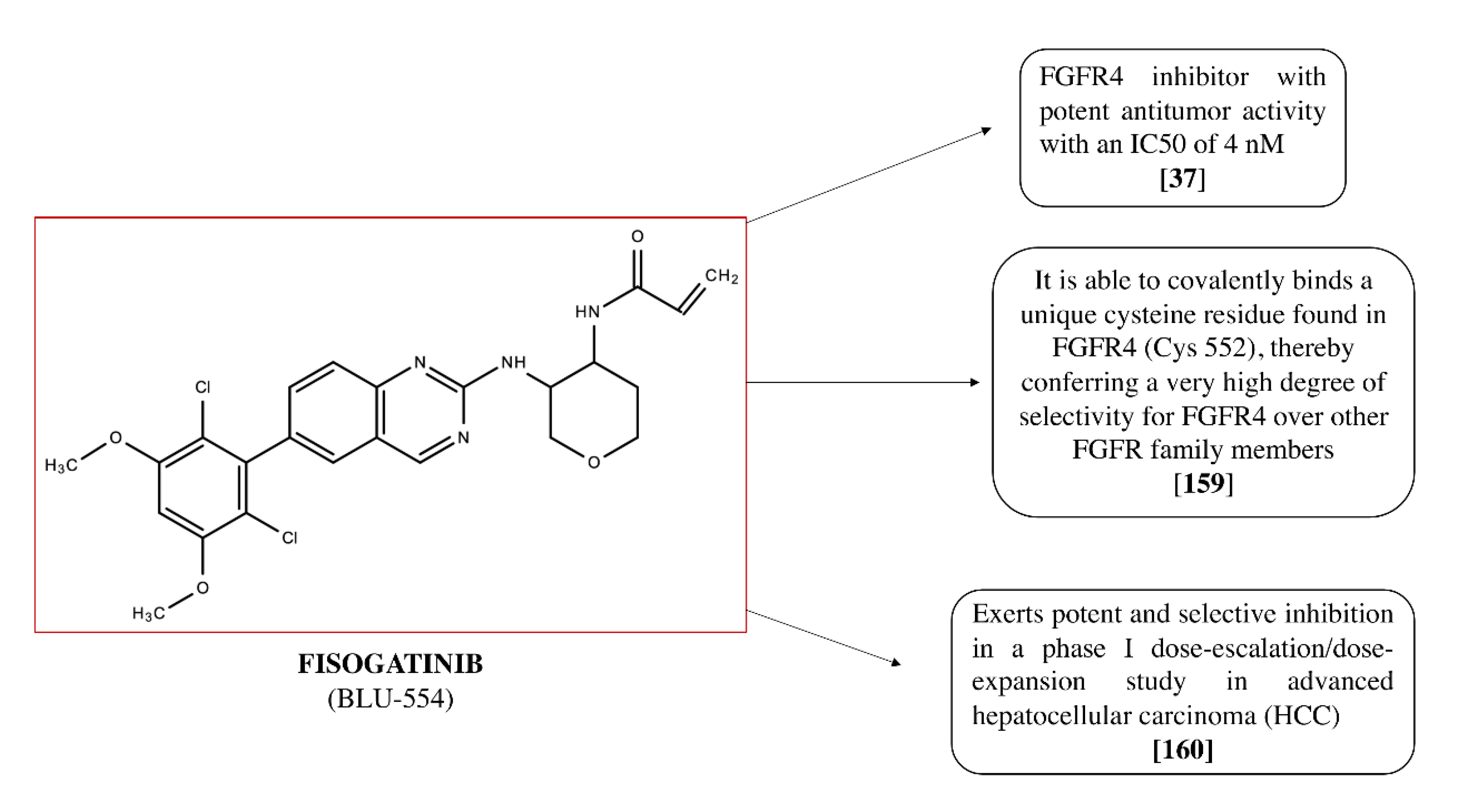
4.1. Fisogatinib
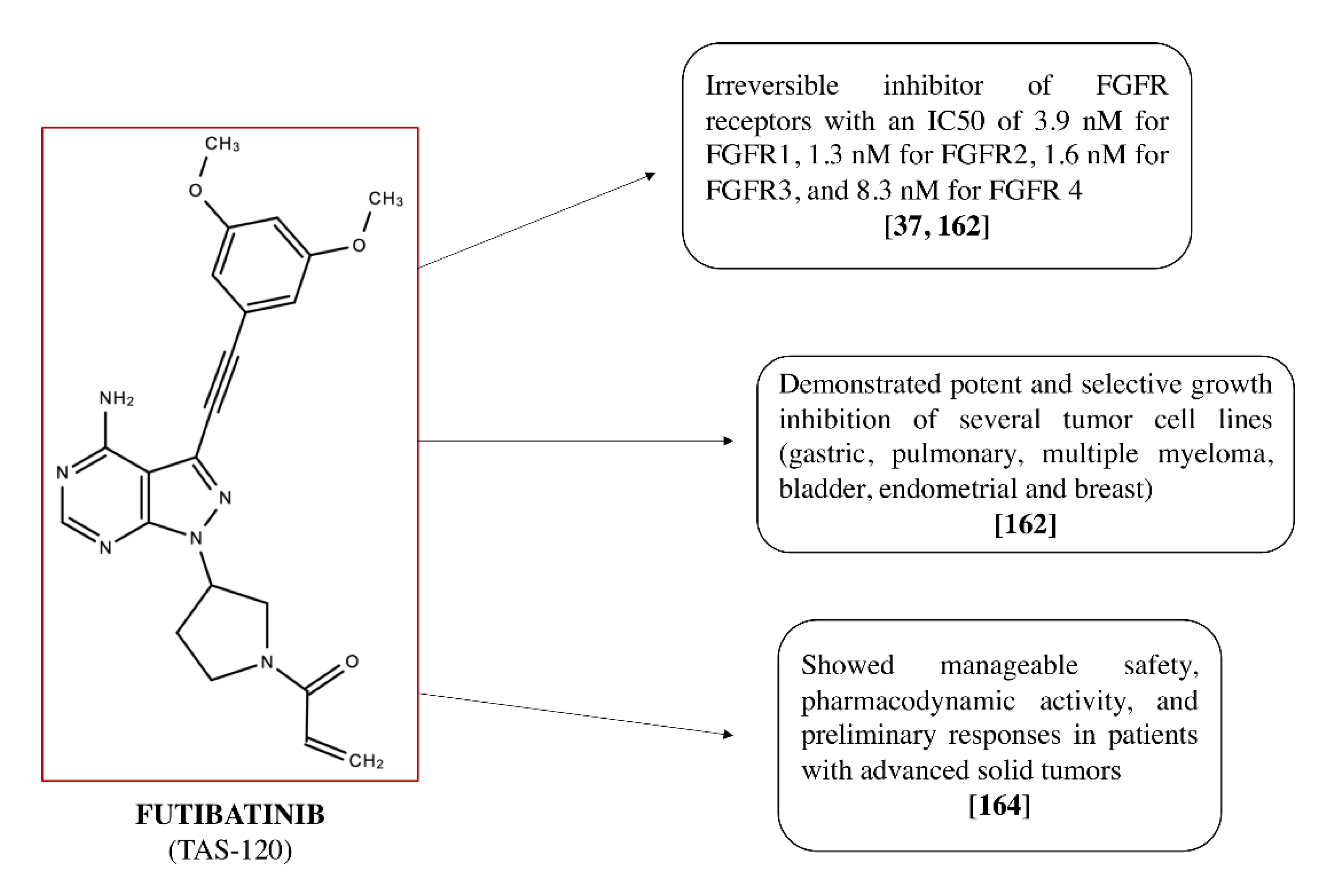
4.2. Futibatinib
4.3. AZD4547
4.4. Infigratinib
5. Conclusions
6. Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Kheirollahi, M.; Dashti, S.; Khalaj, Z.; Nazemroaia, F.; Mahzouni, P. Brain tumors: Special characters for research and banking. Adv. Biomed. Res. 2015, 4, 4. [Google Scholar] [CrossRef] [PubMed]
- Komori, T. The 2016 WHO Classification of Tumours of the Central Nervous System: The Major Points of Revision. Neurol. Med. Chir. 2017, 57, 301–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zacharaki, E.I.; Wang, S.; Chawla, S.; Soo Yoo, D.; Wolf, R.; Melhem, E.R.; Davatzikos, C. Classification of brain tumor type and grade using MRI texture and shape in a machine learning scheme. Magn. Reson. Med. 2009, 62, 1609–1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thakkar, J.P.; Dolecek, T.A.; Horbinski, C.; Ostrom, Q.T.; Lightner, D.D.; Barnholtz-Sloan, J.S.; Villano, J.L. Epidemiologic and molecular prognostic review of glioblastoma. Cancer Epidemiol. Biomark. Prev. 2014, 23, 1985–1996. [Google Scholar] [CrossRef] [Green Version]
- Alentorn, A.; Hoang-Xuan, K.; Mikkelsen, T. Presenting signs and symptoms in brain tumors. Handb. Clin. Neurol. 2016, 134, 19–26. [Google Scholar] [CrossRef]
- Esquenazi, Y.; Lo, V.P.; Lee, K. Critical Care Management of Cerebral Edema in Brain Tumors. J. Intensive Care Med. 2017, 32, 15–24. [Google Scholar] [CrossRef]
- Chen, D.Y.; Chen, C.C.; Crawford, J.R.; Wang, S.G. Tumor-related epilepsy: Epidemiology, pathogenesis and management. J. Neurooncol. 2018, 139, 13–21. [Google Scholar] [CrossRef]
- Hadidchi, S.; Surento, W.; Lerner, A.; Liu, C.J.; Gibbs, W.N.; Kim, P.E.; Shiroishi, M.S. Headache and Brain Tumor. Neuroimaging Clin. N. Am. 2019, 29, 291–300. [Google Scholar] [CrossRef]
- Perkins, A.; Liu, G. Primary Brain Tumors in Adults: Diagnosis and Treatment. Am. Fam. Physician 2016, 93, 211–217. [Google Scholar]
- Karlinska, A.G.; Gromadzka, G.; Karlinski, M.A.; Czlonkowska, A. The activity of malignancy may determine stroke pattern in cancer patients. J. Stroke Cereb. Dis. 2015, 24, 778–783. [Google Scholar] [CrossRef]
- Dietrich, J.; Rao, K.; Pastorino, S.; Kesari, S. Corticosteroids in brain cancer patients: Benefits and pitfalls. Expert Rev. Clin. Pharm. 2011, 4, 233–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nahaczewski, A.E.; Fowler, S.B.; Hariharan, S. Dexamethasone therapy in patients with brain tumors—A focus on tapering. J. Neurosci. Nurs. 2004, 36, 340–343. [Google Scholar] [CrossRef] [PubMed]
- Arvanitis, C.D.; Ferraro, G.B.; Jain, R.K. The blood-brain barrier and blood-tumour barrier in brain tumours and metastases. Nat. Rev. Cancer 2020, 20, 26–41. [Google Scholar] [CrossRef] [PubMed]
- van Tellingen, O.; Yetkin-Arik, B.; de Gooijer, M.C.; Wesseling, P.; Wurdinger, T.; de Vries, H.E. Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug Resist. Updates 2015, 19, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [Green Version]
- Kaina, B.; Christmann, M. DNA repair in personalized brain cancer therapy with temozolomide and nitrosoureas. DNA Repair 2019, 78, 128–141. [Google Scholar] [CrossRef]
- Schreck, K.C.; Grossman, S.A. Role of Temozolomide in the Treatment of Cancers Involving the Central Nervous System. Oncology (Williston Park) 2018, 32, 555–560. [Google Scholar]
- Ma, W.; Li, N.; An, Y.; Zhou, C.; Bo, C.; Zhang, G. Effects of Temozolomide and Radiotherapy on Brain Metastatic Tumor: A Systematic Review and Meta-Analysis. World Neurosurg. 2016, 92, 197–205. [Google Scholar] [CrossRef]
- Parasramka, S.; Talari, G.; Rosenfeld, M.; Guo, J.; Villano, J.L. Procarbazine, lomustine and vincristine for recurrent high-grade glioma. Cochrane Database Syst. Rev. 2017, 7, CD011773. [Google Scholar] [CrossRef]
- Buckner, J.C.; Shaw, E.G.; Pugh, S.L.; Chakravarti, A.; Gilbert, M.R.; Barger, G.R.; Coons, S.; Ricci, P.; Bullard, D.; Brown, P.D.; et al. Radiation plus Procarbazine, CCNU, and Vincristine in Low-Grade Glioma. N. Engl. J. Med. 2016, 374, 1344–1355. [Google Scholar] [CrossRef]
- van den Bent, M.J.; Brandes, A.A.; Taphoorn, M.J.; Kros, J.M.; Kouwenhoven, M.C.; Delattre, J.Y.; Bernsen, H.J.; Frenay, M.; Tijssen, C.C.; Grisold, W.; et al. Adjuvant procarbazine, lomustine, and vincristine chemotherapy in newly diagnosed anaplastic oligodendroglioma: Long-term follow-up of EORTC brain tumor group study 26951. J. Clin. Oncol. 2013, 31, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Grunert, M.; Kassubek, R.; Danz, B.; Klemenz, B.; Hasslacher, S.; Stroh, S.; Schneele, L.; Langhans, J.; Strobele, S.; Barry, S.E.; et al. Radiation and Brain Tumors: An Overview. Crit. Rev. Oncog. 2018, 23, 119–138. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, R.S.; Englander, Z.K.; Canoll, P.; Bruce, J.N. Extent of Resection in Glioma-A Review of the Cutting Edge. World Neurosurg. 2017, 103, 538–549. [Google Scholar] [CrossRef] [PubMed]
- Mut, M. Surgical treatment of brain metastasis: A review. Clin. Neurol. Neurosurg. 2012, 114, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Debinski, W. Receptor-Targeted Glial Brain Tumor Therapies. Int. J. Mol. Sci. 2018, 19, 3326. [Google Scholar] [CrossRef] [Green Version]
- Ornitz, D.M.; Itoh, N. Fibroblast growth factors. Genome Biol. 2001, 2, reviews3005.1. [Google Scholar] [CrossRef] [Green Version]
- Yun, Y.R.; Won, J.E.; Jeon, E.; Lee, S.; Kang, W.; Jo, H.; Jang, J.H.; Shin, U.S.; Kim, H.W. Fibroblast growth factors: Biology, function, and application for tissue regeneration. J. Tissue Eng. 2010, 2010, 218142. [Google Scholar] [CrossRef]
- Armelin, H.A. Pituitary extracts and steroid hormones in the control of 3T3 cell growth. Proc. Natl. Acad. Sci. USA 1973, 70, 2702–2706. [Google Scholar] [CrossRef] [Green Version]
- Gospodarowicz, D. Localisation of a fibroblast growth factor and its effect alone and with hydrocortisone on 3T3 cell growth. Nature 1974, 249, 123–127. [Google Scholar] [CrossRef]
- Katoh, M.; Katoh, M. FGF signaling network in the gastrointestinal tract (review). Int. J. Oncol. 2006, 29, 163–168. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Tian, L.; Yang, F.; Tong, W.; Jia, R.; Zou, Y.; Yin, L.; Li, L.; He, C.; Liang, X.; et al. Tannic Acid Accelerates Cutaneous Wound Healing in Rats Via Activation of the ERK 1/2 Signaling Pathways. Adv. Wound Care (New Rochelle) 2019, 8, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.M.; Su, F.; Kalyana-Sundaram, S.; Khazanov, N.; Ateeq, B.; Cao, X.; Lonigro, R.J.; Vats, P.; Wang, R.; Lin, S.F.; et al. Identification of targetable FGFR gene fusions in diverse cancers. Cancer Discov. 2013, 3, 636–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, N.; Grose, R. Fibroblast growth factor signalling: From development to cancer. Nat. Rev. Cancer 2010, 10, 116–129. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.; Zhou, Z.; Chen, Z.; Xu, G.; Chen, Y. Fibroblast Growth Factor Receptors (FGFRs): Structures and Small Molecule Inhibitors. Cells 2019, 8, 614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mossahebi-Mohammadi, M.; Quan, M.; Zhang, J.S.; Li, X. FGF Signaling Pathway: A Key Regulator of Stem Cell Pluripotency. Front. Cell Dev. Biol. 2020, 8, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goetz, R.; Mohammadi, M. Exploring mechanisms of FGF signalling through the lens of structural biology. Nat. Rev. Mol. Cell Biol. 2013, 14, 166–180. [Google Scholar] [CrossRef] [Green Version]
- Roskoski, R., Jr. The role of fibroblast growth factor receptor (FGFR) protein-tyrosine kinase inhibitors in the treatment of cancers including those of the urinary bladder. Pharmacol. Res. 2020, 151, 104567. [Google Scholar] [CrossRef]
- Wang, J.F.; Shen, M.; Fong, G.H.; Hill, D.J. A soluble fibroblast growth factor receptor is released from HL-60 promyelocytic leukemia cells: Implications for paracrine growth control. Growth Factors 2000, 17, 203–214. [Google Scholar] [CrossRef]
- Harmer, N.J. Insights into the role of heparan sulphate in fibroblast growth factor signalling. Biochem. Soc. Trans. 2006, 34, 442–445. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, L.; Trueb, B. Evolution of the fusogenic activity of the receptor FGFRL1. Arch. Biochem. Biophys. 2017, 625–626, 54–64. [Google Scholar] [CrossRef]
- Farrell, B.; Breeze, A.L. Structure, activation and dysregulation of fibroblast growth factor receptor kinases: Perspectives for clinical targeting. Biochem. Soc. Trans. 2018, 46, 1753–1770. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Liang, X.; Beaudet, J.M.; Lee, Y.; Linhardt, R.J. The Effects of Metal Ions on Heparin/Heparin Sulfate-Protein Interactions. J. Biomed. Technol. Res. 2014, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meneghetti, M.C.; Hughes, A.J.; Rudd, T.R.; Nader, H.B.; Powell, A.K.; Yates, E.A.; Lima, M.A. Heparan sulfate and heparin interactions with proteins. J. R. Soc. Interface 2015, 12, 0589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kucinska, M.; Porebska, N.; Lampart, A.; Latko, M.; Knapik, A.; Zakrzewska, M.; Otlewski, J.; Opalinski, L. Differential regulation of fibroblast growth factor receptor 1 trafficking and function by extracellular galectins. Cell Commun. Signal. 2019, 17, 65. [Google Scholar] [CrossRef] [Green Version]
- Touat, M.; Ileana, E.; Postel-Vinay, S.; Andre, F.; Soria, J.C. Targeting FGFR Signaling in Cancer. Clin. Cancer Res. 2015, 21, 2684–2694. [Google Scholar] [CrossRef] [Green Version]
- Eswarakumar, V.P.; Lax, I.; Schlessinger, J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005, 16, 139–149. [Google Scholar] [CrossRef]
- Schlessinger, J. Common and distinct elements in cellular signaling via EGF and FGF receptors. Science 2004, 306, 1506–1507. [Google Scholar] [CrossRef]
- Sarabipour, S.; Hristova, K. Mechanism of FGF receptor dimerization and activation. Nat. Commun. 2016, 7, 10262. [Google Scholar] [CrossRef]
- Ornitz, D.M.; Itoh, N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 215–266. [Google Scholar] [CrossRef] [Green Version]
- Shalaby, A.A.; Presneau, N.; Idowu, B.D.; Thompson, L.; Briggs, T.R.; Tirabosco, R.; Diss, T.C.; Flanagan, A.M. Analysis of the fibroblastic growth factor receptor-RAS/RAF/MEK/ERK-ETS2/brachyury signalling pathway in chordomas. Mod. Pathol. 2009, 22, 996–1005. [Google Scholar] [CrossRef] [Green Version]
- Latko, M.; Czyrek, A.; Porebska, N.; Kucinska, M.; Otlewski, J.; Zakrzewska, M.; Opalinski, L. Cross-Talk between Fibroblast Growth Factor Receptors and Other Cell Surface Proteins. Cells 2019, 8, 455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiselyov, V.V.; Soroka, V.; Berezin, V.; Bock, E. Structural biology of NCAM homophilic binding and activation of FGFR. J. Neurochem. 2005, 94, 1169–1179. [Google Scholar] [CrossRef] [PubMed]
- Doherty, P.; Ashton, S.V.; Moore, S.E.; Walsh, F.S. Morphoregulatory activities of NCAM and N-cadherin can be accounted for by G protein-dependent activation of L- and N-type neuronal Ca2+ channels. Cell 1991, 67, 21–33. [Google Scholar] [CrossRef]
- Williams, E.J.; Furness, J.; Walsh, F.S.; Doherty, P. Activation of the FGF receptor underlies neurite outgrowth stimulated by L1, N-CAM, and N-cadherin. Neuron 1994, 13, 583–594. [Google Scholar] [CrossRef]
- Saffell, J.L.; Williams, E.J.; Mason, I.J.; Walsh, F.S.; Doherty, P. Expression of a dominant negative FGF receptor inhibits axonal growth and FGF receptor phosphorylation stimulated by CAMs. Neuron 1997, 18, 231–242. [Google Scholar] [CrossRef] [Green Version]
- Meiri, K.F.; Saffell, J.L.; Walsh, F.S.; Doherty, P. Neurite outgrowth stimulated by neural cell adhesion molecules requires growth-associated protein-43 (GAP-43) function and is associated with GAP-43 phosphorylation in growth cones. J. Neurosci. 1998, 18, 10429–10437. [Google Scholar] [CrossRef]
- Ronn, L.C.; Doherty, P.; Holm, A.; Berezin, V.; Bock, E. Neurite outgrowth induced by a synthetic peptide ligand of neural cell adhesion molecule requires fibroblast growth factor receptor activation. J. Neurochem. 2000, 75, 665–671. [Google Scholar] [CrossRef]
- Niethammer, P.; Delling, M.; Sytnyk, V.; Dityatev, A.; Fukami, K.; Schachner, M. Cosignaling of NCAM via lipid rafts and the FGF receptor is required for neuritogenesis. J. Cell Biol. 2002, 157, 521–532. [Google Scholar] [CrossRef]
- Doherty, P.; Walsh, F.S. CAM-FGF receptor interactions: A model for axonal growth. Mol. Cell. Neurosci. 1996, 8, 99–111. [Google Scholar] [CrossRef]
- Peluso, J.J. N-cadherin-mediated cell contact regulates ovarian surface epithelial cell survival. Biol. Signals Recept. 2000, 9, 115–121. [Google Scholar] [CrossRef]
- Erez, N.; Zamir, E.; Gour, B.J.; Blaschuk, O.W.; Geiger, B. Induction of apoptosis in cultured endothelial cells by a cadherin antagonist peptide: Involvement of fibroblast growth factor receptor-mediated signalling. Exp. Cell Res. 2004, 294, 366–378. [Google Scholar] [CrossRef] [PubMed]
- Suyama, K.; Shapiro, I.; Guttman, M.; Hazan, R.B. A signaling pathway leading to metastasis is controlled by N-cadherin and the FGF receptor. Cancer Cell 2002, 2, 301–314. [Google Scholar] [CrossRef] [Green Version]
- Cavallaro, U.; Niedermeyer, J.; Fuxa, M.; Christofori, G. N-CAM modulates tumour-cell adhesion to matrix by inducing FGF-receptor signalling. Nat. Cell Biol. 2001, 3, 650–657. [Google Scholar] [CrossRef] [PubMed]
- Bottcher, R.T.; Niehrs, C. Fibroblast growth factor signaling during early vertebrate development. Endocr. Rev. 2005, 26, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Mege, R.M. N-Cadherin and Fibroblast Growth Factor Receptors crosstalk in the control of developmental and cancer cell migrations. Eur. J. Cell Biol. 2016, 95, 415–426. [Google Scholar] [CrossRef]
- Hulit, J.; Suyama, K.; Chung, S.; Keren, R.; Agiostratidou, G.; Shan, W.; Dong, X.; Williams, T.M.; Lisanti, M.P.; Knudsen, K.; et al. N-cadherin signaling potentiates mammary tumor metastasis via enhanced extracellular signal-regulated kinase activation. Cancer Res. 2007, 67, 3106–3116. [Google Scholar] [CrossRef] [Green Version]
- Qian, X.; Anzovino, A.; Kim, S.; Suyama, K.; Yao, J.; Hulit, J.; Agiostratidou, G.; Chandiramani, N.; McDaid, H.M.; Nagi, C.; et al. N-cadherin/FGFR promotes metastasis through epithelial-to-mesenchymal transition and stem/progenitor cell-like properties. Oncogene 2014, 33, 3411–3421. [Google Scholar] [CrossRef] [Green Version]
- Colombo, F.; Meldolesi, J. L1-CAM and N-CAM: From Adhesion Proteins to Pharmacological Targets. Trends Pharm. Sci. 2015, 36, 769–781. [Google Scholar] [CrossRef]
- Sytnyk, V.; Leshchyns’ka, I.; Schachner, M. Neural Cell Adhesion Molecules of the Immunoglobulin Superfamily Regulate Synapse Formation, Maintenance, and Function. Trends Neurosci. 2017, 40, 295–308. [Google Scholar] [CrossRef]
- Sanchez-Heras, E.; Howell, F.V.; Williams, G.; Doherty, P. The fibroblast growth factor receptor acid box is essential for interactions with N-cadherin and all of the major isoforms of neural cell adhesion molecule. J. Biol. Chem. 2006, 281, 35208–35216. [Google Scholar] [CrossRef] [Green Version]
- Francavilla, C.; Cattaneo, P.; Berezin, V.; Bock, E.; Ami, D.; de Marco, A.; Christofori, G.; Cavallaro, U. The binding of NCAM to FGFR1 induces a specific cellular response mediated by receptor trafficking. J. Cell Biol. 2009, 187, 1101–1116. [Google Scholar] [CrossRef] [PubMed]
- Christensen, C.; Lauridsen, J.B.; Berezin, V.; Bock, E.; Kiselyov, V.V. The neural cell adhesion molecule binds to fibroblast growth factor receptor 2. FEBS Lett. 2006, 580, 3386–3390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francavilla, C.; Loeffler, S.; Piccini, D.; Kren, A.; Christofori, G.; Cavallaro, U. Neural cell adhesion molecule regulates the cellular response to fibroblast growth factor. J. Cell Sci. 2007, 120, 4388–4394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amoureux, M.C.; Coulibaly, B.; Chinot, O.; Loundou, A.; Metellus, P.; Rougon, G.; Figarella-Branger, D. Polysialic acid neural cell adhesion molecule (PSA-NCAM) is an adverse prognosis factor in glioblastoma, and regulates olig2 expression in glioma cell lines. BMC Cancer 2010, 10, 91. [Google Scholar] [CrossRef] [PubMed]
- Ligon, K.L.; Huillard, E.; Mehta, S.; Kesari, S.; Liu, H.; Alberta, J.A.; Bachoo, R.M.; Kane, M.; Louis, D.N.; Depinho, R.A.; et al. Olig2-regulated lineage-restricted pathway controls replication competence in neural stem cells and malignant glioma. Neuron 2007, 53, 503–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bribian, A.; Barallobre, M.J.; Soussi-Yanicostas, N.; de Castro, F. Anosmin-1 modulates the FGF-2-dependent migration of oligodendrocyte precursors in the developing optic nerve. Mol. Cell. Neurosci. 2006, 33, 2–14. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gonzalez, D.; Clemente, D.; Coelho, M.; Esteban, P.F.; Soussi-Yanicostas, N.; de Castro, F. Dynamic roles of FGF-2 and Anosmin-1 in the migration of neuronal precursors from the subventricular zone during pre- and postnatal development. Exp. Neurol. 2010, 222, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Murcia-Belmonte, V.; Esteban, P.F.; Garcia-Gonzalez, D.; De Castro, F. Biochemical dissection of Anosmin-1 interaction with FGFR1 and components of the extracellular matrix. J. Neurochem. 2010, 115, 1256–1265. [Google Scholar] [CrossRef]
- Mohanan, V.; Temburni, M.K.; Kappes, J.C.; Galileo, D.S. L1CAM stimulates glioma cell motility and proliferation through the fibroblast growth factor receptor. Clin. Exp. Metastasis 2013, 30, 507–520. [Google Scholar] [CrossRef]
- Bale, T.A. FGFR- gene family alterations in low-grade neuroepithelial tumors. Acta Neuropathol. Commun. 2020, 8, 21. [Google Scholar] [CrossRef] [Green Version]
- Rand, V.; Huang, J.; Stockwell, T.; Ferriera, S.; Buzko, O.; Levy, S.; Busam, D.; Li, K.; Edwards, J.B.; Eberhart, C.; et al. Sequence survey of receptor tyrosine kinases reveals mutations in glioblastomas. Proc. Natl. Acad. Sci. USA 2005, 102, 14344–14349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, R.M.; Goriely, A.; Wall, S.A.; Roberts, I.S.; Wilkie, A.O. Fibroblast growth factor receptor 2, gain-of-function mutations, and tumourigenesis: Investigating a potential link. J. Pathol. 2005, 207, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Chae, Y.K.; Ranganath, K.; Hammerman, P.S.; Vaklavas, C.; Mohindra, N.; Kalyan, A.; Matsangou, M.; Costa, R.; Carneiro, B.; Villaflor, V.M.; et al. Inhibition of the fibroblast growth factor receptor (FGFR) pathway: The current landscape and barriers to clinical application. Oncotarget 2017, 8, 16052–16074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dienstmann, R.; Rodon, J.; Prat, A.; Perez-Garcia, J.; Adamo, B.; Felip, E.; Cortes, J.; Iafrate, A.J.; Nuciforo, P.; Tabernero, J. Genomic aberrations in the FGFR pathway: Opportunities for targeted therapies in solid tumors. Ann. Oncol. 2014, 25, 552–563. [Google Scholar] [CrossRef]
- Preusser, M.; Berghoff, A.S.; Berger, W.; Ilhan-Mutlu, A.; Dinhof, C.; Widhalm, G.; Dieckmann, K.; Wohrer, A.; Hackl, M.; von Deimling, A.; et al. High rate of FGFR1 amplifications in brain metastases of squamous and non-squamous lung cancer. Lung Cancer 2014, 83, 83–89. [Google Scholar] [CrossRef]
- Freier, K.; Schwaenen, C.; Sticht, C.; Flechtenmacher, C.; Muhling, J.; Hofele, C.; Radlwimmer, B.; Lichter, P.; Joos, S. Recurrent FGFR1 amplification and high FGFR1 protein expression in oral squamous cell carcinoma (OSCC). Oral Oncol. 2007, 43, 60–66. [Google Scholar] [CrossRef]
- Gorringe, K.L.; Jacobs, S.; Thompson, E.R.; Sridhar, A.; Qiu, W.; Choong, D.Y.; Campbell, I.G. High-resolution single nucleotide polymorphism array analysis of epithelial ovarian cancer reveals numerous microdeletions and amplifications. Clin. Cancer Res. 2007, 13, 4731–4739. [Google Scholar] [CrossRef] [Green Version]
- Simon, R.; Richter, J.; Wagner, U.; Fijan, A.; Bruderer, J.; Schmid, U.; Ackermann, D.; Maurer, R.; Alund, G.; Knonagel, H.; et al. High-throughput tissue microarray analysis of 3p25 (RAF1) and 8p12 (FGFR1) copy number alterations in urinary bladder cancer. Cancer Res. 2001, 61, 4514–4519. [Google Scholar]
- Missiaglia, E.; Selfe, J.; Hamdi, M.; Williamson, D.; Schaaf, G.; Fang, C.; Koster, J.; Summersgill, B.; Messahel, B.; Versteeg, R.; et al. Genomic imbalances in rhabdomyosarcoma cell lines affect expression of genes frequently altered in primary tumors: An approach to identify candidate genes involved in tumor development. Genes Chromosomes Cancer 2009, 48, 455–467. [Google Scholar] [CrossRef]
- Jackson, C.C.; Medeiros, L.J.; Miranda, R.N. 8p11 myeloproliferative syndrome: A review. Hum. Pathol. 2010, 41, 461–476. [Google Scholar] [CrossRef]
- Dutt, A.; Salvesen, H.B.; Chen, T.H.; Ramos, A.H.; Onofrio, R.C.; Hatton, C.; Nicoletti, R.; Winckler, W.; Grewal, R.; Hanna, M.; et al. Drug-sensitive FGFR2 mutations in endometrial carcinoma. Proc. Natl. Acad. Sci. USA 2008, 105, 8713–8717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, E.J.; Jung, E.J.; Min, S.Y.; Kim, M.A.; Kim, W.H. Fibroblast growth factor receptor 2 gene amplification status and its clinicopathologic significance in gastric carcinoma. Hum. Pathol. 2012, 43, 1559–1566. [Google Scholar] [CrossRef] [PubMed]
- Hunter, D.J.; Kraft, P.; Jacobs, K.B.; Cox, D.G.; Yeager, M.; Hankinson, S.E.; Wacholder, S.; Wang, Z.; Welch, R.; Hutchinson, A.; et al. A genome-wide association study identifies alleles in FGFR2 associated with risk of sporadic postmenopausal breast cancer. Nat. Genet. 2007, 39, 870–874. [Google Scholar] [CrossRef] [PubMed]
- Tchaicha, J.H.; Akbay, E.A.; Altabef, A.; Mikse, O.R.; Kikuchi, E.; Rhee, K.; Liao, R.G.; Bronson, R.T.; Sholl, L.M.; Meyerson, M.; et al. Kinase domain activation of FGFR2 yields high-grade lung adenocarcinoma sensitive to a Pan-FGFR inhibitor in a mouse model of NSCLC. Cancer Res. 2014, 74, 4676–4684. [Google Scholar] [CrossRef] [Green Version]
- Tanizaki, J.; Ercan, D.; Capelletti, M.; Dodge, M.; Xu, C.; Bahcall, M.; Tricker, E.M.; Butaney, M.; Calles, A.; Sholl, L.M.; et al. Identification of Oncogenic and Drug-Sensitizing Mutations in the Extracellular Domain of FGFR2. Cancer Res. 2015, 75, 3139–3146. [Google Scholar] [CrossRef] [Green Version]
- Arai, Y.; Totoki, Y.; Hosoda, F.; Shirota, T.; Hama, N.; Nakamura, H.; Ojima, H.; Furuta, K.; Shimada, K.; Okusaka, T.; et al. Fibroblast growth factor receptor 2 tyrosine kinase fusions define a unique molecular subtype of cholangiocarcinoma. Hepatology 2014, 59, 1427–1434. [Google Scholar] [CrossRef]
- Dieci, M.V.; Arnedos, M.; Andre, F.; Soria, J.C. Fibroblast growth factor receptor inhibitors as a cancer treatment: From a biologic rationale to medical perspectives. Cancer Discov. 2013, 3, 264–279. [Google Scholar] [CrossRef] [Green Version]
- Katoh, M.; Nakagama, H. FGF receptors: Cancer biology and therapeutics. Med. Res. Rev. 2014, 34, 280–300. [Google Scholar] [CrossRef]
- Li, S.Q.; Cheuk, A.T.; Shern, J.F.; Song, Y.K.; Hurd, L.; Liao, H.; Wei, J.S.; Khan, J. Targeting wild-type and mutationally activated FGFR4 in rhabdomyosarcoma with the inhibitor ponatinib (AP24534). PLoS ONE 2013, 8, e76551. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Stockton, D.W.; Ittmann, M. The fibroblast growth factor receptor-4 Arg388 allele is associated with prostate cancer initiation and progression. Clin. Cancer Res. 2004, 10, 6169–6178. [Google Scholar] [CrossRef] [Green Version]
- Bange, J.; Prechtl, D.; Cheburkin, Y.; Specht, K.; Harbeck, N.; Schmitt, M.; Knyazeva, T.; Muller, S.; Gartner, S.; Sures, I.; et al. Cancer progression and tumor cell motility are associated with the FGFR4 Arg(388) allele. Cancer Res. 2002, 62, 840–847. [Google Scholar] [PubMed]
- Desnoyers, L.R.; Pai, R.; Ferrando, R.E.; Hotzel, K.; Le, T.; Ross, J.; Carano, R.; D’Souza, A.; Qing, J.; Mohtashemi, I.; et al. Targeting FGF19 inhibits tumor growth in colon cancer xenograft and FGF19 transgenic hepatocellular carcinoma models. Oncogene 2008, 27, 85–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrom, Q.T.; Bauchet, L.; Davis, F.G.; Deltour, I.; Fisher, J.L.; Langer, C.E.; Pekmezci, M.; Schwartzbaum, J.A.; Turner, M.C.; Walsh, K.M.; et al. The epidemiology of glioma in adults: A “state of the science” review. Neuro Oncol. 2014, 16, 896–913. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Smith-Cohn, M.; Cohen, A.L.; Colman, H. Glioma Subclassifications and Their Clinical Significance. Neurotherapeutics 2017, 14, 284–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gusyatiner, O.; Hegi, M.E. Glioma epigenetics: From subclassification to novel treatment options. Semin. Cancer Biol. 2018, 51, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Ferris, S.P.; Hofmann, J.W.; Solomon, D.A.; Perry, A. Characterization of gliomas: From morphology to molecules. Virchows Arch. 2017, 471, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Wesche, J.; Haglund, K.; Haugsten, E.M. Fibroblast growth factors and their receptors in cancer. Biochem. J. 2011, 437, 199–213. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.T.; Hutter, B.; Jager, N.; Korshunov, A.; Kool, M.; Warnatz, H.J.; Zichner, T.; Lambert, S.R.; Ryzhova, M.; Quang, D.A.; et al. Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat. Genet. 2013, 45, 927–932. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Research, N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [Green Version]
- Frattini, V.; Trifonov, V.; Chan, J.M.; Castano, A.; Lia, M.; Abate, F.; Keir, S.T.; Ji, A.X.; Zoppoli, P.; Niola, F.; et al. The integrated landscape of driver genomic alterations in glioblastoma. Nat. Genet. 2013, 45, 1141–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapoor, M.; Gupta, V. Astrocytoma. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Freeman, M.R. Specification and morphogenesis of astrocytes. Science 2010, 330, 774–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanfield, C.L.; Germann, W.J.; Niles, M.J.; Cannon, J.G. Principles of Human Physiology; Benjamin Cummings USA: San Francisco, CA, USA, 2011. [Google Scholar]
- Furnari, F.B.; Fenton, T.; Bachoo, R.M.; Mukasa, A.; Stommel, J.M.; Stegh, A.; Hahn, W.C.; Ligon, K.L.; Louis, D.N.; Brennan, C.; et al. Malignant astrocytic glioma: Genetics, biology, and paths to treatment. Genes Dev. 2007, 21, 2683–2710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrom, Q.T.; Gittleman, H.; Fulop, J.; Liu, M.; Blanda, R.; Kromer, C.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2008–2012. Neuro Oncol. 2015, 17 (Suppl. 4), iv1–iv62. [Google Scholar] [CrossRef]
- Sievert, A.J.; Fisher, M.J. Pediatric low-grade gliomas. J. Child. Neurol. 2009, 24, 1397–1408. [Google Scholar] [CrossRef] [Green Version]
- Ryall, S.; Tabori, U.; Hawkins, C. Pediatric low-grade glioma in the era of molecular diagnostics. Acta Neuropathol. Commun. 2020, 8, 30. [Google Scholar] [CrossRef]
- Wen, P.Y.; Kesari, S. Malignant gliomas in adults. N. Engl. J. Med. 2008, 359, 492–507. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, S.; Ghosh, Z. A systemic insight into astrocytoma biology across different grades. J. Cell. Physiol. 2019, 234, 4243–4255. [Google Scholar] [CrossRef]
- Pecina-Slaus, N.; Kafka, A.; Varosanec, A.M.; Markovic, L.; Krsnik, Z.; Njiric, N.; Mrak, G. Expression patterns of Wnt signaling component, secreted frizzledrelated protein 3 in astrocytoma and glioblastoma. Mol. Med. Rep. 2016, 13, 4245–4251. [Google Scholar] [CrossRef] [Green Version]
- Wesseling, P.; Capper, D. WHO 2016 Classification of gliomas. Neuropathol. Appl. Neurobiol. 2018, 44, 139–150. [Google Scholar] [CrossRef]
- Miller, J.J.; Shih, H.A.; Andronesi, O.C.; Cahill, D.P. Isocitrate dehydrogenase-mutant glioma: Evolving clinical and therapeutic implications. Cancer 2017, 123, 4535–4546. [Google Scholar] [CrossRef] [PubMed]
- Waitkus, M.S.; Diplas, B.H.; Yan, H. Isocitrate dehydrogenase mutations in gliomas. Neuro Oncol. 2016, 18, 16–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Eckel-Passow, J.E.; Lachance, D.H.; Molinaro, A.M.; Walsh, K.M.; Decker, P.A.; Sicotte, H.; Pekmezci, M.; Rice, T.; Kosel, M.L.; Smirnov, I.V.; et al. Glioma Groups Based on 1p/19q, IDH, and TERT Promoter Mutations in Tumors. N. Engl. J. Med. 2015, 372, 2499–2508. [Google Scholar] [CrossRef] [Green Version]
- Killela, P.J.; Reitman, Z.J.; Jiao, Y.; Bettegowda, C.; Agrawal, N.; Diaz, L.A., Jr.; Friedman, A.H.; Friedman, H.; Gallia, G.L.; Giovanella, B.C.; et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc. Natl. Acad. Sci. USA 2013, 110, 6021–6026. [Google Scholar] [CrossRef] [Green Version]
- Lew, E.D.; Furdui, C.M.; Anderson, K.S.; Schlessinger, J. The precise sequence of FGF receptor autophosphorylation is kinetically driven and is disrupted by oncogenic mutations. Sci Signal. 2009, 2, ra6. [Google Scholar] [CrossRef] [Green Version]
- Parker, B.C.; Engels, M.; Annala, M.; Zhang, W. Emergence of FGFR family gene fusions as therapeutic targets in a wide spectrum of solid tumours. J. Pathol. 2014, 232, 4–15. [Google Scholar] [CrossRef]
- Sie, M.; den Dunnen, W.F.; Lourens, H.J.; Meeuwsen-de Boer, T.G.; Scherpen, F.J.; Zomerman, W.W.; Kampen, K.R.; Hoving, E.W.; de Bont, E.S. Growth-factor-driven rescue to receptor tyrosine kinase (RTK) inhibitors through Akt and Erk phosphorylation in pediatric low grade astrocytoma and ependymoma. PLoS ONE 2015, 10, e0122555. [Google Scholar] [CrossRef]
- Trisolini, E.; Wardighi, D.E.; Giry, M.; Bernardi, P.; Boldorini, R.L.; Mokhtari, K.; Sanson, M. Actionable FGFR1 and BRAF mutations in adult circumscribed gliomas. J. Neurooncol. 2019, 145, 241–245. [Google Scholar] [CrossRef]
- Lehtinen, B.; Raita, A.; Kesseli, J.; Annala, M.; Nordfors, K.; Yli-Harja, O.; Zhang, W.; Visakorpi, T.; Nykter, M.; Haapasalo, H.; et al. Clinical association analysis of ependymomas and pilocytic astrocytomas reveals elevated FGFR3 and FGFR1 expression in aggressive ependymomas. BMC Cancer 2017, 17, 310. [Google Scholar] [CrossRef] [Green Version]
- Granberg, K.J.; Annala, M.; Lehtinen, B.; Kesseli, J.; Haapasalo, J.; Ruusuvuori, P.; Yli-Harja, O.; Visakorpi, T.; Haapasalo, H.; Nykter, M.; et al. Strong FGFR3 staining is a marker for FGFR3 fusions in diffuse gliomas. Neuro Oncol. 2017, 19, 1206–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frattini, V.; Pagnotta, S.M.; Tala; Fan, J.J.; Russo, M.V.; Lee, S.B.; Garofano, L.; Zhang, J.; Shi, P.; Lewis, G.; et al. A metabolic function of FGFR3-TACC3 gene fusions in cancer. Nature 2018, 553, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Lasorella, A.; Sanson, M.; Iavarone, A. FGFR-TACC gene fusions in human glioma. Neuro Oncol. 2017, 19, 475–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, H.; Lebrun, D.G.; Yang, J.; Zhu, V.F.; Li, M. Deregulated signaling pathways in glioblastoma multiforme: Molecular mechanisms and therapeutic targets. Cancer Investig. 2012, 30, 48–56. [Google Scholar] [CrossRef] [Green Version]
- Sasmita, A.O.; Wong, Y.P.; Ling, A.P.K. Biomarkers and therapeutic advances in glioblastoma multiforme. Asia Pac. J. Clin. Oncol. 2018, 14, 40–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razavi, S.M.; Lee, K.E.; Jin, B.E.; Aujla, P.S.; Gholamin, S.; Li, G. Immune Evasion Strategies of Glioblastoma. Front. Surg. 2016, 3, 11. [Google Scholar] [CrossRef]
- Weathers, S.P.; Gilbert, M.R. Current challenges in designing GBM trials for immunotherapy. J. Neurooncol. 2015, 123, 331–337. [Google Scholar] [CrossRef]
- Wrensch, M.; Minn, Y.; Chew, T.; Bondy, M.; Berger, M.S. Epidemiology of primary brain tumors: Current concepts and review of the literature. Neuro Oncol. 2002, 4, 278–299. [Google Scholar] [CrossRef]
- Omuro, A.; DeAngelis, L.M. Glioblastoma and other malignant gliomas: A clinical review. JAMA 2013, 310, 1842–1850. [Google Scholar] [CrossRef]
- Alifieris, C.; Trafalis, D.T. Glioblastoma multiforme: Pathogenesis and treatment. Pharmacol. Ther. 2015, 152, 63–82. [Google Scholar] [CrossRef]
- Guan, X.; Hasan, M.N.; Maniar, S.; Jia, W.; Sun, D. Reactive Astrocytes in Glioblastoma Multiforme. Mol. Neurobiol. 2018, 55, 6927–6938. [Google Scholar] [CrossRef] [PubMed]
- Crespo, I.; Vital, A.L.; Nieto, A.B.; Rebelo, O.; Tao, H.; Lopes, M.C.; Oliveira, C.R.; French, P.J.; Orfao, A.; Tabernero, M.D. Detailed characterization of alterations of chromosomes 7, 9, and 10 in glioblastomas as assessed by single-nucleotide polymorphism arrays. J. Mol. Diagn. 2011, 13, 634–647. [Google Scholar] [CrossRef] [PubMed]
- Wemmert, S.; Ketter, R.; Rahnenfuhrer, J.; Beerenwinkel, N.; Strowitzki, M.; Feiden, W.; Hartmann, C.; Lengauer, T.; Stockhammer, F.; Zang, K.D.; et al. Patients with high-grade gliomas harboring deletions of chromosomes 9p and 10q benefit from temozolomide treatment. Neoplasia 2005, 7, 883–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Jiang, C.-C.; Ho-Keung, N.; Pang, J.C.; Tong, C.Y. Chromosome 17P may harbor multiple tumor suppressor genes associated with primary glioblastoma multiforme. Chin. J. Cancer Res. 2002, 14, 60–63. [Google Scholar] [CrossRef]
- Jimenez-Pascual, A.; Siebzehnrubl, F.A. Fibroblast Growth Factor Receptor Functions in Glioblastoma. Cells 2019, 8, 715. [Google Scholar] [CrossRef] [Green Version]
- Babina, I.S.; Turner, N.C. Advances and challenges in targeting FGFR signalling in cancer. Nat. Rev. Cancer 2017, 17, 318–332. [Google Scholar] [CrossRef]
- Singh, D.; Chan, J.M.; Zoppoli, P.; Niola, F.; Sullivan, R.; Castano, A.; Liu, E.M.; Reichel, J.; Porrati, P.; Pellegatta, S.; et al. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science 2012, 337, 1231–1235. [Google Scholar] [CrossRef] [Green Version]
- Hierro, C.; Rodon, J.; Tabernero, J. Fibroblast Growth Factor (FGF) Receptor/FGF Inhibitors: Novel Targets and Strategies for Optimization of Response of Solid Tumors. Semin. Oncol. 2015, 42, 801–819. [Google Scholar] [CrossRef]
- Peset, I.; Vernos, I. The TACC proteins: TACC-ling microtubule dynamics and centrosome function. Trends Cell Biol. 2008, 18, 379–388. [Google Scholar] [CrossRef]
- Costa, R.; Carneiro, B.A.; Taxter, T.; Tavora, F.A.; Kalyan, A.; Pai, S.A.; Chae, Y.K.; Giles, F.J. FGFR3-TACC3 fusion in solid tumors: Mini review. Oncotarget 2016, 7, 55924–55938. [Google Scholar] [CrossRef] [Green Version]
- Kwiatkowska, A.; Symons, M. Signaling Determinants of Glioma Cell Invasion. Adv. Exp. Med. Biol. 2020, 1202, 129–149. [Google Scholar] [CrossRef]
- Al-Koussa, H.; Atat, O.E.; Jaafar, L.; Tashjian, H.; El-Sibai, M. The Role of Rho GTPases in Motility and Invasion of Glioblastoma Cells. Anal. Cell. Pathol. 2020, 2020, 9274016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxena, A.; Ali, I.U. Increased expression of genes from growth factor signaling pathways in glioblastoma cell lines. Oncogene 1992, 7, 243–247. [Google Scholar] [PubMed]
- Takano, S.; Gately, S.; Engelhard, H.; Tsanaclis, A.M.; Brem, S. Suramin inhibits glioma cell proliferation in vitro and in the brain. J. Neurooncol. 1994, 21, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Chen, Z.; Hu, Y.D.; Wei, H.; Li, D.; Ji, H.; Wang, D.L. Autocrine factors sustain glioblastoma stem cell self-renewal. Oncol. Rep. 2009, 21, 419–424. [Google Scholar] [PubMed]
- Allerstorfer, S.; Sonvilla, G.; Fischer, H.; Spiegl-Kreinecker, S.; Gauglhofer, C.; Setinek, U.; Czech, T.; Marosi, C.; Buchroithner, J.; Pichler, J.; et al. FGF5 as an oncogenic factor in human glioblastoma multiforme: Autocrine and paracrine activities. Oncogene 2008, 27, 4180–4190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatlen, M.A.; Schmidt-Kittler, O.; Sherwin, C.A.; Rozsahegyi, E.; Rubin, N.; Sheets, M.P.; Kim, J.L.; Miduturu, C.; Bifulco, N.; Brooijmans, N.; et al. Acquired On-Target Clinical Resistance Validates FGFR4 as a Driver of Hepatocellular Carcinoma. Cancer Discov. 2019, 9, 1686–1695. [Google Scholar] [CrossRef] [Green Version]
- Kim, R.D.; Sarker, D.; Meyer, T.; Yau, T.; Macarulla, T.; Park, J.W.; Choo, S.P.; Hollebecque, A.; Sung, M.W.; Lim, H.Y.; et al. First-in-Human Phase I Study of Fisogatinib (BLU-554) Validates Aberrant FGF19 Signaling as a Driver Event in Hepatocellular Carcinoma. Cancer Discov. 2019, 9, 1696–1707. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Sparidans, R.; El-Lari, M.; Wang, Y.; Lebre, M.C.; Beijnen, J.H.; Schinkel, A.H. P-glycoprotein (ABCB1/MDR1) limits brain accumulation and Cytochrome P450-3A (CYP3A) restricts oral availability of the novel FGFR4 inhibitor fisogatinib (BLU-554). Int. J. Pharm. 2020, 573, 118842. [Google Scholar] [CrossRef]
- Sootome, H.; Fujita, H.; Ito, K.; Ochiiwa, H.; Fujioka, Y.; Ito, K.; Miura, A.; Sagara, T.; Ito, S.; Ohsawa, H.; et al. Futibatinib is a novel irreversible FGFR 1–4 inhibitor that shows selective antitumor activity against FGFR-deregulated tumors. Cancer Res. 2020, 80, 4986–4997. [Google Scholar] [CrossRef]
- Kalyukina, M.; Yosaatmadja, Y.; Middleditch, M.J.; Patterson, A.V.; Smaill, J.B.; Squire, C.J. TAS-120 Cancer Target Binding: Defining Reactivity and Revealing the First Fibroblast Growth Factor Receptor 1 (FGFR1) Irreversible Structure. ChemMedChem 2019, 14, 494–500. [Google Scholar] [CrossRef] [PubMed]
- Bahleda, R.; Meric-Bernstam, F.; Goyal, L.; Tran, B.; He, Y.; Yamamiya, I.; Benhadji, K.A.; Matos, I.; Arkenau, H.T. Phase I, first-in-human study of futibatinib, a highly selective, irreversible FGFR1-4 inhibitor in patients with advanced solid tumors. Ann. Oncol. 2020, 31, 1405–1412. [Google Scholar] [CrossRef]
- Gavine, P.R.; Mooney, L.; Kilgour, E.; Thomas, A.P.; Al-Kadhimi, K.; Beck, S.; Rooney, C.; Coleman, T.; Baker, D.; Mellor, M.J.; et al. AZD4547: An orally bioavailable, potent, and selective inhibitor of the fibroblast growth factor receptor tyrosine kinase family. Cancer Res. 2012, 72, 2045–2056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andre, F.; Ranson, M.; Dean, E.; Varga, A.; Van der Noll, R.; Stockman, P.K.; Ghiorghiu, D.; Kilgour, E.; Smith, P.D.; Macpherson, M. Abstract LB-145: Results of a Phase I Study of AZD4547, an Inhibitor of Fibroblast Growth Factor Receptor (FGFR), in Patients with Advanced Solid Tumors. In Proceedings of the AACR 104th Annual Meeting 2013, Washington, DC, USA, 6–10 April 2013. [Google Scholar]
- Guagnano, V.; Furet, P.; Spanka, C.; Bordas, V.; Le Douget, M.; Stamm, C.; Brueggen, J.; Jensen, M.R.; Schnell, C.; Schmid, H. Discovery of 3-(2,6-dichloro-3,5-dimethoxy-phenyl)-1-{6-[4-(4-ethyl-piperazin-1-yl)-phenylamino]-pyrimidin-4-yl}-1-methyl-urea (NVP-BGJ398), a potent and selective inhibitor of the fibroblast growth factor receptor family of receptor tyrosine kinase. J. Med. Chem. 2011, 54, 7066–7083. [Google Scholar] [CrossRef] [PubMed]
- Guagnano, V.; Kauffmann, A.; Wohrle, S.; Stamm, C.; Ito, M.; Barys, L.; Pornon, A.; Yao, Y.; Li, F.; Zhang, Y.; et al. FGFR genetic alterations predict for sensitivity to NVP-BGJ398, a selective pan-FGFR inhibitor. Cancer Discov. 2012, 2, 1118–1133. [Google Scholar] [CrossRef] [Green Version]
- Konecny, G.E.; Kolarova, T.; O’Brien, N.A.; Winterhoff, B.; Yang, G.; Qi, J.; Qi, Z.; Venkatesan, N.; Ayala, R.; Luo, T.; et al. Activity of the fibroblast growth factor receptor inhibitors dovitinib (TKI258) and NVP-BGJ398 in human endometrial cancer cells. Mol. Cancer Ther. 2013, 12, 632–642. [Google Scholar] [CrossRef] [Green Version]
- Tabernero, J.; Bahleda, R.; Dienstmann, R.; Infante, J.R.; Mita, A.; Italiano, A.; Calvo, E.; Moreno, V.; Adamo, B.; Gazzah, A.; et al. Phase I Dose-Escalation Study of JNJ-42756493, an Oral Pan-Fibroblast Growth Factor Receptor Inhibitor, in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2015, 33, 3401–3408. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FGFRs | Molecular Weight | Residues AA | Malignancy Type | Inhibitor FGFRs | Common FGFR Alterations in Brain Tumors | Mutation of FGFRs in Pilocytic Astrocytoma | Mutation of FGFRs in Glioblastoma |
|---|---|---|---|---|---|---|---|
| FGFR1 | 91.9 kDa [37] | 822 [37] | Glioblastomas Low grade brain gliomas [37] | Futibatinib, Infigratinib AZD4547 [37] | FGFR1-TKD [80] FGFR1-TACC1 Fusion [80] FGFR1 hotspot mutations: p.N546, p.K656 [80] | Residues in αA1: N546K (KD1) [108] Residues in αB1: N544K (KD1) [108] Residues in αA1: K655I (KD2) [108] Residues in αB1: K653I (KD2) [108] Residues in αA1: K656D/E/M/N (KD2) [108] Residues in αB1: K654D/E/M/N (KD2) [108] Residues in αA1: T658P (KD2) [108] Residues in αB1: T656P (KD2) [108] | Residues in αA1: N546K (KD1) [81] Residues in αB1: N544K (KD1) [81] Residues in αA1: R576W (KD1) [81] Residues in αB1: R574W (KD1) [81] Residues in αA1: K656E (KD2) [109] Residues in αB1: K654E (KD2) [109] |
| FGFR2 | 92.0 kDa [37] | 821/822 [37] | Glioblastomas, Low grade brain gliomas [37] | Futibatinib, Infigratinib, AZD4547 [37] | FGFR2-CTNNA3 fusion [80] | Residues in IIIb: K660E (KD2) [108] Residues in IIIc: K659E (KD2) [108] | Residues in IIIb: Q212K (IgII) [109] Residues in IIIc: Q212K (IgII) [109] Residues in IIIb: G463E (JM) [82] Residues in IIIc: G462E (JM) [82] |
| FGFR3 | 87.7-88.2 kDa [37] | 806/808 [37] | Glioblastomas, Low grade brain gliomas [37] | Futibatinib, Infigratinib, AZD4547 [37] | FGFR3-TACC3 fusions [80] | Residues in IIIb: E468K (JM) [110] Residues in IIIc: E466K (JM) [110] Residues in IIIb: R605Q (KD2) [111] Residues in IIIc: R603Q (KD2) [111] | |
| FGFR4 | 88.0 kDa [37] | 802 [37] | Glioblastomas, Low grade brain gliomas [37] | Fisogatinib [37] | Residues in P22455-1: Q144E (IgI – IgII) [109] Residues in P22455-2: Q144E (IgI – IgII) [109] Residues in P22455-1: R434Q (JM) [109] Residues in P22455-2: R394Q (JM) [109] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ardizzone, A.; Scuderi, S.A.; Giuffrida, D.; Colarossi, C.; Puglisi, C.; Campolo, M.; Cuzzocrea, S.; Esposito, E.; Paterniti, I. Role of Fibroblast Growth Factors Receptors (FGFRs) in Brain Tumors, Focus on Astrocytoma and Glioblastoma. Cancers 2020, 12, 3825. https://doi.org/10.3390/cancers12123825
Ardizzone A, Scuderi SA, Giuffrida D, Colarossi C, Puglisi C, Campolo M, Cuzzocrea S, Esposito E, Paterniti I. Role of Fibroblast Growth Factors Receptors (FGFRs) in Brain Tumors, Focus on Astrocytoma and Glioblastoma. Cancers. 2020; 12(12):3825. https://doi.org/10.3390/cancers12123825
Chicago/Turabian StyleArdizzone, Alessio, Sarah A. Scuderi, Dario Giuffrida, Cristina Colarossi, Caterina Puglisi, Michela Campolo, Salvatore Cuzzocrea, Emanuela Esposito, and Irene Paterniti. 2020. "Role of Fibroblast Growth Factors Receptors (FGFRs) in Brain Tumors, Focus on Astrocytoma and Glioblastoma" Cancers 12, no. 12: 3825. https://doi.org/10.3390/cancers12123825