 Thomas Knight
Thomas Knight Julie Anne Elizabeth Irving
Julie Anne Elizabeth Irving- Newcastle Cancer Centre at the Northern Institute for Cancer Research, Newcastle University, Newcastle upon Tyne, UK
Deregulation of the Ras/Raf/MEK/extracellular signal-regulated kinase pathway is a common event in childhood acute lymphoblastic leukemia and is caused by point mutation, gene deletion, and chromosomal translocation of a vast array of gene types, highlighting its importance in leukemia biology. Pathway activation can be therapeutically exploited and may guide new therapies needed for relapsed acute lymphoblastic leukemia and other high risk subgroups.
The Ras/Raf/MEK/ERK Pathway
The mitogen-activated protein kinase (MAPK) cascade is a key signaling pathway that regulates diverse cellular functions including cell proliferation, survival, differentiation, angiogenesis, and migration (1–4). Classical activation is initiated by ligand binding to receptor tyrosine kinases (RTK) at the cell surface and via Ras, then Raf, then MEK (mitogen-activated protein kinase kinase), culminates in the regulation of gene transcription in the nucleus by the last pathway component, extracellular signal-regulated kinase (ERK). Since the pathway regulates many cellular functions that are classic hallmarks of cancer, it is not surprising that it is deregulated in numerous cancer types, including childhood acute lymphoblastic leukemia (ALL).
The core of the signaling pathway comprises three dual-specific protein kinases Raf, MEK, ERK, and the G-protein Ras. The RAS family contains three genes HRAS, NRAS, and KRAS that encode 21 kDa proteins. The KRAS transcript undergoes alternate splicing thus resulting in four Ras protein isoforms (HRas, NRas, KRas4A, KRas4B). Ras proteins are monomeric membrane-associated GTPases, which communicate binary on or off messages to downstream effector proteins by cycling between an active GTP bound and inactive GDP bound state. Ras activation is catalyzed by guanine nucleotide exchange factors (GEFs), such as son of sevenless (SOS), which displace GDP and allow GTP to preferentially occupy the vacant nucleotide binding site due to its relative abundance in the cytoplasm. Ras proteins exhibit intrinsic low-level hydrolytic activity and are negatively regulated by GTPase activating proteins (GAPs) such as neurofibromin (Nf1), which stimulate GTP hydrolysis and formation of inactive RAS–GDP (Figure 1).
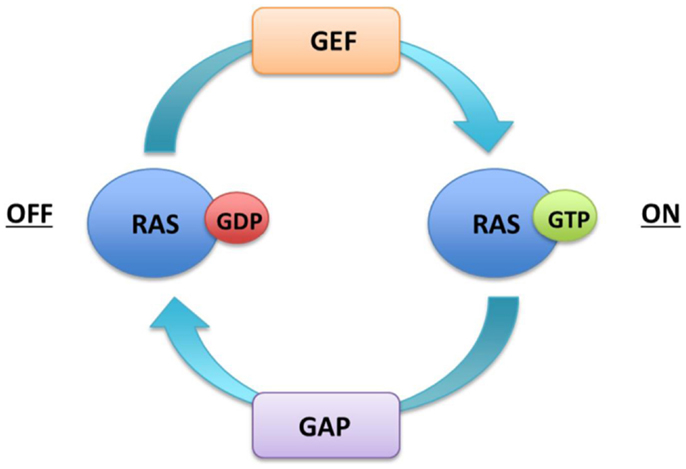
Figure 1. Regulation of Ras activity.
All four isoforms of Ras are widely expressed, although the relative degree of expression varies between tissues and developmental stage. They are highly homologous with differences in sequence largely limited to the C-terminal hypervariable region. Ras proteins must be localized to the inner surface of the plasma membrane to interact with upstream activators and downstream effectors to allow correct signaling. The immature Ras molecule has a hydrophilic globular structure that requires a series of post-translational modifications. These include addition of a 15-carbon farnesyl isoprenoid lipid at the C terminus that greatly improves the affinity of Ras for plasma membranes and palmitoylation that mediates vesicular transport to the cell surface. Palmitoylation is a reversible modification liable to degeneration creating a palmitoylation–depalmitoylation cycle in which Ras is recycled between membrane and Golgi. An exception to this is KRas4B, which translocates directly to the plasma membrane after farnesylation in a process postulated to involve chaperone proteins.
Activation of a RTK stimulates autophosphorylation of its intracellular SH2 domain that recruits growth factor receptor-bound protein 2 (Grb2), which serves to localize GEFs to the membrane to initiate Ras to exchange GDP for GTP. Ras then induces Raf activation by phosphorylation at specific serine residues resulting in the formation of Raf homo or hetero dimers. All four isoforms of Ras are able to activate all three members of the Raf gene family (A Raf, B Raf, Raf-1). Raf subsequently activates MEK1/2, which display restricted substrate specificity for ERK1/2. ERK is a potent kinase with a diverse range of both nuclear and cytoplasmic substrates. It regulates gene expression by phosphorylating numerous transcription factors including Elk1. The Ras/Raf/MEK/ERK pathway (now abbreviated to Ras pathway) is also implicated in the regulation of apoptosis by enhancing gene expression of pro-survival Bcl-2 family proteins and targeting anti-apoptotic proteins for proteasomal degradation (5). In addition to Raf/MEK/ERK, Ras generates signal output via numerous other effector pathways, including PI3K/Akt/mTOR and RalGEF/RAL. Cross talk between Ras and the PI3K pathway is of particular importance given their prominent role in the regulation of cell growth and survival (2, 6–8).
Mechanisms of Ras Pathway Activation in ALL
Somatic mutation of genes, which activate the Ras pathway are recurrently found in ALL and include integral components of the pathway, upstream activators, and regulatory proteins and include NRAS, KRAS, BRAF, FMS-related tyrosine kinase 3 (FLT3), protein tyrosine phosphatase, non-receptor type 11 (PTPN11), casitas B lineage lymphoma (CBL), and NF1. An overview is shown in Figure 2.
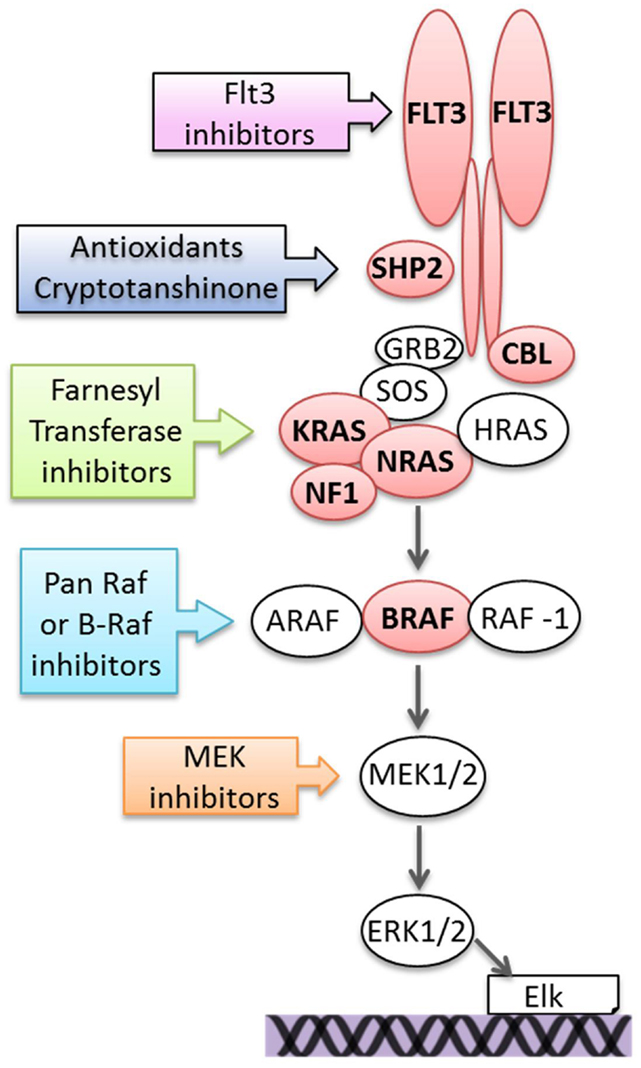
Figure 2. Mutations activating the Ras pathway in ALL and potential therapeutic targeting. Components of the pathway that are mutated in ALL are in red.
NRAS/KRAS (Neuroblastoma RAS Viral Oncogene Homolog/Kirsten Rat Sarcoma Viral Oncogene Homolog)
Mutations in NRAS and KRAS are highly prevalent in ALL (9–25). NRAS mutations are more common in ALL and in other hematological malignancies, contrasting with epithelial malignancies where KRAS mutations predominate (2, 4). HRAS mutations are rare outside the context of urinary tract, cervical, and salivary gland tumors and are not found at a significant level in hematological cancers. The vast majority of mutations in NRAS/KRAS cluster within hotspots at codons 12, 13, and 61 and dramatically reduce the rate of GTP hydrolysis by inhibiting interaction with GAPs and thus are locked in the active GTP bound, leading to constitutive activation. In the largest study to date, 90% of NRAS/KRAS mutations identified in ALL were located within exon 2, with the most commonly occurring genetic aberration involving a G:C to A:T transition (9). While early studies in childhood ALL reported an incidence below 10%, more contemporary studies consistently demonstrate mutations in 15–30% of ALL cases: more in B lineage disease, a preponderance in hyperdiploidy and a rarity in TEL–AML1 subgroups (10, 15, 16, 19). Higher incidences may reflect the use of mutation screening methods such as denaturing high performance liquid chromatography, which are more sensitive than Sanger sequencing and can detect mutations present in only 12–25% of cells (15). Clinical samples harboring NRAS or KRAS mutations invariably demonstrate increased levels of phosphorylated ERK, indicative of activation of the Ras pathway but to varying degrees (15, 26).
FMS-Related Tyrosine Kinase 3
FMS-related tyrosine kinase 3 is an RTK that is preferentially expressed on the surface of hematopoietic progenitors with its ligand is derived from neighboring bone marrow stroma cells. It has been extensively studied in the context of acute myeloid leukemia (AML) in which mutations occur in approximately a third of cases (27). FLT3 mutations occur at lower frequency in ALL, being reported in around 2–9% of cases, and again are often associated with hyperdiploidy (15, 19, 28–34). Mutations are broadly localized to two domains: missense substitutions or small in-frame deletions occur within the tyrosine kinase domain while in-frame insertion or deletions affect the juxta-membrane region of the protein, which has an autoinhibitory function. The latter mutations often affect a 10 amino acid stretch between Tyr 589 and Tyr 599 that has been functionally shown to cause activation if disrupted (35). Mutations cause constitutive activation of Flt3, hypersignaling of the Ras pathway and confer ligand-independent proliferation in vitro, confirming their functional significance (36, 37). Interestingly, in MLL-rearranged ALL, Flt3 is often constitutively activated because of high level expression rather than mutation (38, 39). Other RTKs implicated in deregulated Ras signaling include Mer, which has been shown to be aberrantly expressed in a third of Pre B ALL (40).
Protein Tyrosine Phosphatase, Non-Receptor Type 11
Shp2 encoded by the PTPN11 gene is a member of the protein tyrosine phosphatase family and is highly expressed in hematopoietic cells. Despite its role in protein dephosphorylation, Shp2 plays an overall positive role on cell signaling pathways, including Ras and JAK–STAT pathways. While the mechanism is not clear, postulated targets of Shp2 include Ras GAP binding sites on RTKs or key tyrosyl residues on Sprouty proteins, which inhibit the pathway and more recent evidence implicates a role in increasing production of reactive oxygen species, which hypersensitizes cytokine signaling (41). Somatic mutations of PTPN11 are common in juvenile myelomonocytic leukemia where they occur in around 35% of cases but are also found in ALL at frequencies between 2 and 10% (10, 14, 15, 19, 42). Mutations are usually heterozygote, missense, gain of function mutations sited in the SH2 or phosphatase regions of the protein. In mouse models, PTPN11 mutations cause a myeloproliferative disorder (MPD) but a more recent study showed that the PTPN11 E76K mutation was clearly sufficient to induce acute leukemia and this residue is commonly mutated in ALL (43). The E76K mice leukemias were associated with centrosome amplification and aneuploidy, which is highly relevant given the predominance of PTPN11 mutations in hyperdiploid ALL cases.
BRAF (V-Raf Murine Sarcoma Viral Oncogene Homolog B)
Activating mutations of the serine threonine kinase, BRAF, were first identified in melanoma and cluster at the hot spot V600E site. About two-thirds of melanoma patients have BRAF mutations (44). A first investigation of BRAF mutations in a small Swiss population of pediatric ALL patients found an incidence of 20%, with about half of patients bearing a L597Q mutation (12). This specific mutation was later shown to be transforming in vitro (45). However, mutational screening of larger cohorts of infant, childhood B and T lineage ALL have found only rare instances of BRAF mutations, with the classic V600E mutation seen only once (15, 24, 25, 46, 47). In early thymocyte precursor (ETP) ALL, focal amplification of BRAF was found in one patient, thus amplification of BRAF rather than mutation may be an alternative mechanism of activation (25).
Casitas B Lineage Lymphoma
A more recently reported gene family implicated in Ras pathway activation is the CBL proteins, a highly conserved family of RING finger ubiquitin E3 ligases that target a variety of RTKs for degradation. Screening of CBL in an unselected cohort of ALL patients identified somatic mutations in 1–2% of cases (48, 49). Mutations have also been reported in T ALL and infant ALL and are often associated with acquired uniparental disomy at the CBL gene locus, resulting in a homozygous mutant state (49–51). Functional analyses have shown mutations to be associated with stabilization of RTK receptors in an active state, constitutive activation of the Ras pathway, and cellular sensitivity to MEK inhibitor (MEKi) treatment.
Other
The NF1 gene encodes neurofibromin (Nf1), a GAP that inhibits Ras signaling by stimulating hydrolysis of active RAS–GTP into inactive RAS–GDP. Inactivation of Nf1 by gene microdeletion and mutation have been found at low frequency in both T and B lineage ALL (52, 53). In mouse models, somatic inactivation of NF1 in hematopoietic cells induces an MPD (54). Chromosomal translocation events, including BCR/ABL and those involving the MLL locus, also cause constitutive activation of the Ras pathway (55–57).
The diverse routes exploited by leukemia cells to activate the Ras pathway highlights the importance of the pathway in leukemogenesis and progression. Several studies report on the mutual exclusivity of Ras pathway mutations in that few patients appear to have more than one mutation, suggesting that one pathway activating event precludes the need for a second (10, 14, 15, 19). However, this finding is not universal and it may be that more sensitive mutation detection methodologies such as high coverage next generation sequencing and allele-specific PCR will reveal more co-existing Ras pathway mutations within one leukemia, but it is likely that they are present in separate cell populations (24).
Are Ras Pathway Mutations an Initiating or Secondary Event?
There are various inherited developmental disorders caused by germline mutations in components of the Ras signaling pathway, so called Rasopathies that have an increased risk of hematological malignancies, including ALL (58). In addition, there is mounting evidence from a variety of mouse models that genes that activate the Ras pathway including NRAS/KRAS, NF1, and PTPN11 are initiating events in the development of ALL, giving rise to both B and T lineage ALL (43, 59–64). The majority of studies have focused on KRAS and NRAS mutant forms and suggest that oncogenic Ras alone is insufficient to drive leukemogenesis and cooperating genetic events are necessary for full-blown leukemia. In the context of T ALL, NOTCH mutations are often this cooperating genetic event (59, 63, 65, 66). Hematopoietic stem/progenitor cell populations from KRASG12D mice show similar basal levels of phosphorylated ERK relative to wild type but generate an exaggerated response to growth factor stimulation suggesting mutant Ras renders them more sensitive to mitogenic signals rather than causing unbridled saturation of the pathway (67). A recent publication by Li et al. shows that in hematopoietic stem cells, NRAS mutations have a bimodal effect, increasing the likelihood of cell division in some cells and decreasing it in others, thus generating an expanding, more rapidly dividing cell population and another with long-term self-renewal (68). This may allow mutated cells to have long-term clonal dominance over wild type cells.
However, there are several lines of evidence in primary samples suggesting that Ras pathway mutations occur as a second, cooperative genetic “hit” as opposed to the initiating event, at least in some patients. For example, there are several reports of mutations existing in only a minor population of leukemia cells at diagnosis and a recent report suggests that KRAS mutations are commonly found at very low level in a significant number of diagnostic ALL who were genotyped as Ras pathway wild type by standard mutation screening (10, 15, 26). PTPN11 and RAS mutations have been shown to be present at diagnosis of ALL but not at relapse, suggesting a secondary role (42, 69). In MLL-rearranged ALL, oncogenic KRAS has itself been shown to act as a cooperative lesion and the introduction of KRAS mutations into MLL/AF4 transgenic mice results in a more aggressive leukemic phenotype, which more closely mirrors the human equivalent (70, 71). In addition, Wiemels et al. used allele-specific PCR to backtrack KRAS mutations present in diagnostic hyperdiploid ALL in blood derived from matched neonatal mono-spots (16). These experiments failed to detect the presence of KRAS mutation prenatally but detected the clonal immunoglobulin heavy chain rearrangement specific to the hyperdiploid clone. This suggests a temporal relationship in which KRAS mutation occurs subsequent to the development of the hyperdiploid clone. Taken together, the evidence suggests that similar to AML, Ras pathway activating mutations may act as an early/initiating or cooperating events acquired during disease progression and clearly may have implications for therapies targeting the pathway.
The clear role of NRAS and KRAS mutations in the pathogenesis of many cancer types has prompted both epidemiological and animal studies that examine exposure to certain chemical mutagenic substances with the presence of specific mutations. In the context of ALL, a study by Shu et al. showed that parental occupational exposure to hydrocarbons and mind-altering drugs was related to specific RAS mutations in ALL (11). There was also an association with younger children, which may suggest a preconception or in utero origin of the mutation. Another study identified NRAS and KRAS mutations to be associated with a variety of maternal and paternal exposures but these associations were not corroborated in a subsequent larger study (13, 16).
Ras Pathway Mutations in Newly Diagnosed ALL
While early studies focused solely on mutations in NRAS and KRAS, more comprehensive screens with inclusion of additional genes impacting on the pathway, including PTPN11, FLT3, and CBL, report mutations in up to 35% of newly diagnosed ALL, with a predominance in B lineage ALL (9–11, 13, 14, 16, 18–20, 32, 72–74). Table 1 summarizes the largest studies to date and Figure 3 shows the frequencies of mutated genes identified in a UK diagnostic cohort (15, 48).
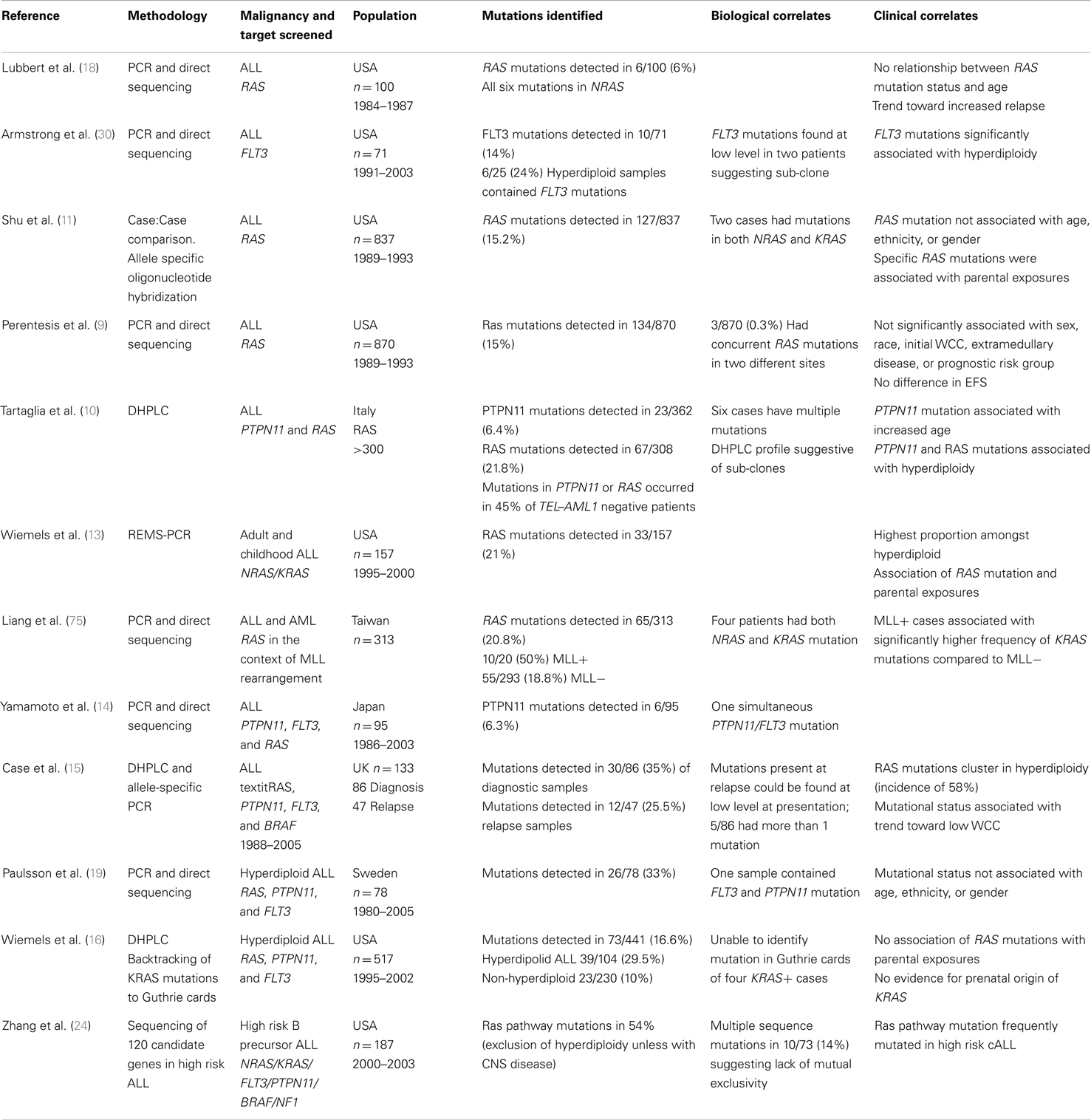
Table 1. Summary of the largest studies of Ras pathway mutations in ALL.
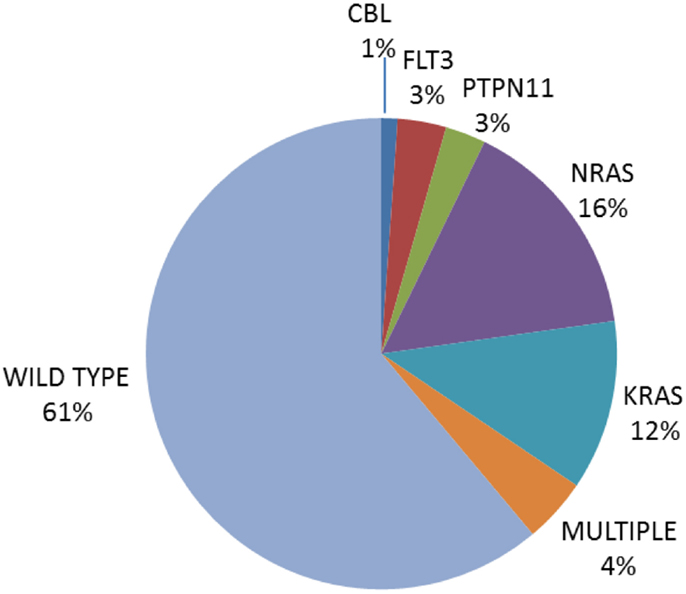
Figure 3. Pie chart of the frequency of Ras pathway mutations in an unselected cohort of 180 UK diagnostic ALL [data from Ref. (15, 48)].
While one early study demonstrated a significantly higher rate of hematological relapse and a trend toward reduced complete remission rates amongst cases harboring NRAS mutations, the association between RAS mutation and adverse outcome has not been replicated in more recent studies, raising the possibility that contemporary chemotherapeutic regimens have negated the effect (9, 12, 18, 74). In the largest investigation of the prognostic significance of NRAS and KRAS mutation that included 870 patients enrolled on an US study, mutation status had no discernable effect on event free survival, disease free survival, or overall survival and there was no relationship between RAS mutations and high risk clinical features (9). A smaller UK study did show a statistically significant association with Ras pathway mutation and some high risk features such as higher presenting white cell count but not others, such as day 8 bone marrow clearance (15). In the US study, when specific RAS mutations were compared individually, mutations in exon 2 of the KRAS gene were associated with younger age at presentation and were less likely to occur in the context of B cell disease when compared to mutations at other locations (9).
Mutations in the Ras pathway clearly show a strong correlation with specific cytogenetic subgroups of ALL and are prevalent in both very poor and very good risk cytogenetic groups, which may neutralize any effect on prognosis when a patient cohort is analyzed as a whole. For example, Ras pathway mutations are consistently associated with high hyperdiploidy, which represents a third of all newly diagnosed children and confers a very favorable prognosis (13, 15, 16, 19, 32, 72, 76). Reported incidences of Ras pathway mutations in this subgroup range from 24% to almost 60%, with a large study reporting mutations in 30% compared to 10% in non-hyperdiploid cases (16). They also occur at high frequency in the context of hypodiploid ALL (23). Hypodiploidy (<45 chromosomes) occurs in around 5% of cALL cases and is associated with a very poor prognosis (77). Hypodiploidy can be further divided by karyotype into three distinct groups, near-haploid (23–29 chromosomes), low hypodiploidy (33–39 chromosomes), and high hypodiploidy (42–45 chromosomes). More than two-thirds of near-haploid cases feature mutations predicted to result in aberrant Ras signaling, in particular intragenic deletion of NF1 (23). Enhanced signaling has been demonstrated in low hypodiploid cases in the absence of known canonical Ras pathway mutations, suggesting that additional mechanisms of pathway activation remain to be identified in this subgroup (23). One possible mechanism is the deregulated expression of a microRNA, MiR335 that targets and regulates levels of ERK2 (78). The high prevalence of pathway mutations in aneuploid leukemias is notable and there is evidence of a possible link between deregulated signaling through the Ras pathway and chromosomal instability (79). Ras pathway mutations have also been identified at high incidence in cases defined as “high risk” and also in the very aggressive, ETP ALL (24, 25). In addition, poor risk chromosomal translocations, including BCR/ABL and those involving the MLL locus are associated with constitutive activation of the Ras pathway (55–57). Interestingly, Ras pathway mutations are rare in the ETV6–RUNX1 cytogenetic subgroup (10, 15).
Recent investigations have questioned the impact of RAS mutation and prognostic significance within the context of specific cytogenetic groups. For example, in MLL-rearranged infant ALL in whom NRAS/KRAS mutations are present in 24–50% of cases, mutations were shown to be an independent prognostic factor associated with an extremely poor outcome, with a 5-year event free survival rates of 0.0% for RAS mutated compared to 32.7% for RAS wildtype (47, 75). Mutations were also shown to be associated with a higher presenting white blood cell count and mutated primary cells were more resistant to glucocorticoids in vitro (47). However, a small study, which focused on hyperdiploid ALL showed no influence of Ras pathway mutation on prognosis (19). Large mutation screening studies of contemporary childhood ALL trials are currently underway and will define whether Ras pathway status has prognostic relevance and if it can enhance current risk stratification strategies.
Ras Pathway Mutations in Relapsed ALL
Mutations in the Ras pathway are common in relapsed cALL, occurring in between 25 and 39% of B lineage cases (15, 26, 69). In the largest study to date, which focused on children with B lineage ALL treated on the ALL-REZ BFM 2002 trial, the presence of Ras pathway mutations was associated with high risk features such as early relapse. For NRAS/KRAS mutations, there was a greater proportion of patients with on-treatment relapse, CNS involvement, and chemo-resistant disease, as evidenced by reduced cytological remission rates (26). However, this did not translate into a significant difference in event free survival. The cytogenetic associations seen in diagnostic cases were mirrored at relapse, with a preponderance of pathway mutations in cases with hyperdiploidy and a relative paucity in TEL–AML1.
An intriguing phenomenon seen in several studies is the finding that matched diagnostic samples, when compared to relapse with known Ras pathway mutations, are frequently wild type by standard mutation detection screening methodologies, but more sensitive assays reveal a minority sub-clone of cells with the same mutation (15, 26, 72). Similar backtracking studies for antigen receptor or copy number lesions dominating at relapse have also identified low level clones within diagnostic samples in the majority of patients (80–82). These observations give insight into the biology of relapse in ALL and in the case of Ras pathway mutations may have therapeutic implications. One model of relapse suggests that resistant leukemic cells preexist by chance as a minority sub-clonal population at diagnosis, evade the cytotoxic effects of chemotherapy, and go on to propagate disease as the dominant relapse clone. In the second, there is therapy-induced acquired resistance that drives the acquisition of de novo mutations and clonal evolution. These models are not mutually exclusive and both may be needed for cells to acquire total resistance to the multi-drug chemotherapeutic barrage used in ALL.
The expansion of a pre-existing mutated RAS sub-clone at relapse suggests that the former model plays a role in relapse and that mutations must confer a degree of resistance to therapy. This hypothesis is supported by in vitro studies in which activation of the Ras pathway in hematopoietic cells is associated with resistance to glucocorticoids and anthracyclines, key drugs used in ALL therapy (47, 83, 84). Resistance is mediated by transcriptional influences on ERK target proteins as well as those regulating the apoptotic regulatory machinery, e.g., Bim. In one anecdotal case with a very high level of minimal residual disease (MRD) at the end of induction, two different KRAS-mutated clones were identified at low level in the diagnostic sample and were enriched during induction therapy to the extent that all of the MRD cells bore KRAS mutations (26). Such cases may benefit from therapies targeting the Ras pathway to eradicate MRD. Importantly, in the same study, low level KRAS mutations were shown to be relatively common in a diagnostic cohort of long-term survivors and are clearly a common clonal event during leukemogenesis that does not necessarily herald relapse. Clearly, other factors must determine survival of these low-level mutated clones during treatment. Importantly Ras pathway mutations found at diagnosis have been shown to be absent at relapse in some patients (42, 69). Whole genome analyses of presentation, MRD, and relapse trios may shed light on the relative contribution of the two models in the biology of relapse and the possible role of MRD-directed therapy to avert relapse.
The Ras/Raf/MEK/ERK Pathway as a Therapeutic Target
Deregulation of the Ras pathway is common across cancer types and thus an attractive target for therapeutic inhibition and such novel therapies may play a role in improving outcome for high risk ALL. Numerous therapeutic strategies targeting the Ras pathway are currently being evaluated in clinical trials.
The Ras protein itself has proved challenging to inhibit directly, although a very recent study reported on the development of a small molecule inhibitor, which specifically decreased viability and induced apoptosis of lung cancer cell lines expressing KRAS G12C, but had no effect on wild type cells or those bearing other KRAS mutations. This important drug development milestone will facilitate the development of other specific mutant inhibitors (85). Early drugs were aimed at disrupting Ras post-translational processing, principally inhibitors of farnesyltransferase (FTase). One of these, Tipifarnib (R-115777) is a competitive, non-peptidomimetic FTase inhibitor, which showed in vitro activity in B and more so in T lineage ALL. However, the small number of RAS mutant positive samples precluded statistical analysis of mutational status on treatment response (86). Phase I clinical trials of Tipifarnib in relapsed or refractory acute leukemia in adults showed some clinical and biological activity in terms of p-ERK inhibition, but the trial included a small number of adult ALL patients, all of which demonstrated disease progression within 7–21 days (87). Dose limiting toxicities included ataxia, confusion, and dysarthria. Targeting FTase is a less than perfect strategy in disrupting Ras pathway signaling as when FTase is inhibited, Nras and Kras can undergo alternate post-translational processing by geranylgeranyltransferase, which allows the crucial cellular trafficking of Ras proteins to continue (88). FTase inhibitors also target a wide variety of other CAAX motif containing peptides and therefore Ras-independent modes of action probably account for the marginal benefit derived in other cancers.
The restricted substrate specificity of MEK1/2 for its sole substrate ERK1/2 has prompted the development of inhibitors of MEK, since one would expect them to be associated with less “off target” activity and inhibit the pathway regardless of the mechanism of upstream activation. There are a number of MEKi in advanced stages of clinical trial including Trametinib (GSK1120212), Pimasertib (MSC1936369B), and Selumetinib (AZD6244, ARRY-142886) (89–92). Selumetinib is a potent, selective, allosteric inhibitor of MEK1/2, which has reached Phase II clinical trials in a range of solid cancers, has a favorable toxicity profile and has demonstrated anti-tumor activity. In general, sensitivity to MEKi is enhanced in tumor cells harboring activating Ras pathway mutations, including ALL cells although this is not universal (15, 48, 66, 92–94). Initial in vivo testing of Selumetinib against mice xenograft models of pediatric B cell ALL showed no significant activity, but RAS mutational status and pathway activation were not assessed (95). However, a recent study clearly demonstrated differential sensitivity in ALL blasts in vitro, with GI50 values being significantly lower in Ras pathway positive cells (mean 250 nM) compared to those that were negative (mean 68 μM). These primary cells included NRAS, KRAS, and FLT3/CBL mutants. These in vitro data were replicated in vivo using NRAS and KRAS mutant ALL primagrafts and pharmacodynamic assessments showed inhibition of p-ERK and induction of apoptosis. Histological analysis of post-mortem brains found extensive meningeal leukemic infiltration in control vehicle treated, but not Selumetinib-treated mice, suggesting that this drug may eradicate CNS ALL. Some activity has also been demonstrated in vivo for KRAS-mutated T ALL with the MEKi, PD0325901 (66). However, similar studies in RAS-mutated hypodiploid ALL showed no activity of MEKi but activity of PI3K inhibitors (23). MEK inhibition has also been studied in NF1-deficient mice (64). Bi-allelic inactivation of NF1 induces an MPD, which can be progressed to AML using retro-viral mutagenesis to induce secondary genetic aberrations. While the initial MPD was relatively resistant to MEK inhibition, the NF1-deficient leukemias were significantly more sensitive, suggesting that cooperating mutations render them highly dependent on Ras signaling (64).
An interesting study has teased apart the relative importance of Raf/MEK/ERK and PI3K/Akt/mTOR pathways in a mouse model of T lineage ALL and shown that both effector pathways are drivers of aberrant growth initiated by KRAS G12D (96). Thus, combining inhibitors of both pathways may be an effective therapeutic strategy and indeed significant synergy has been shown in solid tumors (97, 98). However, in Ras-mutated primary B lineage ALLs, differential activation of the Ras but not the PI3K/Akt pathway is seen (J. Irving, unpublished observations). Nonetheless, as evidenced by imatinib and BCR/ABL positive ALL, targeted therapies are likely to have maximal therapeutic benefit in combination (99). For MEKi, synergism has been demonstrated for PI3K/Akt inhibitors, dexamethasone, Nutlin-3a, which induces the MDM2-p53 axis, and the Bcl-2 and Bcl-x(L) inhibitor, Navitoclax (ABT263) (78, 100–102). In AML cells, enhanced cytotoxicity was achieved in vitro when MEKi were used in combination with Nutlin-3a and PI3K/Akt inhibitors and was related to changes in the relative balance of proapoptotic and anti-apoptotic Bcl-2 family proteins (102, 103). Importantly, standard chemotherapeutic drugs can activate the Ras pathway and contribute to drug resistance, thus MEKi may have potential relevance in Ras pathway induced as well as constitutively activated ALLs (104).
Other potential drugs include Raf inhibitors, specifically Braf inhibitors, given their identification in some ALL patients. For children with FLT3 mutations, small molecule inhibitors of Flt3 may offer clinical benefit and there are several multi-targeted agents in clinical trial including Lestaurtinib, Sorafenib, and AT9283. Promising preclinical data of Flt3 inhibitors in MLL-rearranged ALL have prompted a Phase III trial in which newly diagnosed infants are randomized to receive combination chemotherapy plus or minus Lestaurtinib (39, 105). The results of this trial are not expected until 2018. Recent evidence also suggests that direct targeting of mutant PTPN11 may be possible. A naturally occurring compound, cryptotanshinone, used to treat cardiovascular disease in Asian countries, has been shown to inhibit Shp2 and preclinical studies found that mouse myeloid progenitors and primary leukemia cells bearing the activating PTPN11 E76K mutation were sensitive to this inhibitor (106). In addition, there is increased mitochondrial aerobic metabolism and cytokine hypersensitivity associated with PTPN11 mutations, which can be partially corrected with antioxidants in mutant progenitor cells, suggesting this may be another possible therapeutic approach (41).
Intriguingly, targeting mutated Ras may also be possible with more standard chemotherapeutic agents. Early in vitro studies suggested that Ras-mutated cancer cells may be more sensitive to certain drugs, particularly the nucleoside analog cytarabine. These observations have been confirmed in the clinic, with Ras mutant positive AML patients responding significantly better to high-dose cytarabine (107–111). In the largest study to date, Ras-mutated AML patients receiving high-dose cytarabine during consolidation had a 10-year cumulative relapse incidence of 45% compared to 68% for wild type RAS patients and in the low dose cytarabine arm, comparative figures were 100 versus 80%, respectively (109). The mechanism underlying this preferential sensitivity appears to be mutant Ras synergizing with cytarabine to activate DNA damage checkpoints and via p53 results in increased differentiation and an associated reduction in clonogenicity (112). Similar investigations in childhood ALL trials have not been performed to date.
Summary
Ras pathway activation is common in ALL and is caused by point mutation, gene deletion, and chromosomal translocation of a vast array of gene types, including GTPases, RTKs, phosphatases, and ubiquitin ligases, emphasizing its importance in leukemia biology. Pathway activation can be therapeutically exploited and may define new therapies needed for relapsed ALL and given its high prevalence may offer clinical benefit for a significant number of children. Such therapies may also be used upfront for high risk subgroups. One of the most promising targeted agents, a MEKi, has shown activity in vivo as a single agent in Ras pathway-activated ALL, however, maximal therapeutic benefit is likely to be in combination with other drugs. Clinical trials of MEK inhibitors in multiple relapsed ALL are planned.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Chung E, Kondo M. Role of Ras/Raf/MEK/ERK signaling in physiological hematopoiesis and leukemia development. Immunol Res (2011) 49(1–3):248–68. doi: 10.1007/s12026-010-8187-5
2. Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer (2011) 11(11):761–74. doi:10.1038/nrc3106
3. Ahearn IM, Haigis K, Bar-Sagi D, Philips MR. Regulating the regulator: post-translational modification of RAS. Nat Rev Mol Cell Biol (2012) 13(1):39–51. doi:10.1038/nrm3255
4. Ward AF, Braun BS, Shannon KM. Targeting oncogenic Ras signaling in hematologic malignancies. Blood (2012) 120(17):3397–406. doi:10.1182/blood-2012-05-378596
5. Balmanno K, Cook SJ. Tumour cell survival signalling by the ERK1/2 pathway. Cell Death Differ (2009) 16(3):368–77. doi:10.1038/cdd.2008.148
6. Castellano E, Downward J. RAS interaction with PI3K: more than just another effector pathway. Genes Cancer (2011) 2(3):261–74. doi:10.1177/1947601911408079
7. Mendoza MC, Er EE, Blenis J. The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem Sci (2011) 36(6):320–8. doi:10.1016/j.tibs.2011.03.006
8. Collado M, Serrano M. Senescence in tumours: evidence from mice and humans. Nat Rev Cancer (2010) 10(1):51–7. doi:10.1038/nrc2772
9. Perentesis JP, Bhatia S, Boyle E, Shao Y, Shu XO, Steinbuch M, et al. RAS oncogene mutations and outcome of therapy for childhood acute lymphoblastic leukemia. Leukemia (2004) 18(4):685–92. doi:10.1038/sj.leu.2403272
10. Tartaglia M, Martinelli S, Cazzaniga G, Cordeddu V, Iavarone I, Spinelli M, et al. Genetic evidence for lineage-related and differentiation stage-related contribution of somatic PTPN11 mutations to leukemogenesis in childhood acute leukemia. Blood (2004) 104(2):307–13. doi:10.1182/blood-2003-11-3876
11. Shu XO, Perentesis JP, Wen W, Buckley JD, Boyle E, Ross JA, et al. Parental exposure to medications and hydrocarbons and ras mutations in children with acute lymphoblastic leukemia: a report from the Children’s Oncology Group. Cancer Epidemiol Biomarkers Prev (2004) 13(7):1230–5.
12. Gustafsson B, Angelini S, Sander B, Christensson B, Hemminki K, Kumar R. Mutations in the BRAF and N-ras genes in childhood acute lymphoblastic leukaemia. Leukemia (2005) 19(2):310–2. doi:10.1038/sj.leu.2403589
13. Wiemels JL, Zhang Y, Chang J, Zheng S, Metayer C, Zhang L, et al. RAS mutation is associated with hyperdiploidy and parental characteristics in pediatric acute lymphoblastic leukemia. Leukemia (2005) 19(3):415–9. doi:10.1038/sj.leu.2403641
14. Yamamoto T, Isomura M, Xu Y, Liang J, Yagasaki H, Kamachi Y, et al. PTPN11, RAS and FLT3 mutations in childhood acute lymphoblastic leukemia. Leuk Res (2006) 30(9):1085–9. doi:10.1016/j.leukres.2006.02.004
15. Case M, Matheson E, Minto L, Hassan R, Harrison CJ, Bown N, et al. Mutation of genes affecting the RAS pathway is common in childhood acute lymphoblastic leukemia. Cancer Res (2008) 68(16):6803–9. doi:10.1158/0008-5472.CAN-08-0101
16. Wiemels JL, Kang M, Chang JS, Zheng L, Kouyoumji C, Zhang L, et al. Backtracking RAS mutations in high hyperdiploid childhood acute lymphoblastic leukemia. Blood Cells Mol Dis (2010) 45(3):186–91. doi:10.1016/j.bcmd.2010.07.007
17. Ahuja HG, Foti A, Bar-Eli M, Cline MJ. The pattern of mutational involvement of RAS genes in human hematologic malignancies determined by DNA amplification and direct sequencing. Blood (1990) 75(8):1684–90.
18. Lubbert M, Mirro J Jr, Miller CW, Kahan J, Isaac G, Kitchingman G, et al. N-ras gene point mutations in childhood acute lymphocytic leukemia correlate with a poor prognosis. Blood (1990) 75(5):1163–9.
19. Paulsson K, Horvat A, Strombeck B, Nilsson F, Heldrup J, Behrendtz M, et al. Mutations of FLT3, NRAS, KRAS, and PTPN11 are frequent and possibly mutually exclusive in high hyperdiploid childhood acute lymphoblastic leukemia. Genes Chromosomes Cancer (2008) 47(1):26–33. doi:10.1002/gcc.20502
20. Kawamura M, Ohnishi H, Guo SX, Sheng XM, Minegishi M, Hanada R, et al. Alterations of the p53, p21, p16, p15 and RAS genes in childhood T-cell acute lymphoblastic leukemia. Leuk Res (1999) 23(2):115–26. doi:10.1016/S0145-2126(98)00146-5
21. Kawamura M, Kikuchi A, Kobayashi S, Hanada R, Yamamoto K, Horibe K, et al. Mutations of the p53 and ras genes in childhood t(1;19)-acute lymphoblastic leukemia. Blood (1995) 85(9):2546–52.
22. Terada N, Miyoshi J, Kawa-Ha K, Sasai H, Orita S, Yumura-Yagi K, et al. Alteration of N-ras gene mutation after relapse in acute lymphoblastic leukemia. Blood (1990) 75(2):453–7.
23. Holmfeldt L, Wei L, Diaz-Flores E, Walsh M, Zhang J, Ding L, et al. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat Genet (2013) 45(3):242–52. doi:10.1038/ng.2532
24. Zhang J, Mullighan CG, Harvey RC, Wu G, Chen X, Edmonson M, et al. Key pathways are frequently mutated in high-risk childhood acute lymphoblastic leukemia: a report from the Children’s Oncology Group. Blood (2011) 118(11):3080–7. doi:10.1182/blood-2011-03-341412
25. Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature (2012) 481(7380):157–63. doi:10.1038/nature10725
26. Matheson EC, Minto L, Blair H, Case M, Halsey C, Swidenbank I, et al. RAS pathway mutations are highly prevalent in relapsed childhood acute lymphoblastic leukaemia, are frequently relapse-drivers and confer sensitivity to MEK Inhibition. Blood (2013) 122(21):823.
27. Small D. FLT3 mutations: biology and treatment. Hematology Am Soc Hematol Educ Program (2006):178–84. doi:10.1182/asheducation-2006.1.178
28. Taketani T, Taki T, Sugita K, Furuichi Y, Ishii E, Hanada R, et al. FLT3 mutations in the activation loop of tyrosine kinase domain are frequently found in infant ALL with MLL rearrangements and pediatric ALL with hyperdiploidy. Blood (2004) 103(3):1085–8. doi:10.1182/blood-2003-02-0418
29. Chang P, Kang M, Xiao A, Chang J, Feusner J, Buffler P, et al. FLT3 mutation incidence and timing of origin in a population case series of pediatric leukemia. BMC Cancer (2010) 10:513. doi:10.1186/1471-2407-10-513
30. Armstrong SA, Mabon ME, Silverman LB, Li A, Gribben JG, Fox EA, et al. FLT3 mutations in childhood acute lymphoblastic leukemia. Blood (2004) 103(9):3544–6. doi:10.1182/blood-2003-07-2441
31. Leow S, Kham SK, Ariffin H, Quah TC, Yeoh AE. FLT3 mutation and expression did not adversely affect clinical outcome of childhood acute leukaemia: a study of 531 Southeast Asian children by the Ma-Spore study group. Hematol Oncol (2011) 29(4):211–9. doi:10.1002/hon.987
32. Braoudaki M, Karpusas M, Katsibardi K, Papathanassiou C, Karamolegou K, Tzortzatou-Stathopoulou F. Frequency of FLT3 mutations in childhood acute lymphoblastic leukemia. Med Oncol (2009) 26(4):460–2. doi:10.1007/s12032-008-9146-z
33. Andersson A, Paulsson K, Lilljebjorn H, Lassen C, Strombeck B, Heldrup J, et al. FLT3 mutations in a 10 year consecutive series of 177 childhood acute leukemias and their impact on global gene expression patterns. Genes Chromosomes Cancer (2008) 47(1):64–70. doi:10.1002/gcc.20508
34. Karabacak BH, Erbey F, Bayram I, Yilmaz S, Acipayam C, Kilinc Y, et al. Fms-like tyrosine kinase 3 mutations in childhood acute leukemias and their association with prognosis. Asian Pac J Cancer Prev (2010) 11(4):923–7.
35. Kiyoi H, Ohno R, Ueda R, Saito H, Naoe T. Mechanism of constitutive activation of FLT3 with internal tandem duplication in the juxtamembrane domain. Oncogene (2002) 21(16):2555–63. doi:10.1038/sj.onc.1205332
36. Yamamoto Y, Kiyoi H, Nakano Y, Suzuki R, Kodera Y, Miyawaki S, et al. Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood (2001) 97(8):2434–9. doi:10.1182/blood.V97.8.2434
37. Choudhary C, Schwable J, Brandts C, Tickenbrock L, Sargin B, Kindler T, et al. AML-associated Flt3 kinase domain mutations show signal transduction differences compared with Flt3 ITD mutations. Blood (2005) 106(1):265–73. doi:10.1182/blood-2004-07-2942
38. Armstrong SA, Kung AL, Mabon ME, Silverman LB, Stam RW, Den Boer ML, et al. Inhibition of FLT3 in MLL. Validation of a therapeutic target identified by gene expression based classification. Cancer Cell (2003) 3(2):173–83. doi:10.1016/S1535-6108(03)00003-5
39. Stam RW, den Boer ML, Schneider P, Nollau P, Horstmann M, Beverloo HB, et al. Targeting FLT3 in primary MLL-gene-rearranged infant acute lymphoblastic leukemia. Blood (2005) 106(7):2484–90. doi:10.1182/blood-2004-09-3667
40. Linger RM, Lee-Sherick AB, DeRyckere D, Cohen RA, Jacobsen KM, McGranahan A, et al. Mer receptor tyrosine kinase is a therapeutic target in pre-B-cell acute lymphoblastic leukemia. Blood (2013) 122(9):1599–609. doi:10.1182/blood-2013-01-478156
41. Xu D, Zheng H, Yu WM, Qu CK. Activating mutations in protein tyrosine phosphatase Ptpn11 (Shp2) enhance reactive oxygen species production that contributes to myeloproliferative disorder. PLoS One (2013) 8(5):e63152. doi:10.1371/journal.pone.0063152
42. Molteni CG, Te Kronnie G, Bicciato S, Villa T, Tartaglia M, Basso G, et al. PTPN11 mutations in childhood acute lymphoblastic leukemia occur as a secondary event associated with high hyperdiploidy. Leukemia (2010) 24(1):232–5. doi:10.1038/leu.2009.200
43. Xu D, Liu X, Yu WM, Meyerson HJ, Guo C, Gerson SL, et al. Non-lineage/stage-restricted effects of a gain-of-function mutation in tyrosine phosphatase Ptpn11 (Shp2) on malignant transformation of hematopoietic cells. J Exp Med (2011) 208(10):1977–88. doi:10.1084/jem.20110450
44. Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature (2002) 417(6892):949–54. doi:10.1038/nature00766
45. Hou P, Liu D, Xing M. The T1790A BRAF mutation (L597Q) in childhood acute lymphoblastic leukemia is a functional oncogene. Leukemia (2007) 21(10):2216–8. doi:10.1038/sj.leu.2404761
46. Davidsson J, Lilljebjorn H, Panagopoulos I, Fioretos T, Johansson B. BRAF mutations are very rare in B- and T-cell pediatric acute lymphoblastic leukemias. Leukemia (2008) 22(8):1619–21. doi:10.1038/leu.2008.14
47. Driessen EM, van Roon EH, Spijkers-Hagelstein JA, Schneider P, de Lorenzo P, Valsecchi MG, et al. Frequencies and prognostic impact of RAS mutations in MLL-rearranged acute lymphoblastic leukemia in infants. Haematologica (2013) 98(6):937–44. doi:10.3324/haematol.2012.067983
48. Nicholson L, Knight T, Matheson E, Minto L, Case M, Sanichar M, et al. Casitas B lymphoma mutations in childhood acute lymphoblastic leukemia. Genes Chromosomes Cancer (2012) 51(3):250–6. doi:10.1002/gcc.20949
49. Martinelli S, Checquolo S, Consoli F, Stellacci E, Rossi C, Silvano M, et al. Loss of CBL E3-ligase activity in B-lineage childhood acute lymphoblastic leukaemia. Br J Haematol (2012) 159(1):115–9. doi:10.1111/j.1365-2141.2012.09245.x
50. Saito Y, Aoki Y, Muramatsu H, Makishima H, Maciejewski JP, Imaizumi M, et al. Casitas B-cell lymphoma mutation in childhood T-cell acute lymphoblastic leukemia. Leuk Res (2012) 36(8):1009–15. doi:10.1016/j.leukres.2012.04.018
51. Shiba N, Park MJ, Taki T, Takita J, Hiwatari M, Kanazawa T, et al. CBL mutations in infant acute lymphoblastic leukaemia. Br J Haematol (2012) 156(5):672–4. doi:10.1111/j.1365-2141.2011.08900.x
52. Balgobind BV, Van Vlierberghe P, van den Ouweland AM, Beverloo HB, Terlouw-Kromosoeto JN, van Wering ER, et al. Leukemia-associated NF1 inactivation in patients with pediatric T-ALL and AML lacking evidence for neurofibromatosis. Blood (2008) 111(8):4322–8. doi:10.1182/blood-2007-06-095075
53. Mullighan CG, Goorha S, Radtke I, Miller CB, Coustan-Smith E, Dalton JD, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature (2007) 446(7137):758–64. doi:10.1038/nature05690
54. Le DT, Kong N, Zhu Y, Lauchle JO, Aiyigari A, Braun BS, et al. Somatic inactivation of Nf1 in hematopoietic cells results in a progressive myeloproliferative disorder. Blood (2004) 103(11):4243–50. doi:10.1182/blood-2003-08-2650
55. Nakanishi H, Nakamura T, Canaani E, Croce CM. ALL1 fusion proteins induce deregulation of EphA7 and ERK phosphorylation in human acute leukemias. Proc Natl Acad Sci U S A (2007) 104(36):14442–7. doi:10.1073/pnas.0703211104
56. Mandanas RA, Leibowitz DS, Gharehbaghi K, Tauchi T, Burgess GS, Miyazawa K, et al. Role of p21 RAS in p210 bcr-abl transformation of murine myeloid cells. Blood (1993) 82(6):1838–47.
57. Pendergast AM, Quilliam LA, Cripe LD, Bassing CH, Dai Z, Li N, et al. BCR-ABL-induced oncogenesis is mediated by direct interaction with the SH2 domain of the GRB-2 adaptor protein. Cell (1993) 75(1):175–85. doi:10.1016/0092-8674(93)90689-N
58. Schubbert S, Shannon K, Bollag G. Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer (2007) 7(4):295–308. doi:10.1038/nrc2175
59. Sabnis AJ, Cheung LS, Dail M, Kang HC, Santaguida M, Hermiston ML, et al. Oncogenic Kras initiates leukemia in hematopoietic stem cells. PLoS Biol (2009) 7(3):e59. doi:10.1371/journal.pbio.1000059
60. Wang J, Liu Y, Tan LX, Lo JC, Du J, Ryu MJ, et al. Distinct requirements of hematopoietic stem cell activity and Nras G12D signaling in different cell types during leukemogenesis. Cell Cycle (2011) 10(17):2836–9. doi:10.4161/cc.10.17.17195
61. Kindler T, Cornejo MG, Scholl C, Liu J, Leeman DS, Haydu JE, et al. K-RasG12D-induced T-cell lymphoblastic lymphoma/leukemias harbor Notch1 mutations and are sensitive to gamma-secretase inhibitors. Blood (2008) 112(8):3373–82. doi:10.1182/blood-2008-03-147587
62. Zhang J, Wang J, Liu Y, Sidik H, Young KH, Lodish HF, et al. Oncogenic Kras-induced leukemogeneis: hematopoietic stem cells as the initial target and lineage-specific progenitors as the potential targets for final leukemic transformation. Blood (2009) 113(6):1304–14. doi:10.1182/blood-2008-01-134262
63. Wang J, Liu Y, Li Z, Wang Z, Tan LX, Ryu MJ, et al. Endogenous oncogenic Nras mutation initiates hematopoietic malignancies in a dose- and cell type-dependent manner. Blood (2011) 118(2):368–79. doi:10.1182/blood-2010-12-326058
64. Lauchle JO, Kim D, Le DT, Akagi K, Crone M, Krisman K, et al. Response and resistance to MEK inhibition in leukaemias initiated by hyperactive Ras. Nature (2009) 461(7262):411–4. doi:10.1038/nature08279
65. Kong G, Du J, Liu Y, Meline B, Chang YI, Ranheim EA, et al. Notch1 gene mutations target KRAS G12D-expressing CD8+ cells and contribute to their leukemogenic transformation. J Biol Chem (2013) 288(25):18219–27. doi:10.1074/jbc.M113.475376
66. Dail M, Li Q, McDaniel A, Wong J, Akagi K, Huang B, et al. Mutant Ikzf1, KrasG12D, and Notch1 cooperate in T lineage leukemogenesis and modulate responses to targeted agents. Proc Natl Acad Sci U S A (2010) 107(11):5106–11. doi:10.1073/pnas.1001064107
67. Van Meter ME, Diaz-Flores E, Archard JA, Passegue E, Irish JM, Kotecha N, et al. K-RasG12D expression induces hyperproliferation and aberrant signaling in primary hematopoietic stem/progenitor cells. Blood (2007) 109(9):3945–52. doi:10.1182/blood-2006-09-047530
68. Li Q, Bohin N, Wen T, Ng V, Magee J, Chen SC, et al. Oncogenic Nras has bimodal effects on stem cells that sustainably increase competitiveness. Nature (2013) 504(7478):143–7. doi:10.1038/nature12830
69. Mullighan CG, Zhang J, Kasper LH, Lerach S, Payne-Turner D, Phillips LA, et al. CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature (2011) 471(7337):235–9. doi:10.1038/nature09727
70. Tamai H, Miyake K, Takatori M, Miyake N, Yamaguchi H, Dan K, et al. Activated K-Ras protein accelerates human MLL/AF4-induced leukemo-lymphomogenicity in a transgenic mouse model. Leukemia (2011) 25(5):888–91. doi:10.1038/leu.2011.15
71. Chen W, Li Q, Hudson WA, Kumar A, Kirchhof N, Kersey JH. A murine Mll-AF4 knock-in model results in lymphoid and myeloid deregulation and hematologic malignancy. Blood (2006) 108(2):669–77. doi:10.1182/blood-2005-08-3498
72. Davidsson J, Paulsson K, Lindgren D, Lilljebjorn H, Chaplin T, Forestier E, et al. Relapsed childhood high hyperdiploid acute lymphoblastic leukemia: presence of preleukemic ancestral clones and the secondary nature of microdeletions and RTK-RAS mutations. Leukemia (2010) 24(5):924–31. doi:10.1038/leu.2010.39
73. Terada N, Smith TJ, Stork LC, Odom LF, Gelfand EW. N-ras gene mutations in childhood acute non-lymphoblastic leukemia. Leuk Res (1991) 15(10):935–41. doi:10.1016/0145-2126(91)90170-X
74. Yokota S, Nakao M, Horiike S, Seriu T, Iwai T, Kaneko H, et al. Mutational analysis of the N-ras gene in acute lymphoblastic leukemia: a study of 125 Japanese pediatric cases. Int J Hematol (1998) 67(4):379–87. doi:10.1016/S0925-5710(98)00015-2
75. Liang DC, Shih LY, Fu JF, Li HY, Wang HI, Hung IJ, et al. K-Ras mutations and N-Ras mutations in childhood acute leukemias with or without mixed-lineage leukemia gene rearrangements. Cancer (2006) 106(4):950–6. doi:10.1002/cncr.21687
76. Moorman AV, Richards SM, Martineau M, Cheung KL, Robinson HM, Jalali GR, et al. Outcome heterogeneity in childhood high-hyperdiploid acute lymphoblastic leukemia. Blood (2003) 102(8):2756–62. doi:10.1182/blood-2003-04-1128
77. Harrison CJ, Moorman AV, Broadfield ZJ, Cheung KL, Harris RL, Reza Jalali G, et al. Three distinct subgroups of hypodiploidy in acute lymphoblastic leukaemia. Br J Haematol (2004) 125(5):552–9. doi:10.1111/j.1365-2141.2004.04948.x
78. Yan J, Jiang N, Huang G, Tay JL, Lin B, Bi C, et al. Deregulated MIR335 that targets MAPK1 is implicated in poor outcome of paediatric acute lymphoblastic leukaemia. Br J Haematol (2013) 163(1):93–103. doi:10.1111/bjh.12489
79. Kamata T, Pritchard C. Mechanisms of aneuploidy induction by RAS and RAF oncogenes. Am J Cancer Res (2011) 1(7):955–71.
80. Choi S, Henderson MJ, Kwan E, Beesley AH, Sutton R, Bahar AY, et al. Relapse in children with acute lymphoblastic leukemia involving selection of a preexisting drug-resistant subclone. Blood (2007) 110(2):632–9. doi:10.1182/blood-2007-01-067785
81. Mullighan CG, Phillips LA, Su X, Ma J, Miller CB, Shurtleff SA, et al. Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science (2008) 322(5906):1377–80. doi:10.1126/science.1164266
82. Anderson K, Lutz C, van Delft FW, Bateman CM, Guo Y, Colman SM, et al. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature (2011) 469(7330):356–61. doi:10.1038/nature09650
83. Garza AS, Miller AL, Johnson BH, Thompson EB. Converting cell lines representing hematological malignancies from glucocorticoid-resistant to glucocorticoid-sensitive: signaling pathway interactions. Leuk Res (2009) 33(5):717–27. doi:10.1016/j.leukres.2008.10.006
84. McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EW, Chang F, et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta (2007) 1773(8):1263–84. doi:10.1016/j.bbamcr.2006.10.001
85. Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature (2013) 503(7477):548–51. doi:10.1038/nature12796
86. Goemans BF, Zwaan CM, Harlow A, Loonen AH, Gibson BE, Hahlen K, et al. In vitro profiling of the sensitivity of pediatric leukemia cells to tipifarnib: identification of T-cell ALL and FAB M5 AML as the most sensitive subsets. Blood (2005) 106(10):3532–7. doi:10.1182/blood-2005-04-1640
87. Karp JE, Lancet JE, Kaufmann SH, End DW, Wright JJ, Bol K, et al. Clinical and biologic activity of the farnesyltransferase inhibitor R115777 in adults with refractory and relapsed acute leukemias: a phase 1 clinical-laboratory correlative trial. Blood (2001) 97(11):3361–9. doi:10.1182/blood.V97.11.3361
88. Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer (2003) 3(1):11–22. doi:10.1038/nrc969
89. Gilmartin AG, Bleam MR, Groy A, Moss KG, Minthorn EA, Kulkarni SG, et al. GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clin Cancer Res (2011) 17(5):989–1000. doi:10.1158/1078-0432.CCR-10-2200
90. Kim K, Kong SY, Fulciniti M, Li X, Song W, Nahar S, et al. Blockade of the MEK/ERK signalling cascade by AS703026, a novel selective MEK1/2 inhibitor, induces pleiotropic anti-myeloma activity in vitro and in vivo. Br J Haematol (2010) 149(4):537–49. doi:10.1111/j.1365-2141.2010.08127.x
91. Bennouna J, Lang I, Valladares-Ayerbes M, Boer K, Adenis A, Escudero P, et al. A phase II, open-label, randomised study to assess the efficacy and safety of the MEK1/2 inhibitor AZD6244 (ARRY-142886) versus capecitabine monotherapy in patients with colorectal cancer who have failed one or two prior chemotherapeutic regimens. Invest New Drugs (2011) 29(5):1021–8. doi:10.1007/s10637-010-9392-8
92. Davies BR, Logie A, McKay JS, Martin P, Steele S, Jenkins R, et al. AZD6244 (ARRY-142886), a potent inhibitor of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase 1/2 kinases: mechanism of action in vivo, pharmacokinetic/pharmacodynamic relationship, and potential for combination in preclinical models. Mol Cancer Ther (2007) 6(8):2209–19.
93. Yeh TC, Marsh V, Bernat BA, Ballard J, Colwell H, Evans RJ, et al. Biological characterization of ARRY-142886 (AZD6244), a potent, highly selective mitogen-activated protein kinase kinase 1/2 inhibitor. Clin Cancer Res (2007) 13(5):1576–83. doi:10.1158/1078-0432.CCR-06-1150
94. Meng XW, Chandra J, Loegering D, Van Becelaere K, Kottke TJ, Gore SD, et al. Central role of Fas-associated death domain protein in apoptosis induction by the mitogen-activated protein kinase kinase inhibitor CI-1040 (PD184352) in acute lymphocytic leukemia cells in vitro. J Biol Chem (2003) 278(47):47326–39. doi:10.1074/jbc.M304793200
95. Kolb EA, Gorlick R, Houghton PJ, Morton CL, Neale G, Keir ST, et al. Initial testing (stage 1) of AZD6244 (ARRY-142886) by the Pediatric Preclinical Testing Program. Pediatr Blood Cancer (2010) 55(4):668–77. doi:10.1002/pbc.22576
96. Shieh A, Ward AF, Donlan KL, Harding-Theobald ER, Xu J, Mullighan CG, et al. Defective K-Ras oncoproteins overcome impaired effector activation to initiate leukemia in vivo. Blood (2013) 121(24):4884–93. doi:10.1182/blood-2012-05-432252
97. Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med (2008) 14(12):1351–6. doi:10.1038/nm.1890
98. Haagensen EJ, Kyle S, Beale GS, Maxwell RJ, Newell DR. The synergistic interaction of MEK and PI3K inhibitors is modulated by mTOR inhibition. Br J Cancer (2012) 106(8):1386–94. doi:10.1038/bjc.2012.70
99. Schultz KR, Bowman WP, Aledo A, Slayton WB, Sather H, Devidas M, et al. Improved early event-free survival with imatinib in Philadelphia chromosome-positive acute lymphoblastic leukemia: a Children’s Oncology Group Study. J Clin Oncol (2009) 27(31):5175–81. doi:10.1200/JCO.2008.21.2514
100. Corcoran RB, Cheng KA, Hata AN, Faber AC, Ebi H, Coffee EM, et al. Synthetic lethal interaction of combined BCL-XL and MEK inhibition promotes tumor regressions in KRAS mutant cancer models. Cancer Cell (2013) 23(1):121–8. doi:10.1016/j.ccr.2012.11.007
101. Rambal AA, Panaguiton ZL, Kramer L, Grant S, Harada H. MEK inhibitors potentiate dexamethasone lethality in acute lymphoblastic leukemia cells through the pro-apoptotic molecule BIM. Leukemia (2009) 23(10):1744–54. doi:10.1038/leu.2009.80
102. Zhang W, Konopleva M, Burks JK, Dywer KC, Schober WD, Yang JY, et al. Blockade of mitogen-activated protein kinase/extracellular signal-regulated kinase and murine double minute synergistically induces apoptosis in acute myeloid leukemia via BH3-only proteins Puma and Bim. Cancer Res (2010) 70(6):2424–34. doi:10.1158/0008-5472.CAN-09-0878
103. Rahmani M, Anderson A, Habibi JR, Crabtree TR, Mayo M, Harada H, et al. The BH3-only protein Bim plays a critical role in leukemia cell death triggered by concomitant inhibition of the PI3K/Akt and MEK/ERK1/2 pathways. Blood (2009) 114(20):4507–16. doi:10.1182/blood-2008-09-177881
104. Steelman LS, Franklin RA, Abrams SL, Chappell W, Kempf CR, Basecke J, et al. Roles of the Ras/Raf/MEK/ERK pathway in leukemia therapy. Leukemia (2011) 25(7):1080–94. doi:10.1038/leu.2011.66
105. Brown P, Levis M, Shurtleff S, Campana D, Downing J, Small D. FLT3 inhibition selectively kills childhood acute lymphoblastic leukemia cells with high levels of FLT3 expression. Blood (2005) 105(2):812–20. doi:10.1182/blood-2004-06-2498
106. Liu W, Yu B, Xu G, Xu WR, Loh ML, Tang LD, et al. Identification of cryptotanshinone as an inhibitor of oncogenic protein tyrosine phosphatase SHP2 (PTPN11). J Med Chem (2013) 56(18):7212–21. doi:10.1021/jm400474r
107. Koo HM, McWilliams MJ, Alvord WG, Vande Woude GF. Ras oncogene-induced sensitization to 1-beta-d-arabinofuranosylcytosine. Cancer Res (1999) 59(24):6057–62.
108. Koo HM, Monks A, Mikheev A, Rubinstein LV, Gray-Goodrich M, McWilliams MJ, et al. Enhanced sensitivity to 1-beta-d-arabinofuranosylcytosine and topoisomerase II inhibitors in tumor cell lines harboring activated ras oncogenes. Cancer Res (1996) 56(22):5211–6.
109. Neubauer A, Maharry K, Mrozek K, Thiede C, Marcucci G, Paschka P, et al. Patients with acute myeloid leukemia and RAS mutations benefit most from postremission high-dose cytarabine: a Cancer and Leukemia Group B study. J Clin Oncol (2008) 26(28):4603–9. doi:10.1200/JCO.2007.14.0418
110. Ahmad EI, Gawish HH, Al Azizi NM, Elhefni AM. The prognostic impact of K-RAS mutations in adult acute myeloid leukemia patients treated with high-dose cytarabine. Onco Targets Ther (2011) 4:115–21. doi:10.2147/OTT.S12602
111. Illmer T, Thiede C, Fredersdorf A, Stadler S, Neubauer A, Ehninger G, et al. Activation of the RAS pathway is predictive for a chemosensitive phenotype of acute myelogenous leukemia blasts. Clin Cancer Res (2005) 11(9):3217–24. doi:10.1158/1078-0432.CCR-04-2232
Keywords: acute lymphoblastic leukemia, Ras/Raf/MEK/ERK pathway, targeted therapy
Citation: Knight T and Irving JAE (2014) Ras/Raf/MEK/ERK pathway activation in childhood acute lymphoblastic leukemia and its therapeutic targeting. Front. Oncol. 4:160. doi: 10.3389/fonc.2014.00160
Received: 11 March 2014; Accepted: 06 June 2014;
Published online: 24 June 2014.
Edited by:
Mignon Lee-Cheun Loh, University of California San Francisco, USAReviewed by:
John Moppett, Bristol Royal Hospital for Children, UKDavid Anthony Rodeberg, East Carolina University Brody School of Medicine, USA
Copyright: © 2014 Knight and Irving. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Julie Anne Elizabeth Irving, Newcastle Cancer Centre at the Northern Institute for Cancer Research, Newcastle University, Paul O’Gorman Building, Framlington Place, Newcastle upon Tyne NE2 4HH, UK e-mail: j.a.e.irving@ncl.ac.uk