 Bin Ma
Bin Ma Michael O. Hottiger
Michael O. Hottiger- 1School of Biomedical Engineering, Shanghai Jiao Tong University, Shanghai, China
- 2Renji Hospital Clinical Stem Cell Research Center, Shanghai Jiao Tong University School of Medicine, Shanghai, China
- 3Department of Molecular Mechanisms of Disease, University of Zurich, Zurich, Switzerland
Besides its important role in embryonic development and homeostatic self-renewal in adult tissues, Wnt/β-catenin signaling exerts both anti-inflammatory and proinflammatory functions. This is, at least partially, due to either repressing or enhancing the NF-κB pathway. Similarly, the NF-κB pathway either positively or negatively regulates Wnt/β-catenin signaling. Different components of the two pathways are involved in this crosstalk, forming a complex regulatory network. This review summarizes our current understanding of the molecular mechanisms underlying the cross-regulation between the two pathways and discusses their involvement in inflammation and inflammation-associated diseases such as cancer.
Introduction
Wnt/β-catenin and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling both are conserved pathways that regulate a variety of biological processes throughout the development and adult lifetime of mammals. Aberrations within these two pathways result in a wide range of pathologies, such as cancer, inflammatory and immune diseases, as well as metabolic diseases (1–4). The Wnt/β-catenin and NF-κB signaling pathways regulate, through independent cascades, the expression of different subsets of target genes controlling cell proliferation, cell survival, and differentiation. In addition to these shared functions, Wnt/β-catenin signaling is crucial for development and tissue regeneration, whereas NF-κB is a key master of inflammation. Recent findings suggest that the two signaling pathways cross-regulate each of their activities and functions.
Crosstalk of signaling pathways extends the functions of individual pathways and results in a more complex regulatory network, inherent to the diversity and homeostasis of biological systems. Wnt/β-catenin pathway components modulate inflammatory and immune responses via the interaction with NF-κB (5). Reciprocally, NF-κB also influences the activity of Wnt/β-catenin signaling pathway. Depending on the cellular or tissue context, both positive and negative cross-regulation has been observed. The crosstalk between these two pathways can thus significantly influence the progression of inflammation and cancer. Intensive research has revealed NF-κB signaling as an attractive target for the treatment of inflammatory diseases and inflammation-associated cancer (6–8). Members of the Wnt/β-catenin pathway also serve as potential therapeutic targets for many types of cancer (9). Understanding the molecular basis for the cross-regulation thus helps elucidating the underlying pathophysiological mechanisms for inflammation-involved diseases and for developing more specific and effective therapeutic options against these diseases.
In this review, we summarize the current evidence for both positive and negative regulation of NF-κB-mediated inflammation by Wnt/β-catenin signaling and elaborate on the underlying molecular mechanisms. We also describe the reciprocal regulation of Wnt/β-catenin signaling by the NF-κB pathway and novel models for the cooperation of these two pathways in regulating gene transcription. The major components involved in the cross-regulation are discussed. We sought to describe the complexity of the crosstalk between Wnt/β-catenin and NF-κB signaling to link it with the biological importance for inflammation and cancer, and to discuss its potential impact on the development of new therapeutic options.
Wnt/β-Catenin Signaling Pathway
Wnt proteins are lipid-modified in the endoplasmic reticulum (ER), traffic through the Golgi to the plasma membrane, and are secreted into the extracellular space (1). Both the human and mouse genome harbors 19 Wnt genes (10). These Wnt proteins are structurally and functionally conserved, and selectively expressed in certain cell types. Extracellular Wnt proteins activate either the β-catenin-dependent, canonical signaling pathway through engagement of the co-receptors frizzled (FZD) and low-density lipoprotein receptor-related protein (LRP) or the β-catenin-independent, non-canonical pathway via various receptors such as FZD, receptor tyrosine kinase (Ryk), and receptor tyrosine kinase-like orphan receptor (Ror). β-Catenin is the central mediator of the canonical signaling cascade and functions as an adhesion molecule at the plasma membrane (11). In the absence of Wnt stimulation, β-catenin in the cytoplasm is constitutively targeted for degradation by the destruction complex consisting of adenoma polyposis coli (APC), axin, glycogen synthase kinase 3 (GSK-3), and casein kinase I (CKI) (Figure 1). This complex binds to cytosolic β-catenin and facilitates the latter’s sequential phosphorylation by CKI (at S45) and GSK-3 (at S33/S37/T41). Phosphorylated β-catenin is then recognized and ubiquitinated by β-transducing repeat-containing protein (βTrCP), which tags it for degradation by the proteasome. In the absence of Wnt stimulation, the cytoplasmic levels of β-catenin are thus tightly controlled by this degradation complex. Upon binding of Wnt proteins to the FZD receptor and LRP5/6 co-receptor, the intracellular phosphoprotein disheveled (DVL) is activated, causing the inactivation of the degradation complex and cytoplasmic accumulation of β-catenin. After translocation into the nucleus, β-catenin associates with T cell factor/lymphoid enhancer factor (TCF/LEF) transcription factors and promotes the transcription of its target genes. There are four TCF/LEF transcription factor members in vertebrates: TCF1, TCF3, TCF4, and LEF1. They all contain a β-catenin interaction domain at the N-terminus and recognize the same consensus DNA-binding sequences but are structurally and functionally somewhat different (12). Interestingly, the β-catenin:TCF/LEF machinery not only activates gene expression but also directly represses the transcription of certain target genes (13). Additional co-factors, e.g., cAMP response element-binding protein (CREB)-binding protein (CBP)/p300 (14) and ADP-ribosyltransferase diphtheria toxin-like 1 (ARTD1, also known as PARP1) (15), are involved in the transcriptional regulation of Wnt signaling.
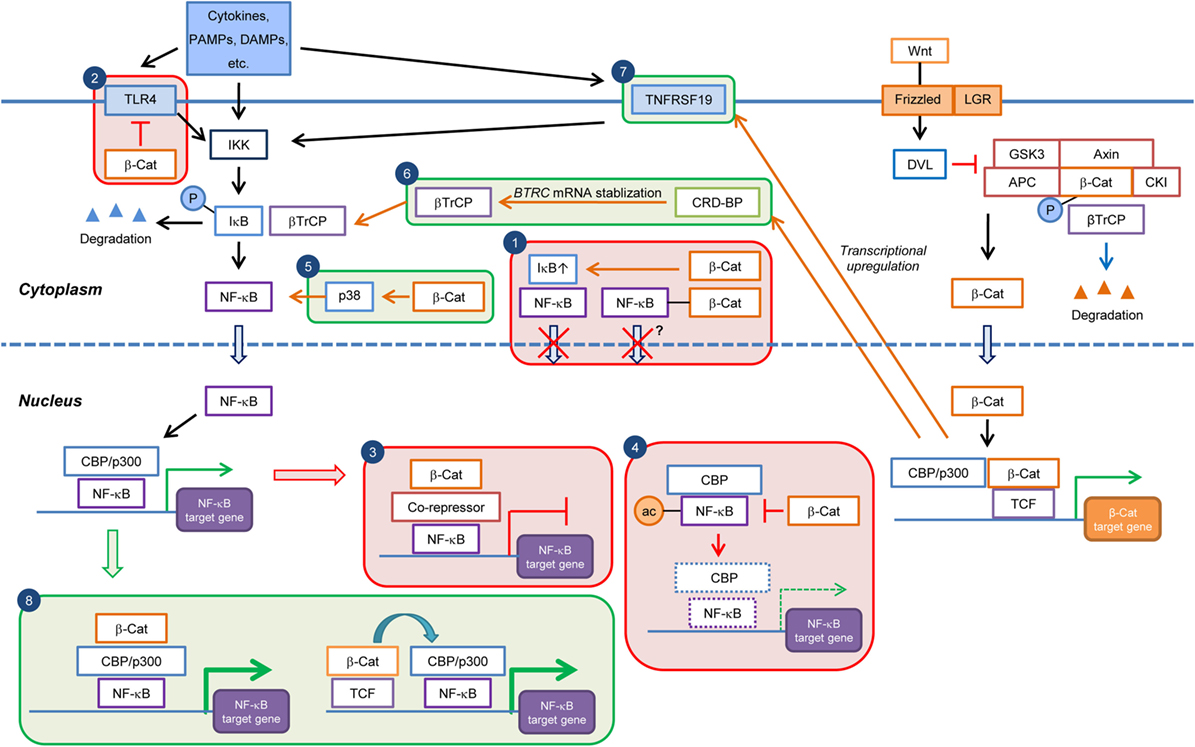
Figure 1. Current knowledge on the cell type/context-dependent regulation of NF-κB signaling by Wnt/β-catenin pathway. Boxes in red and green colors indicate negative and positive regulations, respectively. Box 1: inhibition of NF-κB nuclear translocation by β-catenin-mediated sequestration or upregulation of IκB; Box 2: downregulation of TLR4 expression by β-catenin; Box 3: repression of NF-κB target gene expression through recruitment of corepressor at NF-κB-binding elements; Box 4: downregulation of NF-κB target gene expression through inhibition of CBP-mediated acetylation of NF-κB; Box 5: induction of p38 activity and p38-mediated NF-κB activation by β-catenin; Box 6: promotion of βTrCP-mediated IκB degradation through transcriptional upregulation of CRDBP by Wnt/β-catenin and CRD-BP-mediated BTRC (βTrCP) mRNA stabilization; Box 7: induction of NF-κB activity through transcriptional upregulation of TNFRSF19 mRNA; and Box 8: enhancement of NF-κB target gene expression through cooperation of β-catenin:TCF/LEF and NF-κB transcriptional complexes. PAMPs, pathogen-associated molecular patterns; DAMPs, danger-associated molecular pattern molecules; β-Cat, β-catenin; P, phospho; ac, acetyl.
Expression of Wnt/β-catenin signaling target genes regulates stemness (e.g., NANOG and OCT4), proliferation (e.g., CCND1 and MYC), differentiation (e.g., SOX9 and RUNX2), and immune responses (e.g., CTLA4 and TNFSF9), revealing a broad control of organismal and cellular functions by the Wnt/β-catenin pathway (http://web.stanford.edu/group/nusselab/cgi-bin/wnt/). Aberrant constitutive activation of Wnt/β-catenin signaling caused by mutations in genes, such as APC or CTNNB1 (encoding β-catenin), is involved in tumorigenesis of many organs including intestine, stomach, liver, ovaries, and pancreas (9, 16). Repression of the Wnt pathway by extracellular inhibitors, such as Dickkopf1 (DKK1), Wnt inhibitory factor 1 (WIF1), or secreted frizzled-related proteins (SFRPs), has also been observed in human cancers (16, 17).
Many chemical inhibitors against the Wnt/β-catenin pathway, targeting various components such as porcupine, DVL, tankyrase, or TCF/LEF, have been developed (9). Some reduce Wnt ligand activity and/or the β-catenin protein level, while some directly block transcriptional activity. Wnt/β-catenin signaling has been reported to crosstalk with many other signaling pathways, including NF-κB, mothers against decapentaplegic homolog 3 (Smad3), Notch, forkhead box O (FOXO), and hypoxia-inducible factor-1α (HIF-1α), extending the spectrum of biological functions of this pathway (18–22).
NF-κB Signaling Pathway
The NF-κB transcription factor family consists of five members trapped in the cytoplasm under non-activated conditions: NF-κB1 (p50/p105), NF-κB2 (p52/p100), RelA (p65), RelB, and c-Rel (23). They all possess a structurally conserved amino-terminal Rel-homology domain (RHD), which contains the dimerization, nuclear localization, and DNA-binding domains. RelA, RelB, and c-Rel (but not p50 or p52) also have a transactivation domain that promotes gene transcription. Upon activation, the NF-κB subunits form either a homo- or heterodimer. NF-κB signaling is divided into the NF-κB essential modulator (NEMO)-dependent, canonical pathway and the NEMO-independent, non-canonical pathway. Various signals, including cytokines, growth factors, microbial products, stress-inducing stimuli such as radiation and oxidative stress, and engagement of T cell receptor (TCR), induce canonical NF-κB signaling (4, 8, 24). These stimuli activate membrane-bound receptors, including the tumor necrosis factor receptor superfamily (TNFRSF) and interleukin-1 receptor (IL-1R)/Toll-like receptor (TLR) superfamily as well as TCR, or intracellular mediators, culminating in the activation of the inhibitor of kappa B (IκB) kinase (IKK) complex, composed of the scaffold protein NEMO (IKKγ) and two IKK subunits (IKKα and IKKβ). The IKK complex subsequently phosphorylates IκB proteins, resulting in their ubiquitination and subsequent proteasomal degradation. As a result, the NF-κB dimers now translocate to the nucleus, where they regulate the expression of target genes. The main activated NF-κB dimers formed in the canonical pathway are RelA/p50 and RelA/p52 (4). Co-factors, such as CBP/p300 and ARTD1, participate in the regulation of NF-κB target gene transcription (25, 26).
By controlling the expression of a broad spectrum of target genes, such as for cytokines, chemokines, growth factors, immune receptors, transcription factors, and repressors of apoptosis, NF-κB functions as a crucial coordinator of inflammatory and immune responses, as well as cell differentiation, proliferation, and survival. Dysregulation of NF-κB has been implicated in diverse inflammatory disorders such as rheumatoid arthritis, multiple sclerosis, asthma, and inflammatory bowel disease (3, 7, 8). NF-κB also regulates cancer development, either through direct control of cell proliferation and apoptosis or induction of cancer-related inflammation and tumor immunity (6, 7). Thus, NF-κB is an attractive therapeutic target for treating inflammatory diseases and cancers. This review focuses on canonical NF-κB signaling, as it is the main cascade that crosstalks with Wnt/β-catenin.
Regulation of NF-κB Signaling by the Wnt/β-Catenin Pathway
Negative Regulation of NF-κB Signaling by Wnt/β-Catenin
The first compelling evidence that Wnt/β-catenin negatively regulates NF-κB activity came from the observation that overexpressed β-catenin physically interacted with and inhibited NF-κB function in human colon and breast cancer cells (18) (Table 1). In this study, β-catenin was found to form a complex with RelA and p50, leading to a decrease in NF-κB DNA binding and transactivation activity, and target gene expression. Interestingly, the protein–protein interaction between β-catenin and NF-κB was only indirect, as these two proteins did not bind to each other in an in vitro pull down assay using only β-catenin and NF-κB recombinant proteins, while the formation of a complex occurred when cell lysates were supplemented. Activated β-catenin inhibited the expression of the NF-κB target gene FAS, thus possibly contributing to tumorigenesis through repression of Fas-mediated apoptosis. A strong inverse correlation between β-catenin and Fas expression was also identified in primary human colon and breast tumor samples, supporting an in vivo regulation of NF-κB by β-catenin. Further studies also revealed a negative effect of β-catenin on NF-κB activity and expression of downstream target genes in liver, breast, and colon cancer cells (27, 28). However, such correlation was not seen in head and neck cancer (29), indicating a tissue-specific mechanism and/or more complex regulatory network for these genes. The physical interaction and functional inhibition of NF-κB by β-catenin in colorectal cancer cells was demonstrated to require phosphatidylinositide 3-kinase (PI3K) (30). Blockade of PI3K by chemical inhibitors abrogated the formation of β-catenin and NF-κB protein complexes. In resting colorectal cancer cells, β-catenin and NF-κB colocalized in the cytoplasm, and treatment with PI3K inhibitor resulted in the nuclear translocation of NF-κB and membrane retention of β-catenin. However, it is not clear whether PI3K directly serves as a bridging factor between β-catenin and NF-κB or alternatively plays a role in β-catenin-mediated repression of NF-κB activated by different stimuli.
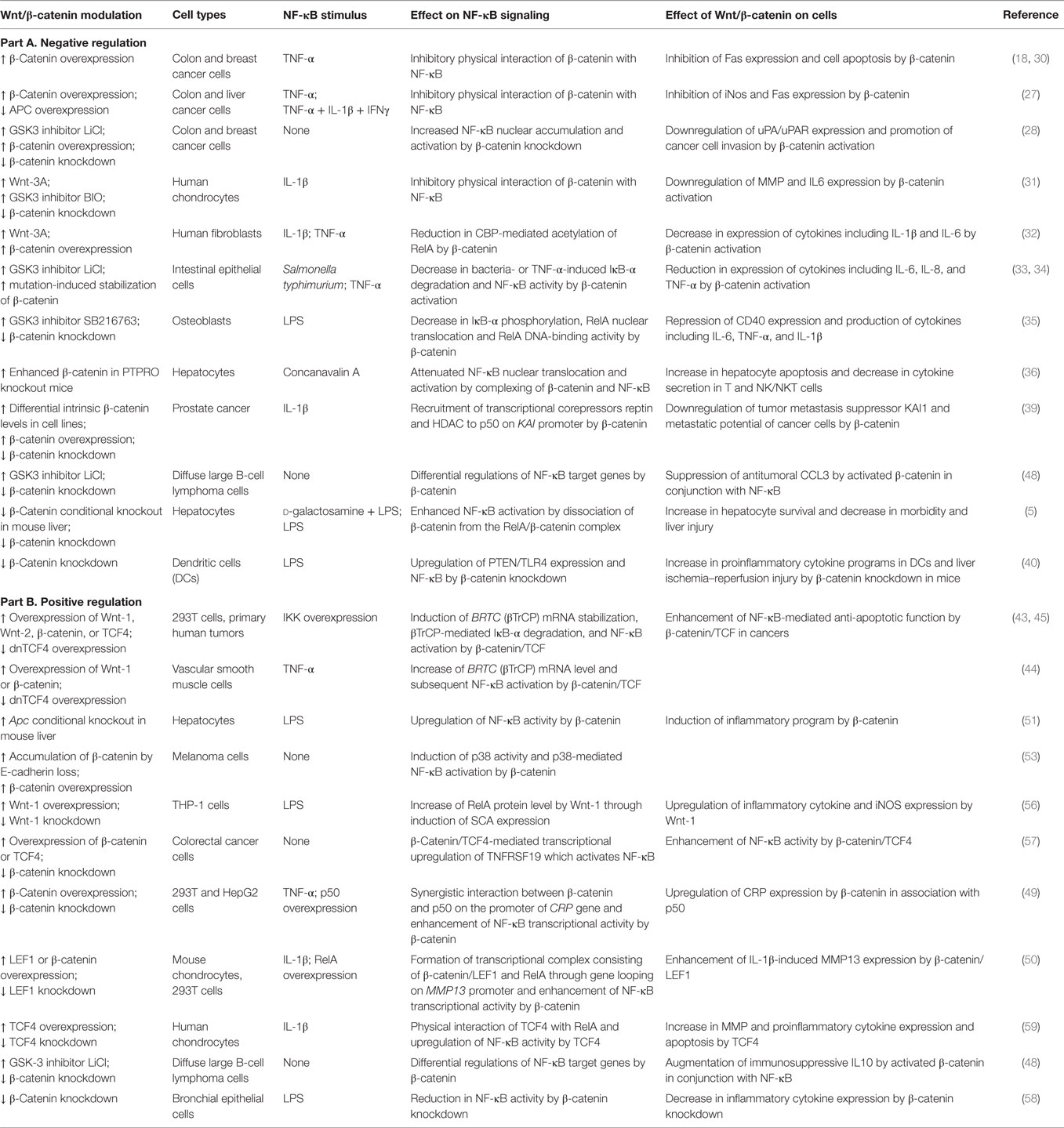
Table 1. Overview of cell type/context-dependent regulatory effects of Wnt/β-catenin pathway on NF-κB signaling.
The inhibitory effect of β-catenin on NF-κB activity has also been found in many non-tumor cell types, including chondrocytes (31), fibroblasts (32), epithelial cells (33, 34), osteoblasts (35), and hepatocytes (5, 36) (Table 1). In human chondrocytes, Wnt-3A stimulation or overexpression of Wnt-7B or β-catenin repressed IL-1β-induced expression of IL6 and matrix metalloproteinase (MMP) genes, including MMP1, MMP3, and MMP13 (31). Physical interaction of β-catenin with RelA and p50 and a reduction in NF-κB reporter activity by Wnt-3A stimulation were observed in this cell type. Notably, a recent study in human fibroblasts showed that IL-1β-induced nuclear translocation of RelA was not directly regulated by Wnt-3A co-stimulation in the initial phase (i.e., 4 h) after NF-κB activation (32). The repression of NF-κB activity by Wnt/β-catenin was rather achieved through a reduction in CBP-mediated acetylation of RelA (Figure 1). The negative effect of Wnt/β-catenin on NF-κB was mediated through CBP, since knockdown of CBP eliminated the downregulation of IL-1β-elicited cytokine expression by Wnt-3A. The selective repression of a subset of proinflammatory cytokine expression by Wnt/β-catenin might be attributed to RelA acetylation-dependent gene expression.
Also in bacteria-colonized intestinal epithelial cells, overexpression of constitutively active mutant β-catenin or activation of β-catenin by GSK-3 inhibition lead to a reduction in NF-κB activity and expression of the target genes IL6, IL8, and TNFA [encoding tumor necrosis factor-α (TNF-α)], indicating an anti-inflammatory function of β-catenin (33, 34). In line with this notion, lipopolysaccharide (LPS) or concanavalin A (ConA)/TNF-α-induced IκB-α phosphorylation and nuclear translocation of RelA were decreased in osteoblasts treated with GSK-3 inhibitor (35) or in hepatocytes with higher β-catenin levels (36). Moreover, cisplatin- or virus-induced nuclear translocation of RelA was also found to be impaired in cochlea cell line HEI-OC1, monkey kidney MARC-145 cells, and pulmonary alveolar macrophages PAM-CD163 treated with a GSK-3 inhibitor (37, 38). Comparably, bacteria or TNF-α-induced IκB-α degradation was decreased in HCT116 epithelial cells overexpressing constitutively active β-catenin (33) (Figure 1). However, IκB-α was not able to complex with β-catenin in these cells, suggesting that IκB-α stabilization is independent of direct β-catenin binding. It is conceivable that a reduction in IκB-α degradation by β-catenin increases the cytoplasmic retention of NF-κB. The discrepancy of the effect of Wnt/β-catenin on IκB-α degradation and NF-κB nuclear translocation between different studies might be due to different cell types used and/or ways to modulate Wnt/β-catenin and NF-κB signaling. Compared to blocking the overall nuclear translocation of NF-κB, which might influence all target genes, the regulatory effect of β-catenin on RelA post-translational modifications provides a more elaborate and selective control.
In a study of the transcriptional regulation of the tumor metastasis suppressor gene KAI1 in prostate cancer cells, β-catenin functioned as corepressor, in association with reptin, to inhibit p50-mediated KAI1 expression (39) (Figure 1). In non-metastatic cells, KAI1 was actively transcribed by p50 and the transcriptional coactivator Tip60. In metastatic cells where β-catenin was upregulated, β-catenin–reptin complex displaced Tip60, bound to p50 at the KAI1 promoter and recruited histone deacetylase (HDAC), eventually leading to transcriptional repression. The balance between the transcriptional coactivator and corepressor complexes determined the expression level of KAI1 and metastatic potential of cancer cells. This cell-type-specific and promoter-specific property underlying β-catenin and NF-κB crosstalk was a unique molecular mechanism for selectively regulating the function of Wnt/β-catenin and might be crucial for cancer metastasis.
A negative regulation of NF-κB signaling by Wnt/β-catenin was also observed in hepatocytes from conditional β-catenin knockout (KO) mice, where lack of β-catenin was found to increase RelA protein levels and LPS-induced NF-κB activation (26). A survival benefit was also observed in d-galactosamine (GalN)/LPS-challenged β-catenin KO mice but not in KO mice with Fas activation, indicating an NF-κB-dependent effect, as the NF-κB-mediated anti-apoptotic effect was enhanced in hepatocytes of GalN/LPS-challenged KO mice, whereas Fas ligand/caspase-mediated apoptosis remained unchanged.
In dendritic cells (DCs), knockdown of β-catenin enhanced TLR4 expression, and LPS-induced NF-κB activation and proinflammatory cytokine programs, providing a potential regulatory mechanism independent of physical interaction of β-catenin with NF-κB (40) (Figure 1). Disruption of β-catenin upregulated phosphatase and tensin homolog deleted on chromosome 10 (PTEN), leading to reduced Akt phosphorylation and Akt-mediated suppression of TLR4. Enhanced TLR4 signaling might thus result in NF-κB activation and increased DC expression of proinflammatory mediators. Elevated inflammatory responses by ablation of β-catenin in DCs were also observed in a mouse model of inflammatory bowel disease, although the dependency of this effect on NF-κB regulation was not investigated (41). In summary, these studies reveal an anti-inflammatory role of β-catenin and that β-catenin signaling programs DCs to a tolerogenic state, limiting the inflammatory response.
These evidences clearly indicate a repressive effect of the Wnt/β-catenin pathway on basal or stimuli-induced NF-κB activity in various cell types via different mechanisms. However, the underlying molecular mechanisms seem to be cell type- and/or gene-specific.
Positive Modulation of NF-κB Activity by Wnt/β-Catenin
While many studies have revealed a negative regulation of NF-κB activity by Wnt/β-catenin, stimulatory effects of Wnt/β-catenin on NF-κB activity have also been reported (Table 1). βTrCP (encoded by the BTRC gene) is an E3 ubiquitin ligase receptor targeting the ubiquitination and subsequent degradation of both β-catenin and IκB-α (42). Wnt/β-catenin activation by overexpression of β-catenin or Wnt proteins elevated βTrCP expression, resulting in enhanced degradation of IκB-α and consequently NF-κB transactivation without affecting IKK activity (43). Induction of βTrCP by β-catenin/TCF also accelerated the degradation of β-catenin, serving as an indirect negative feedback loop. In vascular smooth muscle cells, Wnt-1 activated β-catenin/TCF4-mediated βTrCP expression and NF-κB activity (44). Furthermore, it was demonstrated that βTrCP mRNA was stabilized by β-catenin/TCF via transcriptional upregulation of its target gene CRDBP. Coding region determinant-binding protein (CRD-BP, encoded by the CRDBP gene) was found to bind the coding region of βTrCP mRNA, stabilized βTrCP mRNA, and elevated its protein level (45) (Figure 1). Induced BTRC and CRDBP expression was associated with the activation of both β-catenin and NF-κB in colorectal cancer, implying that integration of these pathways by βTrCP and CRD-BP might contribute to a reduction in tumor cell apoptosis and promotion of tumor metastasis (45, 46). These results suggest that βTrCP and CRD-BP levels are important for coordinating β-catenin/TCF and NF-κB signaling.
It has been demonstrated that stimulation of TLR3 drives breast cancer cells toward a cancer stem cell phenotype, which requires simultaneous activation of the β-catenin and NF-κB signaling pathway, implying a cooperative and synergistic function of β-catenin in TLR3-activated NF-κB signaling (47). A recent study illustrated that the oncogenic mutant scaffold protein CARMA1 causes β-catenin stabilization and connects B-cell receptor (BCR) signaling to NF-κB signaling, thereby coupling β-catenin and NF-κB pathways in diffuse large B-cell lymphomas (48). Increased β-catenin levels were sufficient to induce classical Wnt target gene signatures and additionally were able to augment expression of the immunosuppressive IL10 and to suppress the antitumoral CCL3 in conjunction with NF-κB (Figure 1), thus inducing a favorable tumor microenvironment. These data indicate that the effects of β-catenin on NF-κB target gene expression could be gene-dependent in the same cellular context. In line with this notion, downregulation of proinflammatory cytokine expression and synergistic upregulation of adhesion molecule VCAM1 by Wnt/β-catenin activation were observed in human fibroblasts stimulated with IL-1β (32). Interestingly, a β-catenin:TCF/LEF complex has been shown to bind to promoters of NF-κB target genes, such as CRP (C-reactive protein) and MMP13, and positively regulate gene transcription through gene looping in concert with NF-κB (49, 50) (Figure 1). In 293T and HepG2 cells treated with TNF-α, both β-catenin and p50 were required for CRP expression (49). β-Catenin/TCF4 bound to the CRP gene at a site distant to the p50 binding element and physically interacted with p50 through changes in chromosome conformation as detected by 3C chromosome capture assay. The β-catenin/TCF4 complex might constitute the necessary transcriptional machinery, since p50 lacks a transactivation domain. Similarly, in mouse chondrocytes stimulated with IL-1β, β-catenin/LEF1 together with RelA formed a transcriptional complex through gene looping in the Mmp13 genomic locus to regulate gene transcription (50).
Intriguingly, in contrast to the study in which deletion of β-catenin increased RelA protein levels and target gene expression in hepatocytes from conditional β-catenin KO mice challenged with LPS (5), β-catenin activation in hepatocytes of Apc KO mice also potentiated NF-κB activity as judged from electrophoretic mobility shift assay (EMSA) experiments and an inflammatory program including upregulation of NF-κB-induced chemokines (51). These discrepant findings might be explained by β-catenin and Apc KO leading to the same effect but by different mechanisms. Although β-catenin and Apc KO lead to inactivation and activation of the canonical Wnt pathway, respectively, the elevated NF-κB activity in the liver of Apc KO mice might be caused indirectly by tumor-triggered-inflammation, e.g., recruitment of immune cells and subsequent inflammatory responses in tumor microenvironment. Moreover, Wnt/β-catenin signaling-independent functions of APC has also been proposed, such as control of C-terminal binding protein-1 (CtBP1) degradation, retinoic acid (RA) biosynthesis, and cyclooxygenase-2 (COX-2) expression (52), which may as well influence the inflammatory response. In addition, it is possible that the use of different promoter-driven Cre to generate conditional KO in different cell populations might have invariant effects, as the effect of Wnt/β-catenin activation may differ in different subpopulations of cells. In melanoma cells, E-cadherin loss and cytoplasmic β-catenin accumulation triggered p38-mediated NF-κB activation (53). Similarly, E-cadherin disassembly and concomitant GSK-3β inactivation and β-catenin accumulation induced NF-κB-dependent inducible nitric oxide synthase (iNOS) gene expression in hepatocytes (54). Furthermore, E-cadherin in mesenchymal cells formed a complex with β-catenin and RelA and prevented RelA nuclear translocation, thus downregulating fibronectin gene expression, which is positively regulated by both proteins (55). These evidences suggest that E-cadherin is involved in the interaction of β-catenin and NF-κB. In THP-1 cells, stimulation with recombinant Wnt-1 proteins or overexpression of Wnt-1 induced increased RelA protein levels and production of proinflammatory cytokines, including IL-6 and TNF-α, through induction of scavenger receptor A (SRA) expression, which activated NF-κB in response to LPS, although the involvement of β-catenin was not confirmed (56). In another study using colorectal cancer cells, TNFRSF19 was identified as target gene downstream of the Wnt/β-catenin pathway and TNFRSF19 ligands activated NF-κB signaling, thus revealing an indirect way by which β-catenin positively regulates NF-κB activity (57). In human bronchial epithelial cells, depletion of β-catenin by siRNA reduced LPS-induced NF-κB activation and proinflammatory cytokine expression (58). Overexpression of TCF4 in human chondrocytes augmented NF-κB activity and expression of downstream target genes for several MMPs and proinflammatory cytokines, while LEF1 did not exhibit the same effect (59), suggesting that the synergistic effect of β-catenin and NF-κB transcriptional activity might depend on the TCF/LEF family member depending on the context of genes or cell type. In summary, these studies suggest a potential proinflammatory function of Wnt/β-catenin, which, however, still needs further clarification.
Importantly, the same treatment condition (e.g., with receptor agonists/antagonists or enzyme inhibitors) can influence NF-κB activity differently, depending on whether it is basal or stimulus-induced. For example, it has been shown that hypoxia or prolylhydroxylase inhibition upregulates basal NF-κB activity, while it downregulates IL-1β-induced NF-κB activity in HeLa cells (60, 61). Therefore, although not reported yet, it is possible that under certain conditions, opposing effects of Wnt/β-catenin on NF-κB activity might also be observed between a basal and an induced NF-κB status, or even among different NF-κB-inducing stimuli, as different NF-κB pathway components might be involved under these conditions.
Control of NF-κB Signaling Activity by GSK-3
While GSK-3 inhibition induced a β-catenin-dependent, negative effect on NF-κB activation and expression of subsets of target genes, β-catenin-independent regulation of NF-κB activity by GSK-3 has also been reported (62, 63). These two branches might be induced simultaneously to regulate the same targets and might as well as control different target genes and biological functions. Overexpression of GSK-3β resulted in repression of TNF-α-induced NF-κB activation, while overexpression of β-catenin had a minimal effect on NF-κB activity in endometrial carcinoma cells (62). GSK-3β overexpression stabilized IκB, thereby inhibiting NF-κB signaling. GSK-3β directly phosphorylated RelA and negatively controlled the basal activity of NF-κB (63). In contrast to the negative role of GSK-3 in NF-κB activation, GSK-3β also positively regulated NF-κB activity without changing IκB degradation or NF-κB nuclear translocation in mouse embryonic fibroblasts (MEFs) (64).
In monocytes treated with TLR agonists, GSK-3 inhibition enhanced CREB-mediated IL10 expression but repressed the production of NF-κB-mediated proinflammatory cytokines through differentially affecting the interaction of nuclear RelA and CREB with coactivator CBP (65). GSK-3 inhibition increased binding of CBP to CREB but suppressed the binding of CBP to nuclear RelA. In glioma cells, GSK-3 inhibition caused a drastic decrease in NF-κB activity (66). A study in mice also showed that GSK-3 inhibition significantly reduced TLR-mediated chronic intestinal inflammation (67). However, the involvement of β-catenin in the regulatory function of GSK-3 inhibition was not explored in these studies.
It would be interesting to further investigate if different GSK-3 pools are used in Wnt and NF-κB pathways, respectively, upon induction of these pathways and whether also extracellular Wnt-induced signaling regulates NF-κB activity through GSK-3.
Anti- and Proinflammatory Roles of Wnt/β-Catenin Signaling
Given both repressing and stimulating effects of Wnt/β-catenin signaling on NF-κB activity, it is not surprising that Wnt/β-catenin possesses both anti- and proinflammatory functions at the organismal level. The exact outcome might thus be context-dependent and/or a question of the balance between two contrary functions. So far, most evidence supports that Wnt/β-catenin signaling downregulates production of proinflammatory cytokines, including IL-1β, IL-6, IL-8, and TNF-α, in different cell types and stimulated with various stimuli, such as LPS, cytokines, viruses, and bacteria, regardless of the responsible mechanisms for Wnt/β-catenin-mediated repression of NF-κB activity (31–35, 37, 38).
An anti-inflammatory role of Wnt/β-catenin pathway in vivo has been demonstrated in mice (40, 41). In a mouse model of warm liver ischemia and reperfusion injury (IRI), disruption of β-catenin signaling increased hepatocellular damage and enhanced hepatic DC maturation/function and PTEN/TLR4 local inflammation (40). The protective effect of β-catenin was, at least partly, due to induction of PI3K/Akt signaling and a reduced TLR4-driven inflammatory response in DCs. In agreement with this study, another study also points out an anti-inflammatory role of β-catenin by demonstrating that depletion of β-catenin in DCs leads to enhanced inflammatory responses and disease onset in a mouse model of inflammatory bowel disease (41). In addition, GSK-3 inhibition potently suppressed inflammatory responses in mice challenged with LPS (65). However, a cross-regulation between NF-κB and β-catenin in these specific studies has yet to be proven.
Conversely, a proinflammatory role of Wnt/β-catenin pathway in vivo has been observed in hepatocellular carcinoma in Apc KO mice (51). Oncogenic β-catenin signaling induced an inflammatory program in hepatocytes that involved a direct transcriptional control by β-catenin and activation of the NF-κB pathway. A subset of chemokines was induced by β-catenin activation while none of the canonical proinflammatory cytokines was influenced, implying that oncogenic β-catenin signaling may promote tumor-associated inflammation indirectly through shaping the cellular compositions in the microenvironment. Another study also reported a positive effect of β-catenin in LPS-induced proinflammatory cytokine production in human bronchial epithelial cells (58).
In conclusion, both anti-inflammatory and proinflammatory functions of Wnt/β-catenin pathway were observed, depending on the conditions and regulated through different mechanisms. Wnt/β-catenin also differentially affects NF-κB-mediated subsets of target genes and biological functions (e.g., inflammation, cell proliferation, and apoptosis) in response to different stimuli. It will be crucial to disentangle the precise role of Wnt/β-catenin signaling in inflammation in a cell/tissue- and physiology/pathology-specific context.
Regulation of Wnt/β-Catenin Signaling by the NF-κB Pathway
Crosstalk between Wnt/β-catenin signaling and the NF-κB pathway not only consists of Wnt/β-catenin regulating NF-κB activity but also the reverse effect of NF-κB on Wnt/β-catenin signaling.
Negative Regulation of Wnt/β-Catenin Signaling by NF-κB Pathway
Several studies have reported a negative regulation of Wnt/β-catenin by NF-κB (Table 2). NF-κB has been shown to indirectly regulate the Wnt/β-catenin pathway through regulation of target genes that affect β-catenin activity or stability. Leucine zipper tumor suppressor 2 (LZTS2), a putative tumor suppressor, has been shown to interact with β-catenin and inhibit its nuclear localization and transcriptional activity (68). Intriguingly, NF-κB activation was found to inhibit β-catenin/TCF activity through upregulation of LZTS2 in colon, liver, and breast cancer cells (Figure 2) but downregulate LZTS2 in glioma cells to promote β-catenin/TCF activity, strongly indicating a cell type-specific effect (69). Furthermore, NF-κB inhibited osteogenic differentiation of mesenchymal stem cells (MSCs) by promoting β-catenin degradation through induction of the E3 ubiquitin protein ligases SMAD ubiquitination regulatory factor 1 (Smurf1) and Smurf2 (70). In colon cancer cells, an extract of Polysiphonia japonica downregulated transcriptional activity of Wnt/β-catenin signaling and downstream target gene expression through activation of NF-κB without altering β-catenin levels (71). However, the underlying molecular mechanisms of this finding remain to be elucidated. It is possible that NF-κB activation inhibits the nuclear translocation of β-catenin, as previously described (68), or represses the activity or expression levels of Wnt pathway transcriptional (co-)factors other than β-catenin.
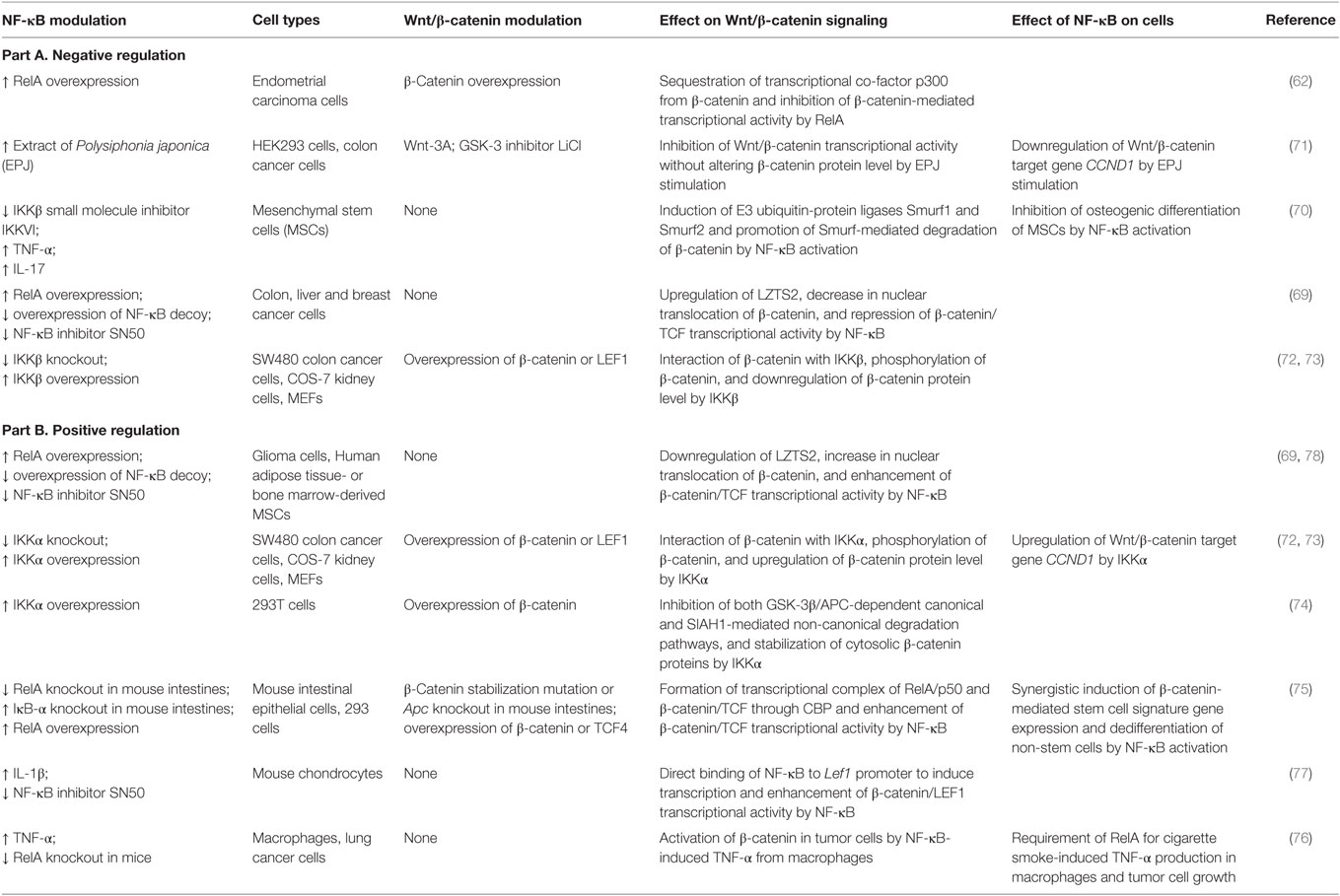
Table 2. Overview of cell type/context-dependent regulatory effects of NF-κB pathway on Wnt/β-catenin signaling.
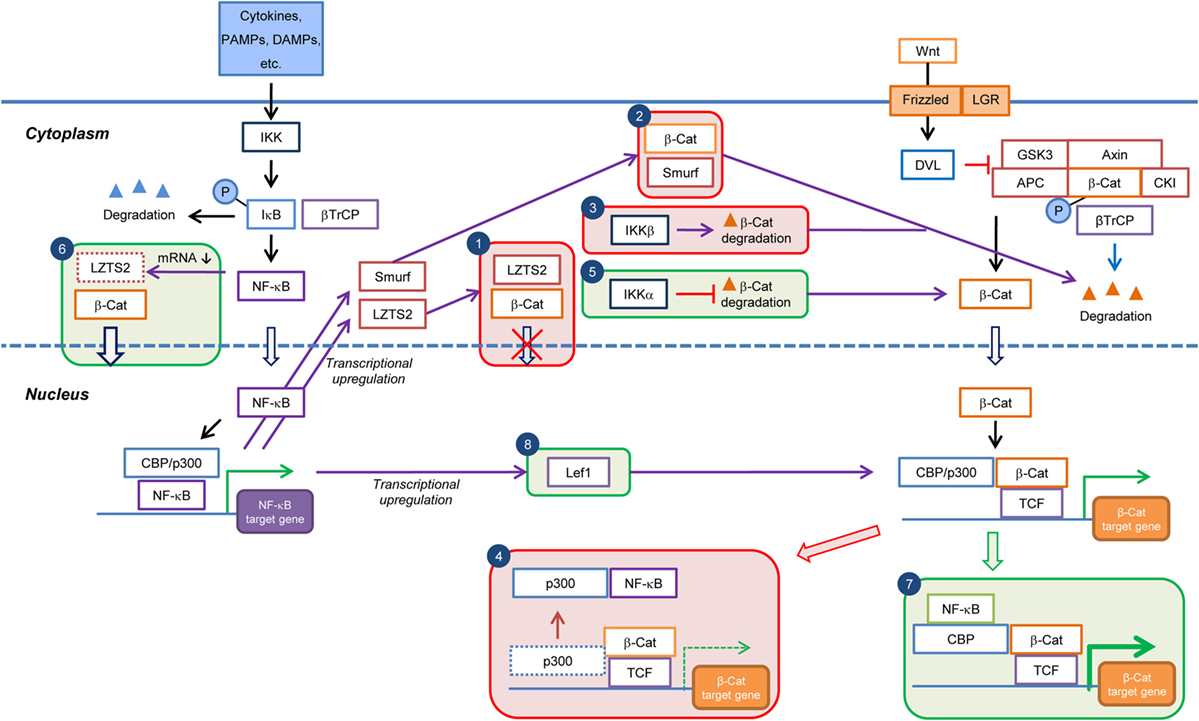
Figure 2. Current knowledge on the cell type/context-dependent regulation of Wnt/β-catenin signaling by NF-κB pathway. Boxes in red and green colors indicate negative and positive regulations, respectively. Box 1: inhibition of β-catenin nuclear translocation through NF-κB-induced transcriptional upregulation of LZTS2; Box 2: promotion of β-catenin degradation through NF-κB-induced transcriptional upregulation of Smurf; Box 3: promotion of β-catenin degradation by IKKβ; Box 4: reduction in Wnt/β-catenin target gene expression through sequestration of transcriptional co-factor p300 from β-catenin by NF-κB; Box 5: inhibition of β-catenin degradation by IKKα; Box 6: increase of β-catenin nuclear translocation by inhibition of LZTS2 expression by NF-κB; Box 7: enhancement of Wnt/β-catenin target gene expression through cooperation of NF-κB and β-catenin:TCF/LEF transcriptional complexes; and Box 8: enhancement of β-catenin:TCF/LEF transcriptional activity through transcriptional upregulation of Lef1 expression by NF-κB. PAMPs, pathogen-associated molecular patterns; DAMPs, danger-associated molecular pattern molecules; β-Cat, β-catenin; P, phospho.
In endometrial carcinoma cells, overexpression of RelA negatively regulated transcriptional activity of β-catenin/TCF (62). In this study, RelA was proposed to inhibit β-catenin-mediated transcription via sequestration of the transcriptional coactivator p300 (Figure 2).
In summary, NF-κB signaling negatively regulates Wnt/β-catenin pathway either indirectly through the functions of NF-κB target genes (e.g., LZTS2 and SMURF) or directly by interfering with the formation of transcriptional complex β-catenin/TCF/p300. The direct mechanism may represent a more rapid and instant way to modulate Wnt/β-catenin signaling activity.
Positive Regulation of Wnt/β-Catenin Signaling by NF-κB Pathway
Several components of the NF-κB signaling pathway, including IKK (72–74) and RelA (75, 76), also seemed to be involved in the positive regulation of Wnt/β-catenin signaling (Table 2). IKKα and IKKβ, the critical activators of the NF-κB pathway, differently regulated β-catenin-dependent transcriptional activity (72). IKKα increased β-catenin turnover, whereas IKKβ had a negative effect on β-catenin levels, although both IKKs interacted with and phosphorylated β-catenin. However, this study did not reveal any conclusive molecular mechanisms for the opposing effects of IKKα and IKKβ on β-catenin protein levels. In agreement with this study, IKKα, but not IKKβ, was found to upregulate β-catenin/TCF transcriptional activity and expression of the downstream target gene CCND1 (encoding cyclin D1) (73). Furthermore, IKKα stabilized cytosolic β-catenin through inhibition of both the GSK-3β/APC-dependent canonical degradation pathway and seven in absentia homolog 1 (SIAH1)-mediated non-canonical degradation pathway (74) (Figure 2).
A novel role for NF-κB as transcriptional co-factors for β-catenin/TCF has been discovered in the study of dedifferentiation-induced intestinal tumorigenesis in mouse models (75). NF-κB activation potentiated Wnt/β-catenin signaling activity and induced dedifferentiation of non-stem cells that acquired tumor-initiating capacity. The RelA/p50 dimer was bound through CBP to the β-catenin/TCF transcription complex at target gene promoters and augmented the expression of a subset of stem cell signature genes such as Lgr5, Ascl2, and Sox9 (Figure 2).
NF-κB also positively regulated Wnt/β-catenin signaling by affecting expression of Wnt/β-catenin pathway genes (Table 2; Figure 2). In mouse chondrocytes, IL-1β-mediated NF-κB activation induced the expression of the Wnt/β-catenin pathway transcription factor Lef1, presumably the transcriptional activity of β-catenin/LEF, providing an indirect mechanism for the control of Wnt/β-catenin signaling pathway activity by NF-κB (77). In human adipose tissue-derived MSCs and bone marrow stromal cells, activated NF-κB repressed the expression of LZTS2, which inhibited nuclear localization and transcriptional activity of β-catenin (78). This is consistent with the observation in glioma but not breast cancer cells or colon and liver tissue, as discussed above (69). Interestingly, the regulation of Wnt/β-catenin signaling by NF-κB can also be observed in between cells. Deletion of RelA largely inhibited cigarette smoke-induced TNF-α production in macrophages as well as TNF-α-mediated β-catenin activation and lung cancer cell growth in mice (76). TNF-α released from macrophages was proposed to activate β-catenin through GSK-3β and Akt signaling in tumor cells, thereby promoting proliferation of the latter.
Evidently, the effects of NF-κB signaling on Wnt/β-catenin activity and the underlying molecular mechanisms are cell type dependent. However, it cannot be excluded that both positive and negative regulations can co-exist in the same cell type, employing different molecular mechanisms, and that the overall observed effect is the sum of two opposing mechanisms.
Wnt/β-Catenin and NF-κB Crosstalk Links Inflammation with Tumorigenesis
Tumor-promoting inflammation has been recognized as one of the hallmarks of cancer (79). NF-κB serves as a critical link between inflammation and cancer through its ability to upregulate the expression of tumor-promoting cytokines, such as IL6 and TNFA, and survival genes, such as BCL2L1 (BCLXL). Besides the direct control of cell proliferation/survival by NF-κB, the crosstalk between NF-κB and Wnt/β-catenin provides another important intracellular basis for inflammation-induced tumorigenesis. Since Wnt/β-catenin signaling has been shown to be oncogenic in a wide range of tumor types, the positive regulation of Wnt/β-catenin by the NF-κB pathway may contribute to cancer development. A convincing example is the synergistic cooperation between NF-κB and β-catenin/TCF4 on the expression of a subset of Wnt target genes in colon cancer (75). In this model, activated NF-κB functions as a transcriptional coactivator in conjunction with β-catenin/TCF4 to induce a set of stem cell signature genes, such as Lgr5, Ascl2, and Sox9, and subsequent tumor cell growth.
The Wnt/β-catenin and NF-κB cross-regulation linking inflammation and tumorigenesis not only occur within cells but also in between cells. In a gastric tumor model, Helicobacter infection-activated macrophages elicited NF-κB-mediated production of TNF-α, which then inactivated GSK-3β and enhanced the oncogenic Wnt/β-catenin signaling in the gastric cancer cells (80). This model provides a missing link of how Helicobacter-induced chronic inflammation prompts oncogenic signaling.
In addition to creating a tumor-favorable microenvironment consisting of various tumor-promoting inflammatory cells (7), NF-κB-mediated inflammation has now been demonstrated to enhance the tumorigenic potential of cancerous cells through upregulation of Wnt/β-catenin signaling, further strengthening the idea that NF-κB may be an attractive therapeutic target for inflammation-associated cancer.
Mutual Regulation of Wnt/β-Catenin and NF-κB Signaling
In most biological processes, crosstalk of Wnt/β-catenin and NF-κB signaling is bidirectional, meaning that both pathways reciprocally regulate each other. Such mutual regulations are critical for either enforcement or limitation of downstream responses. In a hair follicle development model, Wnt/β-catenin and NF-κB signaling was interdependent for their regulatory activities in hair follicle formation (81). Binding of the A1 isoform of the TNF-α family member ectodysplasin (EDA) and its receptor EDAR induced NF-κB nuclear translocation and activation in developing hair follicle placodes. Edar was a direct target of Wnt/β-catenin, and Eda/Edar/NF-κB signaling was required to refine the pattern of Wnt/β-catenin activity and maintain this activity by promoting Wnt10b production at later stages of hair follicle placode development. However, Wnt/β-catenin signaling was initially activated independently of EDA/EDAR/NF-κB activity in primary hair follicle primordia. Maintenance of localized expression of Wnt10b and Wnt10a required NF-κB signaling and Wnt10b was a direct transcriptional target gene of NF-κB, providing a molecular explanation for this observation. Moreover, the Wnt/β-catenin signaling antagonist DKK4 was a target gene of the EDA/EDAR/NF-κB pathway, serving as a negative feedback to limit β-catenin signaling (82). These data reveal a complex interplay and interdependence of Wnt/β-catenin and NF-κB pathways in the initiation and maintenance of primary hair follicle placodes.
Mutations or expression levels of APC and β-catenin levels were found to control the outcome of TLR4 activation by LPS on proliferation of colon cancer cells (83). LPS enhanced cell proliferation in APC mutated or depleted cells but inhibited cell proliferation in APC wild-type cells. It was proposed that high β-catenin levels blocked NF-κB-mediated cell apoptosis and LPS-induced NF-κB promoted β-catenin expression and β-catenin-regulated proliferation in APC mutated or depleted cells where β-catenin degradation was compromised. These data revealed that the reciprocal modulation between β-catenin and NF-κB was involved in the regulation of tumor growth by APC status and TLR4 activation.
Together, the mutual regulation of Wnt/β-catenin and NF-κB signaling is observed in a large number of cell and tissue types and is important for the maintenance of cellular/tissue homeostasis or the reinforcement of cell fate.
Concluding Remarks
Given the crucial biological functions of the Wnt/β-catenin and NF-κB signaling pathways, it is of particular biomedical interest and importance to understand their crosstalk. A lot of efforts in recent years have revealed a complex network of Wnt/β-catenin and NF-κB signaling interaction. These two pathways are interconnected through physical interactions of mediators and regulation of target genes in the convergence. The regulatory effects are often cell/tissue-dependent or even gene-specific, suggesting that cross-regulation needs to be investigated in a context-oriented fashion. Additional efforts using appropriate cell or animal models are required to understand how the Wnt/β-catenin and NF-κB pathways crosstalk in inflammation and inflammation-associated diseases such as cancer. The further knowledge will eventually help design better interventions for the treatment of inflammatory diseases. When interfering with one of the two pathways, the other pathway might also be changed due to their cross-regulation, thereby potentially causing additional, secondary effects not investigated. It is not just a matter of which molecule to target but also how to interfere with it (e.g., blocking activity without altering the protein level or reducing expression affecting both protein level and activity). For example, when treating cancer with Wnt/β-catenin pathway blockers, one could think about whether this might cause undesired proinflammatory effects due to upregulation of NF-κB activity by elevated β-catenin levels, as hinted from the abovementioned evidence. One potential approach to circumvent the negative feedback is to target the transcriptional activity of β-catenin:TCF/LEF or other coactivators, rather that affecting β-catenin protein turnover. Targeting both pathways simultaneously might also be promising in this case, as well as for other inflammation-associated diseases.
Taken together, the existence of a crosstalk between the Wnt/β-catenin and NF-κB signaling pathways provides new opportunities to treat inflammatory and inflammation-associated diseases such as cancer. Although due to the complex interactions, manipulation of this crosstalk might be challenging to achieve a desired and tissue-specific outcome, our increasing knowledge of mechanisms underlying Wnt/β-catenin and NF-κB signaling cross-regulation will hopefully help resolve these obstacles, so that more effective treatments can be designed.
Author Contributions
BM and MOH conceived and wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Stephan Christen provided editorial assistance and critical input during the writing (University of Zurich). NF-κB research in the laboratory of MOH is funded by the Kanton of Zurich and the Swiss National Science Foundation (grant 310030_157019).
Abbreviations
βTrCP, β-transducing repeat-containing protein; APC, adenoma polyposis coli; ARTD1, ADP-ribosyltransferase diphtheria toxin-like 1; BCR, B-cell receptor; CBP, cAMP response element-binding protein-binding protein; CKI, casein kinase I; ConA, concanavalin A; COX-2, cyclooxygenase-2; CRD-BP, coding region determinant-binding protein; CREB, cAMP response element-binding protein; CRP, C-reactive protein; CtBP1, C-terminal binding protein-1; DCs, dendritic cells; DKK1, Dickkopf1; DVL, disheveled; EDA, ectodysplasin; EDAR, ectodysplasin receptor; ER, endoplasmic reticulum; FOXO, forkhead box O; FZD, frizzled; GalN, d-galactosamine; GSK-3, glycogen synthase kinase 3; HDAC, histone deacetylase; HIF-1α, hypoxia-inducible factor-1α; IκB, inhibitor of kappa B; IKK, inhibitor of kappa B kinase; IL-1β, interleukin-1β; IL-1R, interleukin-1 receptor; iNOS, inducible nitric oxide synthase; KO, knockout; LEF, lymphoid enhancer factor; LPS, lipopolysaccharide; LRP, low-density lipoprotein receptor-related protein; LZTS2, leucine zipper tumor suppressor 2; MEFs, mouse embryonic fibroblasts; MMP, matrix metalloproteinase; MSCs, mesenchymal stem cells; NEMO, NF-κB essential modulator; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; PI3K, phosphatidylinositide 3-kinase; PTEN, phosphatase and tensin homolog deleted on chromosome 10; RA, retinoic acid; RHD, Rel-homology domain; Ror, receptor tyrosine kinase-like orphan receptor; Ryk, receptor tyrosine kinase; SFRPs, secreted frizzled-related proteins; SIAH1, seven in absentia homolog 1; Smad3, mothers against decapentaplegic homolog 3; Smurf, SMAD ubiquitination regulatory factor; SRA, scavenger receptor A; TCF, T cell factor; TCR, T cell receptor; TLR, Toll-like receptor; TNF-α, tumor necrosis factor-α; TNFRSF, tumor necrosis factor receptor superfamily; WIF1, Wnt inhibitory factor 1.
References
1. Clevers H. Wnt/beta-catenin signaling in development and disease. Cell (2006) 127(3):469–80. doi:10.1016/j.cell.2006.10.018
2. Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell (2012) 149(6):1192–205. doi:10.1016/j.cell.2012.05.012
3. Tak PP, Firestein GS. NF-kappaB: a key role in inflammatory diseases. J Clin Invest (2001) 107(1):7–11. doi:10.1172/JCI11830
4. Li Q, Verma IM. NF-kappaB regulation in the immune system. Nat Rev Immunol (2002) 2(10):725–34. doi:10.1038/nri910
5. Nejak-Bowen K, Kikuchi A, Monga SP. Beta-catenin-NF-kappaB interactions in murine hepatocytes: a complex to die for. Hepatology (2013) 57(2):763–74. doi:10.1002/hep.26042
6. Karin M. NF-kappaB as a critical link between inflammation and cancer. Cold Spring Harb Perspect Biol (2009) 1(5):a000141. doi:10.1101/cshperspect.a000141
7. Ben-Neriah Y, Karin M. Inflammation meets cancer, with NF-kappaB as the matchmaker. Nat Immunol (2011) 12(8):715–23. doi:10.1038/ni.2060
8. Pasparakis M. Regulation of tissue homeostasis by NF-kappaB signalling: implications for inflammatory diseases. Nat Rev Immunol (2009) 9(11):778–88. doi:10.1038/nri2655
9. Anastas JN, Moon RT. WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer (2013) 13(1):11–26. doi:10.1038/nrc3419
10. van Amerongen R, Nusse R. Towards an integrated view of Wnt signaling in development. Development (2009) 136(19):3205–14. doi:10.1242/dev.033910
11. Brembeck FH, Rosario M, Birchmeier W. Balancing cell adhesion and Wnt signaling, the key role of beta-catenin. Curr Opin Genet Dev (2006) 16(1):51–9. doi:10.1016/j.gde.2005.12.007
12. Arce L, Yokoyama NN, Waterman ML. Diversity of LEF/TCF action in development and disease. Oncogene (2006) 25(57):7492–504. doi:10.1038/sj.onc.1210056
13. Hoverter NP, Waterman ML. A Wnt-fall for gene regulation: repression. Sci Signal (2008) 1(39):e43. doi:10.1126/scisignal.139pe43
14. Hecht A, Vleminckx K, Stemmler MP, van Roy F, Kemler R. The p300/CBP acetyltransferases function as transcriptional coactivators of beta-catenin in vertebrates. EMBO J (2000) 19(8):1839–50. doi:10.1093/emboj/19.8.1839
15. Idogawa M, Yamada T, Honda K, Sato S, Imai K, Hirohashi S. Poly(ADP-ribose) polymerase-1 is a component of the oncogenic T-cell factor-4/beta-catenin complex. Gastroenterology (2005) 128(7):1919–36. doi:10.1053/j.gastro.2005.03.007
16. Chiurillo MA. Role of the Wnt/beta-catenin pathway in gastric cancer: an in-depth literature review. World J Exp Med (2015) 5(2):84–102. doi:10.5493/wjem.v5.i2.84
17. Stewart DJ. Wnt signaling pathway in non-small cell lung cancer. J Natl Cancer Inst (2014) 106(1):djt356. doi:10.1093/jnci/djt356
18. Deng J, Miller SA, Wang HY, Xia W, Wen Y, Zhou BP, et al. Beta-catenin interacts with and inhibits NF-kappa B in human colon and breast cancer. Cancer Cell (2002) 2(4):323–34. doi:10.1016/S1535-6108(02)00154-X
19. Jian H, Shen X, Liu I, Semenov M, He X, Wang XF. Smad3-dependent nuclear translocation of beta-catenin is required for TGF-beta1-induced proliferation of bone marrow-derived adult human mesenchymal stem cells. Genes Dev (2006) 20(6):666–74. doi:10.1101/gad.1388806
20. Rodilla V, Villanueva A, Obrador-Hevia A, Robert-Moreno A, Fernandez-Majada V, Grilli A, et al. Jagged1 is the pathological link between Wnt and Notch pathways in colorectal cancer. Proc Natl Acad Sci U S A (2009) 106(15):6315–20. doi:10.1073/pnas.0813221106
21. Essers MA, de Vries-Smits LM, Barker N, Polderman PE, Burgering BM, Korswagen HC. Functional interaction between beta-catenin and FOXO in oxidative stress signaling. Science (2005) 308(5725):1181–4. doi:10.1126/science.1109083
22. Mazumdar J, O’Brien WT, Johnson RS, LaManna JC, Chavez JC, Klein PS, et al. O2 regulates stem cells through Wnt/beta-catenin signalling. Nat Cell Biol (2010) 12(10):1007–13. doi:10.1038/ncb2102
23. Mitchell S, Vargas J, Hoffmann A. Signaling via the NFkappaB system. Wiley Interdiscip Rev Syst Biol Med (2016) 8(3):227–41. doi:10.1002/wsbm.1331
24. Kingeter LM, Paul S, Maynard SK, Cartwright NG, Schaefer BC. Cutting edge: TCR ligation triggers digital activation of NF-kappaB. J Immunol (2010) 185(8):4520–4. doi:10.4049/jimmunol.1001051
25. Gerritsen ME, Williams AJ, Neish AS, Moore S, Shi Y, Collins T. CREB-binding protein/p300 are transcriptional coactivators of p65. Proc Natl Acad Sci U S A (1997) 94(7):2927–32. doi:10.1073/pnas.94.7.2927
26. Hassa PO, Buerki C, Lombardi C, Imhof R, Hottiger MO. Transcriptional coactivation of nuclear factor-kappaB-dependent gene expression by p300 is regulated by poly(ADP)-ribose polymerase-1. J Biol Chem (2003) 278(46):45145–53. doi:10.1074/jbc.M307957200
27. Du Q, Zhang X, Cardinal J, Cao Z, Guo Z, Shao L, et al. Wnt/beta-catenin signaling regulates cytokine-induced human inducible nitric oxide synthase expression by inhibiting nuclear factor-kappaB activation in cancer cells. Cancer Res (2009) 69(9):3764–71. doi:10.1158/0008-5472.CAN-09-0014
28. Moreau M, Mourah S, Dosquet C. Beta-catenin and NF-kappaB cooperate to regulate the uPA/uPAR system in cancer cells. Int J Cancer (2011) 128(6):1280–92. doi:10.1002/ijc.25455
29. Rodriguez-Pinilla M, Rodriguez-Peralto JL, Hitt R, Sanchez JJ, Sanchez-Verde L, Alameda F, et al. Beta-catenin, Nf-kappaB and FAS protein expression are independent events in head and neck cancer: study of their association with clinical parameters. Cancer Lett (2005) 230(1):141–8. doi:10.1016/j.canlet.2004.12.045
30. Liu J, Liao Y, Ma K, Wang Y, Zhang G, Yang R, et al. PI3K is required for the physical interaction and functional inhibition of NF-kappaB by beta-catenin in colorectal cancer cells. Biochem Biophys Res Commun (2013) 434(4):760–6. doi:10.1016/j.bbrc.2013.03.135
31. Ma B, van Blitterswijk CA, Karperien M. A Wnt/beta-catenin negative feedback loop inhibits interleukin-1-induced matrix metalloproteinase expression in human articular chondrocytes. Arthritis Rheum (2012) 64(8):2589–600. doi:10.1002/art.34425
32. Ma B, Fey M, Hottiger MO. WNT/beta-catenin signaling inhibits CBP-mediated RelA acetylation and expression of proinflammatory NF-kappaB target genes. J Cell Sci (2015) 128(14):2430–6. doi:10.1242/jcs.168542
33. Sun J, Hobert ME, Duan Y, Rao AS, He TC, Chang EB, et al. Crosstalk between NF-kappaB and beta-catenin pathways in bacterial-colonized intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol (2005) 289(1):G129–37. doi:10.1152/ajpgi.00515.2004
34. Duan Y, Liao AP, Kuppireddi S, Ye Z, Ciancio MJ, Sun J. Beta-catenin activity negatively regulates bacteria-induced inflammation. Lab Invest (2007) 87(6):613–24. doi:10.1038/labinvest.3700545
35. Die L, Yan P, Jun Jiang Z, Min Hua T, Cai W, Xing L. Glycogen synthase kinase-3 beta inhibitor suppresses Porphyromonas gingivalis lipopolysaccharide-induced CD40 expression by inhibiting nuclear factor-kappa B activation in mouse osteoblasts. Mol Immunol (2012) 52(1):38–49. doi:10.1016/j.molimm.2012.04.005
36. Jiang R, Chen D, Hou J, Tan Z, Wang Y, Huang X, et al. Survival and inflammation promotion effect of PTPRO in fulminant hepatitis is associated with NF-kappaB activation. J Immunol (2014) 193(10):5161–70. doi:10.4049/jimmunol.1303354
37. Kim SJ, Lim JY, Lee JN, Choe SK, Kim YI, Song SR, et al. Activation of beta-catenin by inhibitors of glycogen synthase kinase-3 ameliorates cisplatin-induced cytotoxicity and pro-inflammatory cytokine expression in HEI-OC1 cells. Toxicology (2014) 320:74–82. doi:10.1016/j.tox.2014.01.013
38. Hao HP, Wen LB, Li JR, Wang Y, Ni B, Wang R, et al. LiCl inhibits PRRSV infection by enhancing Wnt/beta-catenin pathway and suppressing inflammatory responses. Antiviral Res (2015) 117:99–109. doi:10.1016/j.antiviral.2015.02.010
39. Kim JH, Kim B, Cai L, Choi HJ, Ohgi KA, Tran C, et al. Transcriptional regulation of a metastasis suppressor gene by Tip60 and beta-catenin complexes. Nature (2005) 434(7035):921–6. doi:10.1038/nature03452
40. Ke B, Shen XD, Kamo N, Ji H, Yue S, Gao F, et al. Beta-catenin regulates innate and adaptive immunity in mouse liver ischemia-reperfusion injury. Hepatology (2013) 57(3):1203–14. doi:10.1002/hep.26100
41. Manicassamy S, Reizis B, Ravindran R, Nakaya H, Salazar-Gonzalez RM, Wang YC, et al. Activation of beta-catenin in dendritic cells regulates immunity versus tolerance in the intestine. Science (2010) 329(5993):849–53. doi:10.1126/science.1188510
42. Winston JT, Strack P, Beer-Romero P, Chu CY, Elledge SJ, Harper JW. The SCFbeta-TRCP-ubiquitin ligase complex associates specifically with phosphorylated destruction motifs in IkappaBalpha and beta-catenin and stimulates IkappaBalpha ubiquitination in vitro. Genes Dev (1999) 13(3):270–83. doi:10.1101/gad.13.3.270
43. Spiegelman VS, Slaga TJ, Pagano M, Minamoto T, Ronai Z, Fuchs SY. Wnt/beta-catenin signaling induces the expression and activity of betaTrCP ubiquitin ligase receptor. Mol Cell (2000) 5(5):877–82. doi:10.1016/S1097-2765(00)80327-5
44. Wang X, Adhikari N, Li Q, Guan Z, Hall JL. The role of [beta]-transducin repeat-containing protein ([beta]-TrCP) in the regulation of NF-[kappa]B in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol (2004) 24(1):85–90. doi:10.1161/01.ATV.0000104012.40720.c4
45. Noubissi FK, Elcheva I, Bhatia N, Shakoori A, Ougolkov A, Liu J, et al. CRD-BP mediates stabilization of betaTrCP1 and c-myc mRNA in response to beta-catenin signalling. Nature (2006) 441(7095):898–901. doi:10.1038/nature04839
46. Ougolkov A, Zhang B, Yamashita K, Bilim V, Mai M, Fuchs SY, et al. Associations among beta-TrCP, an E3 ubiquitin ligase receptor, beta-catenin, and NF-kappaB in colorectal cancer. J Natl Cancer Inst (2004) 96(15):1161–70. doi:10.1093/jnci/djh219
47. Jia D, Yang W, Li L, Liu H, Tan Y, Ooi S, et al. Beta-catenin and NF-kappaB co-activation triggered by TLR3 stimulation facilitates stem cell-like phenotypes in breast cancer. Cell Death Differ (2015) 22(2):298–310. doi:10.1038/cdd.2014.145
48. Bognar MK, Vincendeau M, Erdmann T, Seeholzer T, Grau M, Linnemann JR, et al. Oncogenic CARMA1 couples NF-kappaB and beta-catenin signaling in diffuse large B-cell lymphomas. Oncogene (2016) 35(32):4269–81. doi:10.1038/onc.2015.493
49. Choi YS, Hur J, Jeong S. Beta-catenin binds to the downstream region and regulates the expression C-reactive protein gene. Nucleic Acids Res (2007) 35(16):5511–9. doi:10.1093/nar/gkm547
50. Yun K, So JS, Jash A, Im SH. Lymphoid enhancer binding factor 1 regulates transcription through gene looping. J Immunol (2009) 183(8):5129–37. doi:10.4049/jimmunol.0802744
51. Anson M, Crain-Denoyelle AM, Baud V, Chereau F, Gougelet A, Terris B, et al. Oncogenic beta-catenin triggers an inflammatory response that determines the aggressiveness of hepatocellular carcinoma in mice. J Clin Invest (2012) 122(2):586–99. doi:10.1172/JCI43937
52. Phelps RA, Broadbent TJ, Stafforini DM, Jones DA. New perspectives on APC control of cell fate and proliferation in colorectal cancer. Cell Cycle (2009) 8(16):2549–56. doi:10.4161/cc.8.16.9278
53. Kuphal S, Poser I, Jobin C, Hellerbrand C, Bosserhoff AK. Loss of E-cadherin leads to upregulation of NFkappaB activity in malignant melanoma. Oncogene (2004) 23(52):8509–19. doi:10.1038/sj.onc.1207831
54. Bandino A, Compagnone A, Bravoco V, Cravanzola C, Lomartire A, Rossetto C, et al. Beta-catenin triggers nuclear factor kappaB-dependent up-regulation of hepatocyte inducible nitric oxide synthase. Int J Biochem Cell Biol (2008) 40(9):1861–71. doi:10.1016/j.biocel.2008.01.029
55. Solanas G, Porta-de-la-Riva M, Agusti C, Casagolda D, Sanchez-Aguilera F, Larriba MJ, et al. E-cadherin controls beta-catenin and NF-kappaB transcriptional activity in mesenchymal gene expression. J Cell Sci (2008) 121(Pt 13):2224–34. doi:10.1242/jcs.021667
56. Zhao W, Sun Z, Wang S, Li Z, Zheng L. Wnt1 participates in inflammation induced by lipopolysaccharide through upregulating scavenger receptor A and NF-kB. Inflammation (2015) 38(4):1700–6. doi:10.1007/s10753-015-0147-8
57. Schon S, Flierman I, Ofner A, Stahringer A, Holdt LM, Kolligs FT, et al. Beta-catenin regulates NF-kappaB activity via TNFRSF19 in colorectal cancer cells. Int J Cancer (2014) 135(8):1800–11. doi:10.1002/ijc.28839
58. Jang J, Ha JH, Chung SI, Yoon Y. Beta-catenin regulates NF-kappaB activity and inflammatory cytokine expression in bronchial epithelial cells treated with lipopolysaccharide. Int J Mol Med (2014) 34(2):632–8. doi:10.3892/ijmm.2014.1807
59. Ma B, Zhong L, van Blitterswijk CA, Post JN, Karperien M. T cell factor 4 is a pro-catabolic and apoptotic factor in human articular chondrocytes by potentiating nuclear factor kappaB signaling. J Biol Chem (2013) 288(24):17552–8. doi:10.1074/jbc.M113.453985
60. Cummins EP, Berra E, Comerford KM, Ginouves A, Fitzgerald KT, Seeballuck F, et al. Prolyl hydroxylase-1 negatively regulates IkappaB kinase-beta, giving insight into hypoxia-induced NFkappaB activity. Proc Natl Acad Sci U S A (2006) 103(48):18154–9. doi:10.1073/pnas.0602235103
61. Scholz CC, Cavadas MA, Tambuwala MM, Hams E, Rodriguez J, von Kriegsheim A, et al. Regulation of IL-1beta-induced NF-kappaB by hydroxylases links key hypoxic and inflammatory signaling pathways. Proc Natl Acad Sci U S A (2013) 110(46):18490–5. doi:10.1073/pnas.1309718110
62. Saegusa M, Hashimura M, Kuwata T, Hamano M, Okayasu I. Crosstalk between NF-kappaB/p65 and beta-catenin/TCF4/p300 signalling pathways through alterations in GSK-3beta expression during trans-differentiation of endometrial carcinoma cells. J Pathol (2007) 213(1):35–45. doi:10.1002/path.2198
63. Buss H, Dorrie A, Schmitz ML, Frank R, Livingstone M, Resch K, et al. Phosphorylation of serine 468 by GSK-3beta negatively regulates basal p65 NF-kappaB activity. J Biol Chem (2004) 279(48):49571–4. doi:10.1074/jbc.C400442200
64. Hoeflich KP, Luo J, Rubie EA, Tsao MS, Jin O, Woodgett JR. Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature (2000) 406(6791):86–90. doi:10.1038/35017574
65. Martin M, Rehani K, Jope RS, Michalek SM. Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat Immunol (2005) 6(8):777–84. doi:10.1038/ni1221
66. Kotliarova S, Pastorino S, Kovell LC, Kotliarov Y, Song H, Zhang W, et al. Glycogen synthase kinase-3 inhibition induces glioma cell death through c-MYC, nuclear factor-kappaB, and glucose regulation. Cancer Res (2008) 68(16):6643–51. doi:10.1158/0008-5472.CAN-08-0850
67. Hofmann C, Dunger N, Scholmerich J, Falk W, Obermeier F. Glycogen synthase kinase 3-beta: a master regulator of toll-like receptor-mediated chronic intestinal inflammation. Inflamm Bowel Dis (2010) 16(11):1850–8. doi:10.1002/ibd.21294
68. Thyssen G, Li TH, Lehmann L, Zhuo M, Sharma M, Sun Z. LZTS2 is a novel beta-catenin-interacting protein and regulates the nuclear export of beta-catenin. Mol Cell Biol (2006) 26(23):8857–67. doi:10.1128/MCB.01031-06
69. Cho HH, Song JS, Yu JM, Yu SS, Choi SJ, Kim DH, et al. Differential effect of NF-kappaB activity on beta-catenin/Tcf pathway in various cancer cells. FEBS Lett (2008) 582(5):616–22. doi:10.1016/j.febslet.2008.01.029
70. Chang J, Liu F, Lee M, Wu B, Ting K, Zara JN, et al. NF-kappaB inhibits osteogenic differentiation of mesenchymal stem cells by promoting beta-catenin degradation. Proc Natl Acad Sci U S A (2013) 110(23):9469–74. doi:10.1073/pnas.1300532110
71. Gwak J, Park S, Cho M, Song T, Cha SH, Kim DE, et al. Polysiphonia japonica extract suppresses the Wnt/beta-catenin pathway in colon cancer cells by activation of NF-kappaB. Int J Mol Med (2006) 17(6):1005–10. doi:10.3892/ijmm.17.6.1005
72. Lamberti C, Lin KM, Yamamoto Y, Verma U, Verma IM, Byers S, et al. Regulation of beta-catenin function by the IkappaB kinases. J Biol Chem (2001) 276(45):42276–86. doi:10.1074/jbc.M104227200
73. Albanese C, Wu K, D’Amico M, Jarrett C, Joyce D, Hughes J, et al. IKKalpha regulates mitogenic signaling through transcriptional induction of cyclin D1 via Tcf. Mol Biol Cell (2003) 14(2):585–99. doi:10.1091/mbc.02-06-0101
74. Carayol N, Wang CY. IKKalpha stabilizes cytosolic beta-catenin by inhibiting both canonical and non-canonical degradation pathways. Cell Signal (2006) 18(11):1941–6. doi:10.1016/j.cellsig.2006.02.014
75. Schwitalla S, Fingerle AA, Cammareri P, Nebelsiek T, Goktuna SI, Ziegler PK, et al. Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell (2013) 152(1–2):25–38. doi:10.1016/j.cell.2012.12.012
76. Li D, Beisswenger C, Herr C, Hellberg J, Han G, Zakharkina T, et al. Myeloid cell RelA/p65 promotes lung cancer proliferation through Wnt/beta-catenin signaling in murine and human tumor cells. Oncogene (2014) 33(10):1239–48. doi:10.1038/onc.2013.75
77. Yun K, Choi YD, Nam JH, Park Z, Im SH. NF-kappaB regulates Lef1 gene expression in chondrocytes. Biochem Biophys Res Commun (2007) 357(3):589–95. doi:10.1016/j.bbrc.2007.03.170
78. Hyun Hwa C, Hye Joon J, Ji Sun S, Yong Chan B, Jin Sup J. Crossregulation of beta-catenin/Tcf pathway by NF-kappaB is mediated by lzts2 in human adipose tissue-derived mesenchymal stem cells. Biochim Biophys Acta (2008) 1783(3):419–28. doi:10.1016/j.bbamcr.2007.08.005
79. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi:10.1016/j.cell.2011.02.013
80. Oguma K, Oshima H, Aoki M, Uchio R, Naka K, Nakamura S, et al. Activated macrophages promote Wnt signalling through tumour necrosis factor-alpha in gastric tumour cells. EMBO J (2008) 27(12):1671–81. doi:10.1038/emboj.2008.105
81. Zhang Y, Tomann P, Andl T, Gallant NM, Huelsken J, Jerchow B, et al. Reciprocal requirements for EDA/EDAR/NF-kappaB and Wnt/beta-catenin signaling pathways in hair follicle induction. Dev Cell (2009) 17(1):49–61. doi:10.1016/j.devcel.2009.05.011
82. Fliniaux I, Mikkola ML, Lefebvre S, Thesleff I. Identification of dkk4 as a target of Eda-A1/Edar pathway reveals an unexpected role of ectodysplasin as inhibitor of Wnt signalling in ectodermal placodes. Dev Biol (2008) 320(1):60–71. doi:10.1016/j.ydbio.2008.04.023
Keywords: Wnt, β-catenin, NF-κB, gene expression, signaling pathways
Citation: Ma B and Hottiger MO (2016) Crosstalk between Wnt/β-Catenin and NF-κB Signaling Pathway during Inflammation. Front. Immunol. 7:378. doi: 10.3389/fimmu.2016.00378
Received: 22 July 2016; Accepted: 08 September 2016;
Published: 22 September 2016
Edited by:
Victor Manuel Baizabal-Aguirre, Universidad Michoacana de San Nicolás de Hidalgo, MexicoReviewed by:
Amit K. Tyagi, The University of Texas MD Anderson Cancer Center, USAJean-Marc Cavaillon, Institut Pasteur, France
Copyright: © 2016 Ma and Hottiger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael O. Hottiger, michael.hottiger@dmmd.uzh.ch