 Chien-Yu Chen
Chien-Yu Chen Jingyu Chen
Jingyu Chen Lina He
Lina He Bangyan L. Stiles
Bangyan L. Stiles- 1Department of Pharmacology and Pharmaceutical Sciences, School of Pharmacy, University of Southern California, Los Angeles, CA, United States
- 2Department of Pathology, Keck School of Medicine, University of Southern California, Los Angeles, CA, United States
Phosphatase and Tensin Homolog deleted on Chromosome 10 (PTEN) is a dual phosphatase with both protein and lipid phosphatase activities. PTEN was first discovered as a tumor suppressor with growth and survival regulatory functions. In recent years, the function of PTEN as a metabolic regulator has attracted significant attention. As the lipid phosphatase that dephosphorylates phosphatidylinositol-3, 4, 5-phosphate (PIP3), PTEN reduces the level of PIP3, a critical 2nd messenger mediating the signal of not only growth factors but also insulin. In this review, we introduced the discovery of PTEN, the PTEN-regulated canonical and nuclear signals, and PTEN regulation. We then focused on the role of PTEN and PTEN-regulated signals in metabolic regulation. This included the role of PTEN in glycolysis, gluconeogenesis, glycogen synthesis, lipid metabolism as well as mitochondrial metabolism. We also included how PTEN and PTEN regulated metabolic functions may act paradoxically toward insulin sensitivity and tumor metabolism and growth. Further understanding of how PTEN regulates metabolism and how such regulations lead to different biological outcomes is necessary for interventions targeting at the PTEN-regulated signals in either cancer or diabetes treatment.
Key Concepts
• Phosphatase and Tensin Homolog deleted on Chromosome 10 (PTEN) is a dual phosphatase with both protein and lipid phosphatase activities.
• PTEN reduces the level of PI-3, 4, 5-P3, a critical 2nd messenger mediating the signal of not only growth factors but also that of insulin.
• In addition to the canonical PI3K/AKT signaling, PTEN also functions in the nucleus.
• PTEN regulates signals in metabolic regulation, includes the role of PTEN in glycolysis, gluconeogenesis, glycogen synthesis, lipid metabolism as well as mitochondrial metabolism.
• PTEN and PTEN regulated metabolic functions act paradoxically toward insulin sensitivity and tumor metabolism and growth.
Introduction
PTEN (phosphatase and tensin homolog deleted on chromosome 10) (also named MMAC1/TEP1) was discovered in 1997 independently by three laboratories as a tumor suppressor of which the expression is often lost in tumors (1–3). Later studies established that PTEN is a negative regulator of a major cell growth and survival signaling pathway, namely the phosphatidylinositol-3-kinase (PI3K)/AKT signaling pathway (4, 5). It is now well established that PTEN plays a role in growth and survival. Studies in recent years also established a role of PTEN in metabolic regulation (6, 7). In this review, we summarized the roles of PTEN as both a tumor suppressor and a metabolic regulator and reviewed the biological functions of PTEN and its downstream target proteins. We also summarized the regulations of PTEN transcriptionally, post-transcriptionally and through regulation of its subcellular localization.
PTEN Function and Its Regulation
PTEN: A Dual Phosphatase
PTEN is encoded on chromosome 10q23, a region where loss of heterozygosity frequently occurs in various types of cancer (8). The protein encoded by PTEN contains 403-amino acid where the amino-terminal region shares sequence homology with the actin filament capping protein TENSIN and the putative tyrosine-protein phosphatase AUXILIN (1, 6). Crystal structure of PTEN revealed a C2 domain that contains the affinity for phospho-lipids on membrane and a phosphatase domain that contains the CX5R signature motif for phosphatases (9) (Figure 1). In vitro, PTEN is capable of dephosphorylating phospho-peptides as well as phospho-lipids. Thus, PTEN is a dual lipid and protein phosphatase. The biological effects of PTEN, however are dominated by its ability to dephosphorylate the lipid substrate phosphatidylinositol-3, 4, 5-triphosphate (PIP3) whereas protein substrates for PTEN are still being discovered (5, 7, 10). The lipid phosphatase motif of PTEN dephosphorylates PIP3 at the 3′ position and converts it back into PIP2 (5), leading to reduced PIP3 production and signals that depends on PIP3 (11) (Figure 2). PI3K functions to catalyze the reaction from PIP2 to PIP3. It achieves this task by phosphorylating the hydroxyl group of the 3rd position on the inositol ring of Phosphatidylinositols (12). The enzymatic function of PTEN thus acts as a negative regulatory signal for the PI3K mitogenic signaling pathway (Figure 2).
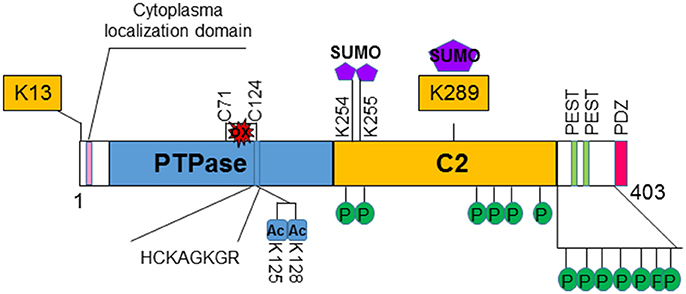
Figure 1. Structure and regulation of PTEN. PTEN is a 403-amino acid protein that shares sequence homology with tensin and auxilin at its amino-terminal. The hosphatase domain (PTPase) contains the signature CX5R (HCKAGKGR) p-loop structure for phosphatases. The C2 domain contains the affinity for phospho-lipids. PTEN also contains two PEST domains and a PDZ domain that may regulate its stability and subcellular localization, respectively. Several post-translational modification sites are discovered on PTEN. Two lysine sites (K13 and K289) are found to be ubiquinated and affects the localization and stability of PTEN. K289 is also sumoylated. Two additional sumoylation (K254 and K255) sites are identified that facilitates the binding of PTEN to the membrane. PTEN is also acetylated on K125 and K128 that decreases the ability of PTEN to inhibit PI3K/AKT activity. Oxidation of PTEN can occur that leads to formation of disulfide bond between C71 and C124 which results to reduced PTEN activity. Clusters of phosphorylation sites are found on PTEN. Phosphorylations of PTEN in general increases its stability but reduces its activity.
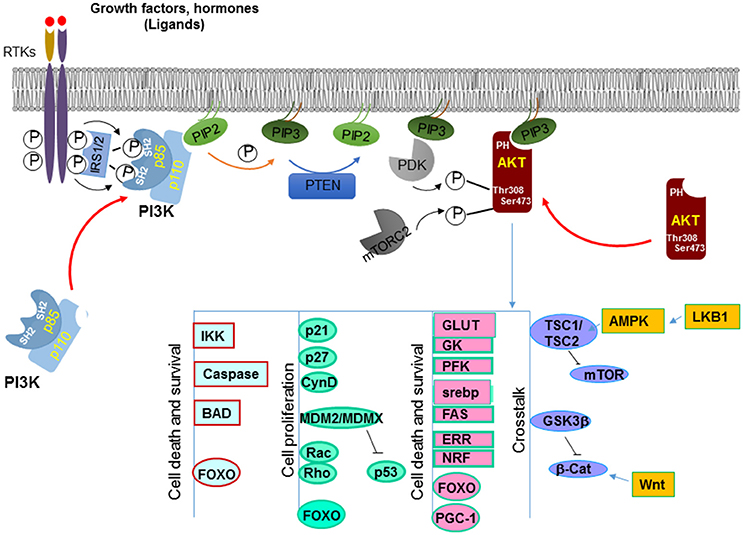
Figure 2. Canonical signals regulated by PTEN. PTEN dephosphorylates PIP3 on the 3′ position at the membrane and converts it back into PIP2. This action antagonize that of PI3K which adds the phosphate onto the 3′ position, leading to increased PIP3 production. PIP3 binds to the pleckstrin homology (PH) domain of downstream proteins (e.g., AKT) and provides a lipid moiety and recruits these proteins to the plasma membrane. AKT is one of the best characterized target of PI3K. Accumulation of PIP3 allows recruitment of AKT to the plasma membrane via direct interaction with its PH domain. AKT is then phosphorylated by two kinases, PDK1 and mTORC2 that leads to its full activation. Activated AKT has a plethora of downstream targets that it phosphorylates. Together these molecules are involved in cell growth and survival, metabolism and crosstalk with a number of other signaling pathways.
While the lipid substrate is well characterized to be PIP3, the identity of the protein substrates for PTEN in vivo has been illusive (13). However, in vitro study has revealed that PTEN is able to regulate cell migration by dephosphorylating itself, providing insights for investigating potential protein substrates for PTEN (14).
Regulation of PTEN
Localization of PTEN
Several non-canonical nuclear localization domains have been found on PTEN (15). A cytoplasmic localization signal has been identified for the N-terminus of PTEN that spans the residue from 19 to 25 (16). Mutations in these residues leads to increased nuclear localization of PTEN with unknown mechanisms. Studies suggest that ubiquitination controls the shuttling of PTEN between cytosol and nucleus (17–19). Monoubiquitination of lysine 289 (K289) is necessary for PTEN to move into the nucleus. Mutation of this site, K289E, is found in familial Cowden's syndrome that carries multiple mutations of the PTEN gene (17, 20, 21). A second ubiquitination site K13 and several other sites may also facilitate the nuclear transportation of PTEN (Figure 1). In addition, the localization of PTEN is regulated by Ca2+-mediated interactions with the major vault protein (MVP) (22, 23). In the nucleus, PTEN is more stable and still capable of inhibiting AKT and inducing cell death. Structural analysis also reviewed that PTEN harbors a PDZ domain and two PEST sequences in the C-terminal region (24). The PDZ domain is thought to regulate PTEN's subcellular localization whereas the two PEST sequences regulate its protein stability (25) (Figure 1).
Interestingly, while monoubiquitination leads to its nuclear shuttling, poly-ubiquitination of PTEN leads to its degradation. Though disagreement exists, an E3 ubiquitin ligase for PTEN has been reported (18, 21). NEDD4-1 was reported to add ubiquitin to both K13 and K289 of PTEN molecule, leading to both mono- and poly-ubiquitination of PTEN. However, others suggest that NEDD4 is dispensable for the regulation of PTEN (21).
Transcriptional Regulation of PTEN
PTEN is also regulated on the transcriptional and post-transcriptional levels. Several transcriptional factors have been reported to control the transcription of PTEN, including the tumor suppressor p53, the early growth response protein 1 (EGR-1), a metabolic regulatory gene peroxisome proliferation-activator receptor γ (PPARγ) [for detail, see 6)] and active transcription factor 2 (ATF2) (26). PTEN is also transcriptionally repressed by SNAIL and SLUG (27). These two zinc finger-like transcriptional factors compete with p53 for PTEN promoter binding. In addition, the nuclear factor kappa B (NFkB), the AP-1 transcription factor subunit c-Jun and the Notch signaling coregulatory CBF-1 (C-promoter binding factor-1) also bind to the PTEN promoter to regulate its transcription (28, 29).
More recently, regulation of PTEN by RNA-RNA interaction is reported that include microRNAs and long noncoding RNAs. Several miRNA including miR-205, miR-122, miR-21, etc. were identified to bind to the 3′untranslated region of PTEN mRNA. Elevated levels of many miRNAs are correlated with a concomitant reduction of PTEN mRNA (30–33). The long noncoding RNA that is encoded by the PTEN pseudogene transcript PTENP1 shares sequence identity with PTEN mRNA (34, 35). This transcript binds to miRNAs that target PTEN, leading to stabilization of PTEN mRNA. The antisense transcript of this pseudogene binds to the promoter of PTEN and negatively regulates the transcription of PTEN (34, 36).
Post-Translational Regulation of PTEN
Post-translationally, PTEN is modified by acetylation, oxidation and phosphorylation in addition to the ubiquitination discussed above [for detail, see 37)] (Figure 1). Phosphorylation of PTEN occurs on several clustered residues in the C-terminal domain of PTEN (38, 39). Several enzymes are responsible for these phosphorylations including casein kinase 2 (CK2), GSK3β, RhoA kinase, and P110δ subunits of PI3K (38–43). Phosphorylation of PTEN generally leads to the stabilization of the molecule but may reduce its activity (38, 39, 44). Recently, it was shown that ataxia-telangiectasia-mutated kinase (ATM) phosphorylates SUMOylated PTEN in response to γ-irradiation which leads to its nuclear exclusion (45). SUMOylation of PTEN typically occurs on K254 and K255 (46) which facilitates its binding to the membrane whereas modification on K289 is involved in its nuclear shuttling due to competition with ubiquitination modification. A couple of lysine residues at the catalytic domain of PTEN, lysine 125 and 128 are acetylated by PCAF (47). These acetylations lead to the diminished ability of PTEN to inhibit downstream events. PTEN is also regulated by the redox status of the cells. Two cysteine residues (124 and 71) form a disulfide bond in response to H2O2 treatment that leads to reduced activity of PTEN (48). Cys 124 is one of the “hot spots” that are often found mutated in human cancers.
PTEN as a Tumor Suppressor
Canonical Signaling Pathways Regulated by PTEN
Accumulation of PIP3 serves as a major signal for growth factor stimulation. PIP3 binds to the pleckstrin homology (PH) domain of downstream proteins (e.g., AKT) and provides a lipid moiety and recruits these proteins to the plasma membrane (49). Binding of PIP3 to the PH domain also changes the confirmation of these proteins so they can later be activated by phosphorylation. By reducing the intracellular levels of PIP3, PTEN inhibits the activation of downstream proteins of the PI3K pathway, including the serine/threonine kinase AKT and the protein kinase C (PKC).
A well-known downstream effector protein of the PTEN signal is AKT (50), which plays a critical role in regulating a number of cellular activities including cell growth, survival, cell migration and differentiation, cell and organ size control, metabolism, et al. [for detailed review, see 51)]. AKT, also known as Protein Kinase B (PKB) is a serine/threonine kinase. Following PI3K activation, accumulation of PIP3 allows recruitment of AKT to the plasma membrane via direct interaction with its PH domain (11). This binding of AKT to PIP3 not only allows AKT to be translocated to the membrane but also exposes sites on AKT where it can be further modified. It has been shown that AKT is phosphorylated by another PH domain-containing kinase 3-phosphoinositide-dependent protein kinase 1 (PDK1) at Thr308 (13, 52). This phosphorylation on Thr308 is important for initial activation of AKT whereas phosphorylation of Ser473 by mTORC2 is required for maximal AKT activation (53) (Figure 2).
Activated AKT phosphorylates a plethora of downstream targets including the regulations of kinases such as glycogen synthase kinases (GSK3α and β) (54), IκB kinases (IKKα and IKKβ) (55), apoptotic factors such as BAD (56), MDM2, a ubiquitin ligase for p53 (57), GTPases like Rac and Rho (58), cell cycle inhibitors p21 and p27 (59), and transcription factors such as forkhead transcription family (FoxO) members (Figure 2) (60–62). Phosphorylation by AKT regulates the functions of these molecules that are important for multiple cellular processes. For instance, phosphorylation of the pro-apoptotic factors BAD, caspases 3 and 9 by AKT renders them inactive and thus promotes cell survival (56, 63, 64). Phosphorylation of p21 on T145 and p27 on T157 leads to their nuclear exclusion and the inability of these cell cycle inhibitors to inhibit cell proliferation (65, 66). Likewise, AKT also directly phosphorylates MDM2 and MDMX (67, 68). The phosphorylated MDM2 and MDMX bind to 14-3-3 proteins, leading to stabilization of the MDM2-MDMX complexes which mediates the degradation of p53 to keep the level of p53 low in the cells.
AKT also phosphorylates forkhead transcriptional factors and induces their binding to 14-3-3 proteins (63). This process blocks their translocation to the nucleus. Several members of the forkhead transcriptional factor family are targets of AKT, including FOXO1 and FOXO3. The binding elements for these forkhead transcriptional factors are widely spread on promoter regions of genes that regulate cell proliferation, survival and metabolic changes (69). For example, FOXO3a binds to the promoters of Bim and PUMA and can initiate apoptosis cascades by inducing the transcription of these death genes (70, 71). FOXO1 transcriptionally activates p21 and p27 and inhibits cell proliferation through these actions (72, 73). Furthermore, these forkhead transcriptional factors are also responsible for many metabolic effects induced by insulin signaling through the PI3K/AKT signaling pathway (74). Additional evidence suggests that the forkhead transcriptional factors may play a key role in the feedback regulation of the Insulin/PI3K/AKT signaling pathway (75).
Two substrates of AKT, GSK3β and tuberous sclerosis complex TSC1/2, play important roles in mediating cross talks between PI3K/AKT signaling pathway and other signaling pathways (Figure 2). GSK3β is phosphorylated by AKT on Serine 21/9 which inhibits its activity (76). GSK3β is an important regulator in Wnt signaling. It phosphorylates β-catenin, resulting in its ubiquitin-mediated degradation. The crosstalk between PTEN and Wnt signaling may underlie some of the effects of PTEN on the regulation of stem cell maintenance (77–79) and G0-G1 cell cycle regulation (80–84). Another substrate of AKT is TSC1/2. TSC1/2 plays a key role in incorporating metabolism and cell size control, together with cell growth and proliferation regulation (85). The heterodimer of TSC1 and TSC2 is essential for suppressing the function of mTOR (mammalian target of rapamycin). TSC2 activity is inhibited when phosphorylated by AKT (86). Therefore, by acting on TSC2, AKT induces the activity of mTOR and the downstream events of mTOR activation that include metabolic changes, protein translation as well as cell proliferation. This regulation of mTOR by AKT-TSC-mediated signal allows the crosstalk of PTEN with another tumor suppressor LKB1 (87).
Nuclear PTEN
In earlier studies, PTEN was reported to be a protein that is exclusively localized in the cytoplasm. However, it is clear now that PTEN can be both cytoplasmic and nuclear (88–90). In more differentiated and resting cells, PTEN is often found in the nucleus even though it was originally identified to be a cytosolic protein (using primarily tumor cells) (90). Nuclear PTEN also plays other roles in addition to its lipid phosphatase activity and the nuclear function of PTEN is important for the ability of PTEN to inhibit tumor development (Figure 3). Nuclear PTEN is reported to play important roles in chromosome stability, DNA repair and cell cycle regulation. In the nucleus, PTEN promotes the stability and transcriptional activity of the tumor suppressor p53 by directly associating with p53 (91–93). Forced expression of PTEN in the nucleus led to MAP kinase-dependent inhibition of cyclin D1 expression (15, 22). Nuclear expression of PTEN results in the dephosphorylation of MAP kinase. Whether this is a direct effect of the protein phosphatase activity of PTEN is not clear. In addition, PTEN is found to be associated with the centromere in the nucleus by direct binding to the centromere specific binding protein C (CENP-C) (94). Disruption of this binding leads to premature centromere separation. In addition, PTEN is also found to collaborate with E2F to induce the expression of Rad51 and thus enhance DNA repair (94). This relationship between PTEN and Rad51 may explain the observation that double-stranded DNA breakage rate is found to be increased when nuclear PTEN function is interrupted. PTEN also interacts with the anaphase-promoting complex (APC) to promote its association with its binding partner which together results in proteolysis of mitotic cyclins (95).
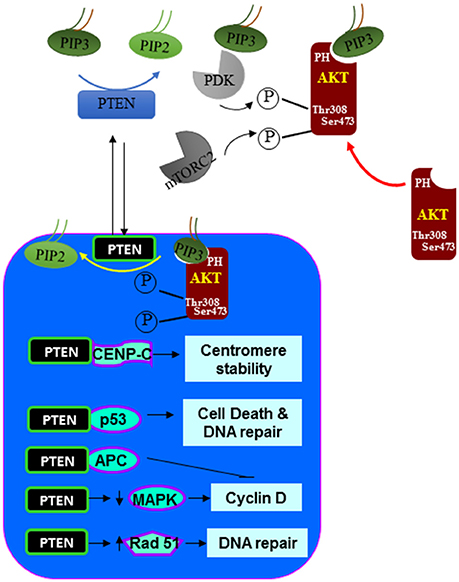
Figure 3. Nuclear signals regulated by PTEN. In addition to the dephosphorylating PIP3 at the plasma membrane. PTEN is also found in the nucleus. In the nucleus, PTEN can act similarly as it does at the plasma membrane by inhibiting the function of AKT. In addition, PTEN also associate with a number of nuclear proteins and regulate other cellular functions such as centromere stability, DNA repair, cell death and proliferation.
PTEN as a Metabolic Regulator
As an important growth and survival regulatory gene, germline deletion of Pten in mice was shown to be embryonic (96–98). Heterozygous mice develop hamartomatous polyps in the colon and tumors in multiple other tissues (99). In human, germline PTEN mutation leads to a number of familial diseases that are characterized by multiple hamartomatous lesions and predisposition to cancer development (100). Conditional deletion of Pten in mouse models has been done in multiple organs. Collectively, these studies confirm the signaling studies verifying PTEN as a tumor suppressor that regulates cell growth and survival. These studies have been comprehensively reviewed previously (6, 101).
While PTEN loss promotes tumorigenesis in multiple organs, genetic studies also indicate that PTEN loss leads to a number of metabolic changes that collectively improve overall insulin sensitivity (Figure 4). PTEN, being a major negative regulator of the PI3K/AKT signaling, is found to play an important role in both lipid and glucose metabolism as well as regulation of mitochondrial functions. Studies in C. elegans and Drosophila have demonstrated a highly conserved signal regulated by PTEN for both growth control and metabolism. In these organisms, the insulin/PI3K pathway negatively regulated by PTEN is used to control dauer formation, metabolism, and life span in response to nutrient availability (102–104).
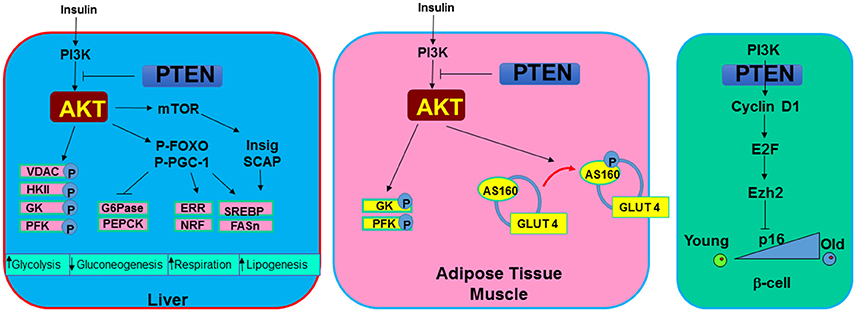
Figure 4. Metabolic signals regulated by PTEN. In metabolic tissues, PTEN and PI3K/AKT signal mediates the action of insulin. In the liver, induction of PI3K/AKT signal or PTEN loss leads to increased phosphorylation of several mitochondrial and glycolytic proteins that collectively increases glycolytic signaling. In addition, G6Pase and PEPCK, two rate limiting enzyme involved in gluconeogenesis are inhibited by the induction of AKT. Cellular respiration is induced with upregulation of PI3K/AKT due to their regulation on ERR and NRF, two mitochondrial biogenesis regulator. SREBP and FASn are induced through this signaling by insulin to induce lipogenesis. In adipose tissue and muscle, AKT also phosphorylates AS160 to mobilize glucose transporter GLUT4 to the membrane in response to hyperglycemia signals. In pancreatic β-cells. PTEN loss is found to rescue the age-onset loss of regenerating ability for β-cells. This phenotype appear to be regulated by the inhibition of PTEN on senescence regulator p16 via E2F and Ezh2 signaling.
Regulation of Glucose Metabolism
Parallel signals for PTEN/PI3K have been reported for mammals as it was in C. elegans and Drosophila. Insulin and insulin-like growth factors (IGF) such as IGF-1 and IGF-2 binds to the insulin and IGF receptors. Binding of insulin and IGF to these receptors either directly induces the activation of PI3K or results in phosphorylation of insulin receptor substrate (IRS) as an adaptor protein to recruit and activate PI3K (10). Through this action and the downstream activation of AKT, adipocytes and myocytes sense the elevated insulin levels and initiate glucose uptake. The serine/threonine kinase AKT phosphorylates a 160-KDa substrate AS160 at Thr642 in adipocytes (105). Phosphorylation of this protein, identified as a GTPase-activating domain for Rab, is found to be responsible for membrane trafficking of GLUT4 induced by the insulin/PI3K signaling. In addition, AKT phosphorylates a variety of targets involved in the regulation of metabolism. Phosphorylation and inhibition of GSK3 not only contribute to regulation of β-catenin and the cell cycle, it also activates glycogen synthase (6). When PTEN is lost and GSK3 is phosphorylated, glycogen was found to accumulate in hepatocytes of the liver-specific Pten null mice (88). In hepatocytes, phosphorylation of FOXO by AKT blocks the transcription of glucose-6-phosphatase (G6Pase) and phosphoenolpyruvate carboxykinase (PEPCK) (74), two rate-limiting enzymes in the process of gluconeogenesis. In addition, AKT was also reported to directly phosphorylate proxisome proliferator-activated receptor gamma co-activator (PGC-1a) at S570 (106). This phosphorylation event was also found to mediate the transcriptional repression of G6Pase and PEPCK. These signals regulated by AKT and blocked by PTEN are important for how metabolic organs like the liver, muscle and adipose tissue respond to elevated insulin signals.
Consistent with these metabolic signals regulated by PTEN, deletion of Pten in the liver led to robust downregulation of PEPCK (88). Moderate downregulation of G6Pase was also reported. In the adipose tissue, deletion of Pten resulted in increased insulin sensitivity and resistance to streptozotocin-induced diabetes (107). Increased GLUT4 membrane localization on adipocytes was observed in these mice.
Regulation of Lipid Metabolism
In addition to glucose metabolism, the PTEN-regulated PI3K signaling also controls lipid metabolism. The sterol receptor element binding protein, SREBP, serves as a key transcriptional factor for genes involved in the biosynthesis of fatty acids and their further incorporation into triglycerides and cholesterol. As a master transcriptional factor controlling the de novo lipogenesis process, SREBP binds to the promoters of many lipogenic enzyme genes, including fatty acid synthase (Fasn) and acetyl-CoA carboxylase (ACC), as well as those controlling the production of NADPH, a reducing equivalent needed for lipid biosynthesis. The PTEN/PI3K/AKT signaling-controlled SREBP expression is mediated through multiple levels including transcriptional and post-translational processing of SREBP. The downstream target of AKT, the forkhead transcriptional factor, FoxO1 regulates SREBP and lipogenesis by repressing SREBP transcription (108). Interestingly, the function of FOXO1 on Fasn expression is dependent on whether PI3K/AKT signal is induced (109). Using rapamycin and siRNA to inhibit mTORC1 and other signals involved in the AKT pathway, it was shown that transcription induction of SREBP1 and lipogenesis is also dependent on TORC1 activity (110). However, this effect was not supported by observations in the TSC1-deficient mice, defective in mTORC1 signaling, which are resistant, rather than sensitive, to high fat diet (HFD)-induced steatosis (111). The processing of SREBP is dependent on two proteins, SREBP cleavage-activating protein (SCAP) and insulin induced gene (Insig). In response to sterol demand, SCAP cleaves SREBP to produce the mature active form of transcriptional factor that moves to the nucleus. Binding of Insig to SCAP prevents this action and thus inhibits the processing of SREBP. While oxysterols suppresses the expression of Insig-1, inhibition of PI3K/AKT activity blocks this inhibition and allows the processing of SREBP (112), consistent with a role of AKT in SREBP processing. This processing is both mTORC1-dependent and mTORC1-independent (111). Thus, both PTEN/PI3K/AKT downstream signaling targets, TORC1 and FoxO1, play critical roles in controlling SREBP expression and lipogenesis. In addition, Maf-1, a central repressor of genes transcribed by RNA pol III is recently found to be regulated by PTEN through AKT2 and mTOR (113). While SREBP binds to the promoter of Fasn and positively regulates its expression, Maf-1 was shown to occupy the promoter and repress the expression of Fasn.
Consistent with these signaling analysis, loss of Pten in the liver led to elevated de novo lipogenesis through robust induction of SREBP and Fasn expression (88). The accumulation of lipid and elevated lipogenesis is a result of activation of AKT2 as deletion of Akt2 completely reversed the phenotype (109, 114). This effect of AKT is both mTOR dependent and independent (111). In addition, FOXO1 gain of function has also been shown to induce lipid synthesis (115).
Regulation of Mitochondrial Metabolism
In recent years, studies attempt to elucidate the molecular signals underlying “Warburg effects” have led to the discoveries of novel roles for PI3K/AKT signaling in mitochondrial function (10). In addition to regulating pro- and anti-apoptotic factors (10), AKT promotes binding of hexokinase II to the mitochondrial voltage dependent anion channel (VDAC) (116). This event, occurring at the mitochondrial outer membrane allows rapid phosphorylation of available glucose molecules and efficient conversion to ATP from glycolysis. AKT was also found to be localized in the inner membranes of the mitochondria (117, 118). In the mitochondria matrix, AKT phosphorylates mitochondrial pool of GSK3β and regulates mitochondrial respiration through phosphorylation of pyruvate dehydrogenase (PDH) (118). In addition, the mitochondrial localized AKT also plays a role in the transcription regulation of mitochondrial DNA. A FOXO3-response element has been found on the promoter of a mitochondrial encoded gene, 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) (119).
In the nucleus, the PI3K/AKT signaling controls mitochondrial gene transcription network through multiple different mechanisms. The forkhead transcriptional factor FOXO3 has been demonstrated to be a transcriptional regulator of mitochondrial genes. In colon cancer cells induced to express a constitutively active form of FOXO3a, a large number of mitochondrial genes are downregulated (120). These genes include Tfam and TFB1M&2M, the nucleus-encoded auxiliary factors for mitochondrial gene transcription. One of the global regulators of metabolism including lipid and glucose metabolism as well as mitochondrial metabolism is PGC-1 (121). As a transcriptional coactivator, members of PGC-1 family of coactivators have the ability to interact with a number of different transcriptional factors including PPARs for fatty acid oxidation, FOXO1 for lipogenesis, FOXO1 and glucocorticoid receptor (GR) for gluconeogenesis, and estrogen-related receptors (ERRs) for mitochondrial function.
The best characterized isoform of ERRs, ERRα is abundantly expressed in high oxidative organs and recognized as a key regulators of adaptive energy metabolism (122). ERRα, itself, is a weak transcriptional factor. Both the activity and expression of ERRα are significantly increased when physically bound by PGC-1α (123). AKT activation was found to control mitochondrial gene transcription by phosphorylating and activating CREB transcriptional factor independent of the cAMP mediated activation of PKA, the common signal that induces CREB phosphorylation (124). When phosphorylated, CREB induces the transcription of PGC-1. Thus, in addition to removing the inhibition of FoxO and phosphorylating PGC-1, activation of AKT also positively induces PGC-1 transcription by phosphorylating CREB. Being the coactivator, PGC-1 robustly increases the transcriptional activity of ERRα to promote transcription of genes encoding mitochondrial function, including TFAM, TFB1M&2M and medium-chain acyl-coA dehydrogenase (MCAD) (124). In hepatocytes, this induction of ERRα leads to increased oxygen consumption and elevated ROS production, likely contributed to the liver injury (and lipid accumulation) phenotypes observed with Pten loss in the liver.
In addition, the AKT substrate consensus sequence has been found on NRF1, another gene involved in mitochondrial gene transcription. In H4IIE hepatoma cells, phosphorylation of NRF1 by AKT is reported to mediate pro-oxidant t-BOOH induced Tfam expression (125). Thus, through directly phosphorylating FOXO and NRF1 or indirectly inducing ERRα expression, AKT controls the gene transcriptional networks of mitochondria. Consistently, over-expression of NRF1 and AKT has been shown to mimic the effect of TFAM to abrogate 1-methyl-4-phenyl-2, 3-dihydropyridinium ion induced mitochondrial damage (126), confirming a signaling relationship between PI3K/AKT/FOXO signal and mitochondrial gene transcription regulation.
Paradoxical Roles of PTEN Regulated Metabolic and Growth Signals on Tumor Growth and Metabolism
Metabolic Sensitivity Regulated by PTEN
In mammals, ectopic expression of PTEN by introduction of bacterial artificial chromosomes (BACs) into the mouse genome led to reduced body size, increased energy expenditure and low body fat content (127, 128). Consistent with the observations in C. elegance and Drosophila, these mice also have a longer tumor free lifespan. This enhanced metabolic phenotype however is paradoxical with the enhanced metabolic functions associated with PTEN loss observed with the tissue specific Pten deletion mice. In adipose tissue, liver, pancreatic β-cells and muscle, deletion of Pten consistently lead to enhanced insulin and metabolic sensitivity as well as resistance to HFD induced diabetes (88, 89, 107, 129–131). In addition to the enhanced ability to transport and metabolize glucose by adipocytes, myocytes and hepatocytes, PTEN loss was associated with enhanced energy expenditure in brown adipose tissue (128). In β-cells, deletion of Pten relieved the suppression of cell cycle re-entry by inhibiting the senescence regulatory gene p16Ink4a through E2F/Ezh2 mechanism (132). This regulation led to the rescue of aging-induced loss of growth potential in β-cells. The enhanced ability of pancreatic islets to respond to hyperglycemic stress led to improved systemic metabolic health (89, 130). Overall, PTEN loss and activation of PI3K/AKT signal lead to the improved ability to handle metabolic stress in mice. The improved metabolic health phenotypes observed with overexpression of PTEN is likely contributed to metabolic adaptation.
Tumor Metabolism Regulated by PTEN
While loss of PTEN leads to improved insulin sensitivity, this metabolic effect has been credited for the tumor suppressing functions of PTEN (7). The metabolic signals regulated by this pathway, including the glycolytic signal such as localization of glucose transporters, activation of hexokinase and phosphofructokinase as well as induction of de novo lipogenesis are among the signals that are recognized as promoting factors for tumorigenesis. Indeed, a number of metabolic enzymes, particularly glycolytic genes have been found to have oncogenic or tumor suppressive functions as manipulation of these genes modulate tumor growth. Particularly, expression of isoform specific metabolic genes appears to be linked to tumorigenesis (133). Association of lipogenic and other lipid metabolic genes with tumors are recognized but more works are needed to understand their contributions to tumorigenesis.
Steatosis Due to PTEN Loss Establishes a Tumor Promoting Environment
During insulin resistance, suppression of hepatic glucose synthesis by insulin is blunted and the persistence of hepatic glucose output leads to postprandial hyperglycemia (134). At the same time, hyperinsulinemia signal in the liver induces lipogenesis, resulting in fatty liver disease that is a hallmark of insulin resistance syndrome. This differential response of gluconeogenesis and lipogenesis to insulin during insulin resistance has been termed “selective hepatic insulin resistance” (135). Mimicking insulin signal in the liver, loss of hepatic PTEN resulted in non-alcoholic steatohepatitis (NASH) while suppressing gluconeogenesis (89, 109, 136, 137). Unlike that observed with insulin resistance, NASH developed in the liver-specific Pten deletion mice is not due to hyperinsulinemia resulting from high circulating glucose levels. Locally enhanced hepatic PI3K/AKT signal actually led to improved ability for the liver to handle glucose, turning the liver into a glucose sink, leading to an improved ability to handle glycemic stress in these mice.
While the increased insulin/PI3K/AKT signal in the liver leads to improved systemic insulin sensitivity (88), the resulting NASH due to increased de novo lipogenesis however forms an environment that results in damage of the liver parenchymal (138–142). When NASH is inhibited via either dietary approach or genetic deletion of a metabolic AKT, Akt2, tumor development is inhibited (79, 114). The NASH thus serves as a tumor promoting event that promotes the development of tumors that arose from the PTEN loss transformed tumor-initiating cells (77–79, 114). How steatosis establishes a tumor environment is being explored currently. In the liver, inflammation as a result of damage to the liver parenchymal was shown to play an important role (Figure 5). Wnt signal produced by macrophages is one of the niche signals established by this NASH environment to promote tumorigenesis (79).
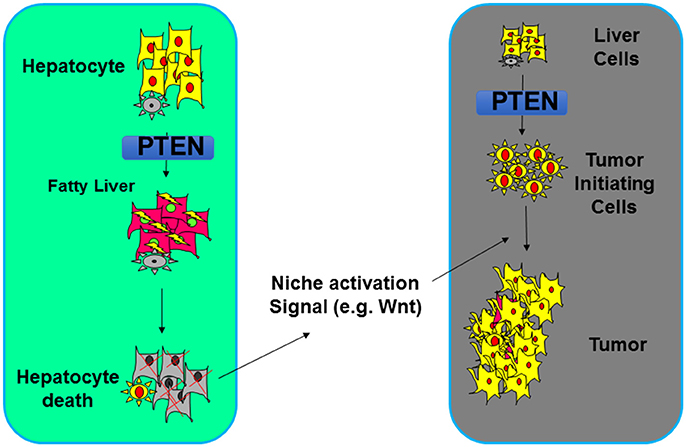
Figure 5. Steatosis regulation by PTEN alters the tumor microenvironment. Deletion of Pten leads to increased lipogenesis and deposition of lipid. In hepatocytes, such lipid accumulation results in hepatocytes death which establishes a niche activation signal (e.g., Wnt). This then leads to the proliferation of tumor initiating cells and tumorigenesis.
Future Considerations
PTEN is a critical regulator of cell growth/survival as well as metabolism. As a metabolic regulator, PTEN controls the metabolism of both glucose and fatty acids. These effects of PTEN through targeting the PI3K/AKT dependent and independent pathways lead to suppressed insulin sensitivity and inhibited cell growth and survival. While the signals by which PTEN regulates growth and survival has been well elucidated, the mechanisms by which PTEN regulates metabolism, particularly lipid and mitochondrial metabolism is not well understood. Future studies to understand the molecular signals that PTEN controls to regulate these cellular functions are necessary for both the cancer and diabetes treatment.
Author Contributions
C-YC and JC drafted the original manuscript. LH proofed the manuscript. BS edited the finalized the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
BS acknowledges support by NIH grants NCI (R01CA154986) as well as support from USC center for Liver Disease (P30DK48522).
References
1. Li DM, Sun H. TEP1, encoded by a candidate tumor suppressor locus, is a novel protein tyrosine phosphatase regulated by transforming growth factor beta. Cancer Res. (1997) 57:2124–9.
2. Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science (1997) 275:1943–7. doi: 10.1126/science.275.5308.1943
3. Liaw D, Marsh DJ, Li J, Dahia Pl, Wang SI, Zheng Z, et al. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet. (1997) 16:64–7. doi: 10.1038/ng0597-64
4. Stiles B, Groszer M, Wang S, Jiao J, Wu H. PTENless means more. Dev Biol. (2004) 273:175–84. doi: 10.1016/j.ydbio.2004.06.008
5. Downes CP, Ross S, Maccario H, Perera N, Davidson L, Leslie NR. Stimulation of PI 3-kinase signaling via inhibition of the tumor suppressor phosphatase, PTEN. Adv Enzyme Regul. (2007) 47:184–94. doi: 10.1016/j.advenzreg.2006.12.018
6. Stiles BL. Phosphatase and tensin homologue deleted on chromosome 10: extending its PTENtacles. Int J Biochem Cell Biol. (2009) 41:757–61. doi: 10.1016/j.biocel.2008.09.022
7. Worby CA, Dixon JE. Pten. Annu Rev Biochem. (2014) 83:641–69. doi: 10.1146/annurev-biochem-082411-113907
8. Pulido R, Baker SJ, Barata JT, Carracedo A, Cid VJ, Chin-Sang ID, et al. A unified nomenclature and amino acid numbering for human PTEN. Sci Signal (2014) 7:pe15. doi: 10.1126/scisignal.2005560
9. Lee JO, Yang H, Georgescu MM, Di Cristofano A, Maehama T, Shi Y, et al. Crystal structure of the PTEN tumor suppressor: implications for its phosphoinositide phosphatase activity and membrane association. Cell (1999) 99:323–34. doi: 10.1016/S0092-8674(00)81663-3
10. Stiles BL. PI-3-K and AKT: onto the mitochondria. Adv Drug Deliv Rev. (2009) 61:1276–82. doi: 10.1016/j.addr.2009.07.017
11. Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer (2002) 2:489–501. doi: 10.1038/nrc839
12. Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene (2008) 27:5497–510. doi: 10.1038/onc.2008.245
13. Knowles MA, Platt FM, Ross RL, Hurst CD. Phosphatidylinositol 3-kinase (PI3K) pathway activation in bladder cancer. Cancer Metastasis Rev. (2009) 28:305–16. doi: 10.1007/s10555-009-9198-3
14. Raftopoulou M, Etienne-Manneville S, Self A, Nicholls S, Hall A. Regulation of cell migration by the C2 domain of the tumor suppressor PTEN. Science (2004) 303:1179–81. doi: 10.1126/science.1092089
15. Chung JH, Eng C. Nuclear-cytoplasmic partitioning of phosphatase and tensin homologue deleted on chromosome 10 (PTEN) differentially regulates the cell cycle and apoptosis. Cancer Res. (2005) 65:8096–100. doi: 10.1158/0008-5472.CAN-05-1888
16. Denning G, Jean-Joseph B, Prince C, Durden DL, Vogt PK. A short N-terminal sequence of PTEN controls cytoplasmic localization and is required for suppression of cell growth. Oncogene (2007) 26:3930–40. doi: 10.1038/sj.onc.1210175
17. Trotman LC, Wang X, Alimonti A, Chen Z, Teruya-Feldstein J, Yang H, et al. Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell (2007) 128:141–56. doi: 10.1016/j.cell.2006.11.040
18. Wang X, Trotman LC, Koppie T, Alimonti A, Chen Z, Gao Z, et al. NEDD4-1 is a proto-oncogenic ubiquitin ligase for PTEN. Cell (2007) 128:129–39. doi: 10.1016/j.cell.2006.11.039
19. Drinjakovic J, Jung H, Campbell DS, Strochlic L, Dwivedy A, Holt C. E. E3 ligase Nedd4 promotes axon branching by downregulating PTEN. Neuron (2010) 65:341–57. doi: 10.1016/j.neuron.2010.01.017
20. Chi SG, Kim HJ, Park BJ, Min HJ, Park JH, Kim YW, et al. Mutational abrogation of the PTEN/MMAC1 gene in gastrointestinal polyps in patients with Cowden disease. Gastroenterology (1998) 115:1084–9. doi: 10.1016/S0016-5085(98)70078-2
21. Fouladkou F, Landry T, Kawabe H, Neeb A, Lu C, Brose N, et al. The ubiquitin ligase Nedd4-1 is dispensable for the regulation of PTEN stability and localization. Proc Natl Acad Sci USA. (2008) 105:8585–90. doi: 10.1073/pnas.0803233105
22. Chung JH, Ostrowski MC, Romigh T, Minaguchi T, Waite KA, Eng C. The ERK1/2 pathway modulates nuclear PTEN-mediated cell cycle arrest by cyclin D1 transcriptional regulation. Hum Mol Genet. (2006) 15:2553–9. doi: 10.1093/hmg/ddl177
23. Minaguchi T, Waite KA, Eng C. Nuclear localization of PTEN is regulated by Ca(2+) through a tyrosil phosphorylation-independent conformational modification in major vault protein. Cancer Res. (2006) 66:11677–82. doi: 10.1158/0008-5472.CAN-06-2240
24. Furnari FB, Huang HJ, Cavenee WK. The phosphoinositol phosphatase activity of PTEN mediates a serum-sensitive G1 growth arrest in glioma cells. Cancer Res. (1998) 58:5002–8.
25. Jiang X, Chen S, Asara JM, Balk SP. Phosphoinositide 3-kinase pathway activation in phosphate and tensin homolog (PTEN)-deficient prostate cancer cells is independent of receptor tyrosine kinases and mediated by the p110beta and p110delta catalytic subunits. J Biol Chem. (2010) 285:14980–9. doi: 10.1074/jbc.M109.085696
26. Shen YH, Zhang L, Gan Y, Wang X, Wang J, Lemaire SA, et al. Up-regulation of PTEN (phosphatase and tensin homolog deleted on chromosome ten) mediates p38 MAPK stress signal-induced inhibition of insulin signaling. a cross-talk between stress signaling and insulin signaling in resistin-treated human endothelial cells. J Biol Chem. (2006) 281:7727–36. doi: 10.1074/jbc.M511105200
27. Uygur B, Abramo K, Leikina E, Vary C, Liaw L, Wu WS. SLUG is a direct transcriptional repressor of PTEN tumor suppressor. Prostate (2015) 75:907–16. doi: 10.1002/pros.22974
28. Hettinger K, Vikhanskaya F, Poh MK, Lee MK, De Belle I, Zhang JT, et al. c-Jun promotes cellular survival by suppression of PTEN. Cell Death Differ. (2007) 14:218–29. doi: 10.1038/sj.cdd.4401946
29. Whelan JT, Forbes SL, Bertrand FE. CBF-1 (RBP-J kappa) binds to the PTEN promoter and regulates PTEN gene expression. Cell Cycle (2007) 6:80–4. doi: 10.4161/cc.6.1.3648
30. Hong L, Lai M, Chen M, Xie C, Liao R, Kang YJ, et al. The miR-17-92 cluster of microRNAs confers tumorigenicity by inhibiting oncogene-induced senescence. Cancer Res. (2010) 70:8547–57. doi: 10.1158/0008-5472.CAN-10-1938
31. Olive V, Jiang I, He L. mir-17-92, a cluster of miRNAs in the midst of the cancer network. Int J Biochem Cell Biol. (2010) 42:1348–54. doi: 10.1016/j.biocel.2010.03.004
32. Lujambio A, Lowe SW. The microcosmos of cancer. Nature (2012) 482:347–55. doi: 10.1038/nature10888
33. Jin HY, Oda H, Lai M, Skalsky RL, Bethel K, Shepherd J, et al. MicroRNA-17~92 plays a causative role in lymphomagenesis by coordinating multiple oncogenic pathways. EMBO J. (2013) 32:2377–91. doi: 10.1038/emboj.2013.178
34. Poliseno L, Salmena L, Zhang J, Carver B, Haveman WJ, Pandolfi PP. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature (2010) 465:1033–8. doi: 10.1038/nature09144
35. Johnsson P, Ackley A, Vidarsdottir L, Lui WO, Corcoran M, Grander D, et al. A pseudogene long-noncoding-RNA network regulates PTEN transcription and translation in human cells. Nat Struct Mol Biol. (2013) 20:440–6. doi: 10.1038/nsmb.2516
36. Sumazin P, Yang X, Chiu HS, Chung WJ, Iyer A, Llobet-Navas D, et al. An extensive microRNA-mediated network of RNA-RNA interactions regulates established oncogenic pathways in glioblastoma. Cell (2011) 147:370–81. doi: 10.1016/j.cell.2011.09.041
37. Leslie NR, Batty IH, Maccario H, Davidson L, Downes CP. Understanding PTEN regulation: PIP2, polarity and protein stability. Oncogene (2008) 27:5464–76. doi: 10.1038/onc.2008.243
38. Torres J, Rodriguez J, Myers MP, Valiente M, Graves JD, Tonks NK, et al. Phosphorylation-regulated cleavage of the tumor suppressor PTEN by caspase-3: implications for the control of protein stability and PTEN-protein interactions. J Biol Chem. (2003) 278:30652–60. doi: 10.1074/jbc.M212610200
39. Al-Khouri AM, Ma Y, Togo SH, Williams S, Mustelin T. Cooperative phosphorylation of the tumor suppressor phosphatase and tensin homologue (PTEN) by casein kinases and glycogen synthase kinase 3beta. J Biol Chem. (2005) 280:35195–202. doi: 10.1074/jbc.M503045200
40. Miller SJ, Lou DY, Seldin DC, Lane WS, Neel BG. Direct identification of PTEN phosphorylation sites. FEBS Lett. (2002) 528:145–53. doi: 10.1016/S0014-5793(02)03274-X
41. Maccario H, Perera NM, Davidson L, Downes CP, Leslie NR. PTEN is destabilized by phosphorylation on Thr366. Biochem J. (2007) 405:439–44. doi: 10.1042/BJ20061837
42. Patsoukis N, Li L, Sari D, Petkova V, Boussiotis VA. PD-1 increases PTEN phosphatase activity while decreasing PTEN protein stability by inhibiting casein kinase 2. Mol Cell Biol. (2013) 33:3091–8. doi: 10.1128/MCB.00319-13
43. Tzenaki N, Aivaliotis M, Papakonstanti EA. Focal adhesion kinase phosphorylates the phosphatase and tensin homolog deleted on chromosome 10 under the control of p110delta phosphoinositide-3 kinase. FASEB J. (2015) 29:4840–52. doi: 10.1096/fj.15-274589
44. Torres J, Pulido R. The tumor suppressor PTEN is phosphorylated by the protein kinase CK2 at its C terminus. implications for PTEN stability to proteasome-mediated degradation. J Biol Chem. (2001) 276:993–8. doi: 10.1074/jbc.M009134200
45. Bassi C, Ho J, Srikumar T, Dowling RJ, Gorrini C, Miller SJ, et al. Nuclear PTEN controls DNA repair and sensitivity to genotoxic stress. Science (2013) 341:395–9. doi: 10.1126/science.1236188
46. Huang J, Yan J, Zhang J, Zhu S, Wang Y, Shi T, et al. SUMO1 modification of PTEN regulates tumorigenesis by controlling its association with the plasma membrane. Nat Commun. (2012) 3:911. doi: 10.1038/ncomms1919
47. Okumura K, Mendoza M, Bachoo RM, Depinho RA, Cavenee WK, Furnari FB. PCAF modulates PTEN activity. J Biol Chem. (2006) 281:26562–8. doi: 10.1074/jbc.M605391200
48. Lee SR, Yang KS, Kwon J, Lee C, Jeong W, Rhee SG. Reversible inactivation of the tumor suppressor PTEN by H2O2. J Biol Chem. (2002) 277:20336–42. doi: 10.1074/jbc.M111899200
49. Dinitto JP, Cronin TC, Lambright DG. Membrane recognition and targeting by lipid-binding domains. Sci STKE (2003) 2003:re16. doi: 10.1126/stke.2132003re16
50. Stiles B, Gilman V, Khanzenzon N, Lesche R, Li A, Qiao R, et al. Essential role of AKT-1/protein kinase B alpha in PTEN-controlled tumorigenesis. Mol Cell Biol. (2002) 22:3842–51. doi: 10.1128/MCB.22.11.3842-3851.2002
51. Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell (2007) 129:1261–74. doi: 10.1016/j.cell.2007.06.009
52. Hresko RC, Mueckler M. mTOR.RICTOR is the Ser473 kinase for Akt/protein kinase B in 3T3-L1 adipocytes. J Biol Chem. (2005) 280:40406–16. doi: 10.1074/jbc.M508361200
53. Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, Cohen P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol. (1997) 7:261–9. doi: 10.1016/S0960-9822(06)00122-9
54. Pap M, Cooper GM. Role of glycogen synthase kinase-3 in the phosphatidylinositol 3-Kinase/Akt cell survival pathway. J Biol Chem. (1998) 273:19929–32. doi: 10.1074/jbc.273.32.19929
55. Gustin JA, Ozes ON, Akca H, Pincheira R, Mayo LD, Li Q, et al. Cell type-specific expression of the IkappaB kinases determines the significance of phosphatidylinositol 3-kinase/Akt signaling to NF-kappa B activation. J Biol Chem. (2004) 279:1615–20. doi: 10.1074/jbc.M306976200
56. Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell (1997) 91:231–41. doi: 10.1016/S0092-8674(00)80405-5
57. Ashcroft M, Ludwig RL, Woods DB, Copeland TD, Weber HO, Macrae EJ, et al. Phosphorylation of HDM2 by Akt. Oncogene (2002) 21:1955–62. doi: 10.1038/sj.onc.1205276
58. Liliental J, Moon SY, Lesche R, Mamillapalli R, Li D, Zheng Y, et al. Genetic deletion of the Pten tumor suppressor gene promotes cell motility by activation of Rac1 and Cdc42 GTPases. Curr Biol. (2000) 10:401–4. doi: 10.1016/S0960-9822(00)00417-6
59. Collado M, Medema RH, Garcia-Cao I, Dubuisson ML, Barradas M, Glassford J, et al. Inhibition of the phosphoinositide 3-kinase pathway induces a senescence-like arrest mediated by p27Kip1. J Biol Chem. (2000) 275:21960–8. doi: 10.1074/jbc.M000759200
60. Biggs WHIII, Meisenhelder J, Hunter T, Cavenee WK, Arden KC. Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc Natl Acad Sci USA. (1999) 96:7421–6. doi: 10.1073/pnas.96.13.7421
61. Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell (1999) 96:857–68. doi: 10.1016/S0092-8674(00)80595-4
62. Guo S, Rena G, Cichy S, He X, Cohen P, Unterman T. Phosphorylation of serine 256 by protein kinase B disrupts transactivation by FKHR and mediates effects of insulin on insulin-like growth factor-binding protein-1 promoter activity through a conserved insulin response sequence. J Biol Chem. (1999) 274:17184–92. doi: 10.1074/jbc.274.24.17184
63. Cardone MH, Roy N, Stennicke HR, Salvesen GS, Franke TF, Stanbridge E, et al. Regulation of cell death protease caspase-9 by phosphorylation. Science (1998) 282:1318–21. doi: 10.1126/science.282.5392.1318
64. Kermer P, Klöcker N, Labes M, Bahr M. Insulin-like growth factor-I protects axotomized rat retinal ganglion cells from secondary death via PI3-K-dependent akt phosphorylation and inhibition of caspase-3 in vivo. J Neurosci. (2000) 20:722–8. doi: 10.1523/JNEUROSCI.20-02-00722.2000
65. Zhou BP, Liao Y, Xia W, Spohn B, Lee MH, Hung MC. Cytoplasmic localization of p21Cip1/WAF1 by Akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nat Cell Biol. (2001) 3:245–52. doi: 10.1038/35060032
66. Fujita N, Sato S, Katayama K, Tsuruo T. Akt-dependent phosphorylation of p27Kip1 promotes binding to 14-3-3 and cytoplasmic localization. J Biol Chem. (2002) 277:28706–13. doi: 10.1074/jbc.M203668200
67. Feng J, Tamaskovic R, Yang Z, Brazil DP, Merlo A, Hess D, et al. Stabilization of Mdm2 via decreased ubiquitination is mediated by protein kinase B/Akt-dependent phosphorylation. J Biol Chem. (2004) 279:35510–7. doi: 10.1074/jbc.M404936200
68. Lopez-Pajares V, Kim MM, Yuan ZM. Phosphorylation of MDMX mediated by Akt leads to stabilization and induces 14-3-3 binding. J Biol Chem. (2008) 283:13707–13. doi: 10.1074/jbc.M710030200
69. Hedrick SM. The cunning little vixen: foxo and the cycle of life and death. Nat Immunol. (2009) 10:1057–63. doi: 10.1038/ni.1784
70. Dijkers PF, Medema RH, Lammers JW, Koenderman L, Coffer PJ. Expression of the pro-apoptotic Bcl-2 family member Bim is regulated by the forkhead transcription factor FKHR-L1. Curr Biol. (2000) 10:1201–4. doi: 10.1016/S0960-9822(00)00728-4
71. You H, Pellegrini M, Tsuchihara K, Yamamoto K, Hacker G, Erlacher M, et al. FOXO3a-dependent regulation of Puma in response to cytokine/growth factor withdrawal. J Exp Med. (2006) 203:1657–63. doi: 10.1084/jem.20060353
72. Stahl M, Dijkers PF, Kops GJ, Lens SM, Coffer PJ, Burgering BM, et al. The forkhead transcription factor FoxO regulates transcription of p27Kip1 and Bim in response to IL-2. J Immunol. (2002) 168:5024–31. doi: 10.4049/jimmunol.168.10.5024
73. Liu R, Wang L, Chen G, Katoh H, Chen C, Liu Y, et al. FOXP3 up-regulates p21 expression by site-specific inhibition of histone deacetylase 2/histone deacetylase 4 association to the locus. Cancer Res. (2009) 69:2252–9. doi: 10.1158/0008-5472.CAN-08-3717
74. Gross DN, Wan M, Birnbaum MJ. The role of FOXO in the regulation of metabolism. Curr Diab Rep. (2009) 9:208–14. doi: 10.1007/s11892-009-0034-5
75. Marr MTII, D'alessio JA, Puig O, Tjian R. IRES-mediated functional coupling of transcription and translation amplifies insulin receptor feedback. Genes Dev. (2007) 21:175–83. doi: 10.1101/gad.1506407
76. Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature (1995) 378:785–9. doi: 10.1038/378785a0
77. Rountree CB, Ding W, He L, Stiles B. Expansion of CD133-expressing liver cancer stem cells in liver-specific phosphatase and tensin homolog deleted on chromosome 10-deleted mice. Stem Cells (2009) 27:290–9. doi: 10.1634/stemcells.2008-0332
78. Ding W, You H, Dang H, Leblanc F, Galicia V, Lu SC, et al. Epithelial-to-mesenchymal transition of murine liver tumor cells promotes invasion. Hepatology (2010) 52:945–53. doi: 10.1002/hep.23748
79. Debebe A, Medina V, Chen CY, Mahajan IM, Jia C, Fu D, et al. Wnt/beta-catenin activation and macrophage induction during liver cancer development following steatosis. Oncogene (2017) 36:6020–9. doi: 10.1038/onc.2017.207
80. Groszer M, Erickson R, Scripture-Adams DD, Lesche R, Trumpp A, Zack JA, et al. Negative regulation of neural stem/progenitor cell proliferation by the Pten tumor suppressor gene in vivo. Science (2001) 294:2186–9. doi: 10.1126/science.1065518
81. Kwon CH, Zhu X, Zhang J, Knoop LL, Tharp R, Smeyne RJ, et al. Pten regulates neuronal soma size: a mouse model of Lhermitte-Duclos disease. Nat Genet. (2001) 29:404–11. doi: 10.1038/ng781
82. Bachman ES, Dhillon H, Zhang CY, Cinti S, Bianco AC, Kobilka BK, et al. betaAR signaling required for diet-induced thermogenesis and obesity resistance. Science (2002) 297:843–5. doi: 10.1126/science.1073160
83. Yilmaz OH, Valdez R, Theisen BK, Guo W, Ferguson DO, Wu H Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature (2006) 441:475–2. doi: 10.1038/nature04703
84. Zhang J, Grindley JC, Yin T, Jayasinghe S, He XC, Ross JT, et al. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature (2006) 441:518–22. doi: 10.1038/nature04747
85. Leung AK, Robson WL. Tuberous sclerosis complex: a review. J Pediatr Health Care (2007) 21:108–14. doi: 10.1016/j.pedhc.2006.05.004
86. Li Y, Corradetti MN, Inoki K, Guan KL. TSC2: filling the GAP in the mTOR signaling pathway. Trends Biochem Sci. (2004) 29:32–8. doi: 10.1016/j.tibs.2003.11.007
87. Jia C, Medina V, Liu C, He L, Qian D, Taojian T, et al. Crosstalk of LKB1- and PTEN-regulated signals in liver morphogenesis and tumor development. Hepatol Commun. (2017) 1:153–67. doi: 10.1002/hep4.1027
88. Stiles B, Wang Y, Stahl A, Bassilian S, Lee WP, Kim YJ, et al. Liver-specific deletion of negative regulator Pten results in fatty liver and insulin hypersensitivity [corrected]. Proc Natl Acad Sci USA. (2004) 101:2082–7. doi: 10.1073/pnas.0308617100
89. Stiles BL, Kuralwalla-Martinez C, Guo W, Gregorian C, Wang Y, Tian J, et al. Selective deletion of Pten in pancreatic beta cells leads to increased islet mass and resistance to STZ-induced diabetes. Mol Cell Biol. (2006) 26:2772–81. doi: 10.1128/MCB.26.7.2772-2781.2006
90. Planchon SM, Waite KA, Eng C. The nuclear affairs of PTEN. J Cell Sci. (2008) 121:249–53. doi: 10.1242/jcs.022459
91. Freeman DJ, Li AG, Wei G, Li HH, Kertesz N, Lesche R, et al. PTEN tumor suppressor regulates p53 protein levels and activity through phosphatase-dependent and -independent mechanisms. Cancer Cell (2003) 3:117–30. doi: 10.1016/S1535-6108(03)00021-7
92. Tang Y, Eng C. p53 down-regulates phosphatase and tensin homologue deleted on chromosome 10 protein stability partially through caspase-mediated degradation in cells with proteasome dysfunction. Cancer Res. (2006a) 66:6139–48. doi: 10.1158/0008-5472.CAN-06-0772
93. Tang Y, Eng C. PTEN autoregulates its expression by stabilization of p53 in a phosphatase-independent manner. Cancer Res. (2006) 66:736–42. doi: 10.1158/0008-5472.CAN-05-1557
94. Shen WH, Balajee AS, Wang J, Wu H, Eng C, Pandolfi PP, et al. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell (2007) 128:157–70. doi: 10.1016/j.cell.2006.11.042
95. Song MS, Carracedo A, Salmena L, Song SJ, Egia A, Malumbres M, et al. Nuclear PTEN regulates the APC-CDH1 tumor-suppressive complex in a phosphatase-independent manner. Cell (2011) 144:187–99. doi: 10.1016/j.cell.2010.12.020
96. Di Cristofano A, Pesce B, Cordon-Cardo C, Pandolfi PP. Pten is essential for embryonic development and tumour suppression. Nat Genet. (1998) 19:348–55. doi: 10.1038/1235
97. Suzuki A, De La Pompa JL, Stambolic V, Elia AJ, Sasaki T, Del Barco Barrantes I, et al. High cancer susceptibility and embryonic lethality associated with mutation of the PTEN tumor suppressor gene in mice. Curr Biol. (1998) 8:1169–78. doi: 10.1016/S0960-9822(07)00488-5
98. Sun H, Lesche R, Li DM, Liliental J, Zhang H, Gao J, et al. PTEN modulates cell cycle progression and cell survival by regulating phosphatidylinositol 3,4,5,-trisphosphate and Akt/protein kinase B signaling pathway. Proc Natl Acad Sci USA. (1999) 96:6199–204. doi: 10.1073/pnas.96.11.6199
99. Podsypanina K, Ellenson LH, Nemes A, Gu J, Tamura M, Yamada KM, et al. Mutation of Pten/Mmac1 in mice causes neoplasia in multiple organ systems. Proc Natl Acad Sci USA. (1999) 96:1563–8. doi: 10.1073/pnas.96.4.1563
100. Eng C, Peacocke M. PTEN and inherited hamartoma-cancer syndromes. Nat Genet. (1998) 19:223. doi: 10.1038/897
101. Knobbe CB, Lapin V, Suzuki A, Mak TW. The roles of PTEN in development, physiology and tumorigenesis in mouse models: a tissue-by-tissue survey. Oncogene (2008) 27:5398–415. doi: 10.1038/onc.2008.238
102. Ogg S, Ruvkun G. The C. elegans PTEN homolog, DAF-18, acts in the insulin receptor-like metabolic signaling pathway. Mol Cell. (1998) 2:887–93. doi: 10.1016/S1097-2765(00)80303-2
103. Goberdhan DC, Paricio N, Goodman EC, Mlodzik M, Wilson C. Drosophila tumor suppressor PTEN controls cell size and number by antagonizing the Chico/PI3-kinase signaling pathway. Genes Dev. (1999) 13:3244–58. doi: 10.1101/gad.13.24.3244
104. Scanga SE, Ruel L, Binari RC, Snow B, Stambolic V, Bouchard D, et al. The conserved PI3′K/PTEN/Akt signaling pathway regulates both cell size and survival in Drosophila. Oncogene (2000) 19:3971–7. doi: 10.1038/sj.onc.1203739
105. Sano H, Kane S, Sano E, Miinea CP, Asara JM, Lane WS, et al. Insulin-stimulated phosphorylation of a Rab GTPase-activating protein regulates GLUT4 translocation. J Biol Chem. (2003) 278:14599–602. doi: 10.1074/jbc.C300063200
106. Xiong S, Salazar G, San Martin A, Ahmad M, Patrushev N, Hilenski L, et al. PGC-1 alpha serine 570 phosphorylation and GCN5-mediated acetylation by angiotensin II drive catalase down-regulation and vascular hypertrophy. J Biol Chem. (2010) 285:2474–87. doi: 10.1074/jbc.M109.065235
107. Kurlawalla-Martinez C, Stiles B, Wang Y, Devaskar SU, Kahn BB, Wu H. Insulin hypersensitivity and resistance to streptozotocin-induced diabetes in mice lacking PTEN in adipose tissue. Mol Cell Biol. (2005) 25:2498–510. doi: 10.1128/MCB.25.6.2498-2510.2005
108. Haeusler RA, Hartil K, Vaitheesvaran B, Arrieta-Cruz I, Knight CM, Cook JR, et al. Integrated control of hepatic lipogenesis versus glucose production requires FoxO transcription factors. Nat Commun. (2014) 5:5190. doi: 10.1038/ncomms6190
109. He L, Hou X, Kanel G, Zeng N, Galicia V, Wang Y, et al. The critical role of AKT2 in hepatic steatosis induced by PTEN loss. Am J Pathol. (2010) 176:2302–8. doi: 10.2353/ajpath.2010.090931
110. Porstmann T, Santos CR, Griffiths B, Cully M, Wu M, Leevers S, et al. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. (2008) 8:224–36. doi: 10.1016/j.cmet.2008.07.007
111. Yecies JL, Zhang HH, Menon S, Liu S, Yecies D, Lipovsky AI, et al. Akt stimulates hepatic SREBP1c and lipogenesis through parallel mTORC1-dependent and independent pathways. Cell Metab. (2011) 14:21–32. doi: 10.1016/j.cmet.2011.06.002
112. Du X, Kristiana I, Wong J, Brown AJ. Involvement of Akt in ER-to-Golgi transport of SCAP/SREBP: a link between a key cell proliferative pathway and membrane synthesis. Mol Biol Cell (2006) 17:2735–45. doi: 10.1091/mbc.e05-11-1094
113. Palian BM, Rohira AD, Johnson SA, He L, Zheng N, Dubeau L, et al. Maf1 is a novel target of PTEN and PI3K signaling that negatively regulates oncogenesis and lipid metabolism. PLoS Genet. (2014) 10:e1004789. doi: 10.1371/journal.pgen.1004789
114. Galicia VA, He L, Dang H, Kanel G, Vendryes C, French BA, et al. Expansion of hepatic tumor progenitor cells in Pten-null mice requires liver injury and is reversed by loss of AKT2. Gastroenterology (2010) 139:2170–82. doi: 10.1053/j.gastro.2010.09.002
115. Matsumoto M, Han S, Kitamura T, Accili D. Dual role of transcription factor FoxO1 in controlling hepatic insulin sensitivity and lipid metabolism. J Clin Invest. (2006) 116:2464–72. doi: 10.1172/JCI27047
116. Gottlob K, Majewski N, Kennedy S, Kandel E, Robey RB, Hay N. Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev. (2001) 15:1406–18. doi: 10.1101/gad.889901
117. Antico Arciuch VG, Galli S, Franco MC, Lam PY, Cadenas E, Carreras MC, et al. (2009). Akt1 intramitochondrial cycling is a crucial step in the redox modulation of cell cycle progression. PLoS ONE 4:e7523. doi: 10.1371/journal.pone.0007523
118. Li C, Li Y, He L, Agarwal AR, Zeng N, Cadenas E, et al. PI3K/AKT signaling regulates bioenergetics in immortalized hepatocytes. Free Radic Biol Med. (2013) 60:29–40. doi: 10.1016/j.freeradbiomed.2013.01.013
119. Nadal A, Marrero PF, Haro D. Down-regulation of the mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase gene by insulin: the role of the forkhead transcription factor FKHRL1. Biochem J. (2002) 366:289–97. doi: 10.1042/bj20020598
120. Ferber EC, Peck B, Delpuech O, Bell GP, East P, Schulze A. FOXO3a regulates reactive oxygen metabolism by inhibiting mitochondrial gene expression. Cell Death Differ. (2012) 19:968–79. doi: 10.1038/cdd.2011.179
121. Di W, Lv J, Jiang S, Lu C, Yang Z, Ma Z, et al. PGC-1: the energetic regulator in cardiac metabolism. Curr Issues Mol Biol. (2018) 28:29–46. doi: 10.21775/cimb.028.029
122. Villena JA, Kralli A. ERRalpha: a metabolic function for the oldest orphan. Trends Endocrinol Metab. (2008) 19:269–76. doi: 10.1016/j.tem.2008.07.005
123. Machida K, Ying Q, Ou J-HJ, Stiles B. Nuclear receptor 3B (NR3B): bridging mitochondrial reprogramming and pluripotency through crosstalk with Nanog. Trends Cell Mol Biol. (2016) 11:97–07.
124. Li Y, He L, Zeng N, Sahu D, Cadenas E, Shearn C, et al. Phosphatase and tensin homolog deleted on chromosome 10 (PTEN) signaling regulates mitochondrial biogenesis and respiration via estrogen-related receptor alpha (ERRalpha). J Biol Chem. (2013) 288:25007–24. doi: 10.1074/jbc.M113.450353
125. Piantadosi CA, Suliman HB. Mitochondrial transcription factor A induction by redox activation of nuclear respiratory factor 1. J Biol Chem. (2006) 281:324–33. doi: 10.1074/jbc.M508805200
126. Piao Y, Kim HG, Oh MS, Pak YK. Overexpression of TFAM, NRF-1 and myr-AKT protects the MPP(+)-induced mitochondrial dysfunctions in neuronal cells. Biochim Biophys Acta (2012) 1820:577–85. doi: 10.1016/j.bbagen.2011.08.007
127. Garcia-Cao I, Song MS, Hobbs RM, Laurent G, Giorgi C, De Boer VC, et al. Systemic elevation of PTEN induces a tumor-suppressive metabolic state. Cell (2012) 149:49–62. doi: 10.1016/j.cell.2012.02.030
128. Ortega-Molina A, Efeyan A, Lopez-Guadamillas E, Munoz-Martin M, Gomez-Lopez G, Canamero M, et al. Pten positively regulates brown adipose function, energy expenditure, and longevity. Cell Metab. (2012) 15:382–94. doi: 10.1016/j.cmet.2012.02.001
129. Wijesekara N, Konrad D, Eweida M, Jefferies C, Liadis N, Giacca A, et al. Muscle-specific Pten deletion protects against insulin resistance and diabetes. Mol Cell Biol. (2005) 25:1135–45. doi: 10.1128/MCB.25.3.1135-1145.2005
130. Yang KT, Bayan JA, Zeng N, Aggarwal R, He L, Peng Z, et al. Adult-onset deletion of Pten increases islet mass and beta cell proliferation in mice. Diabetologia (2014) 57:352–61. doi: 10.1007/s00125-013-3085-8
131. Bayan JA, Peng Z, Zeng N, He L, Chen J, Stiles BL. Crosstalk between activated myofibroblasts and beta cells in injured mouse pancreas. Pancreas (2015) 44:1111–20. doi: 10.1097/MPA.0000000000000431
132. Zeng N, Yang KT, Bayan JA, He L, Aggarwal R, Stiles JW, et al. PTEN controls beta-cell regeneration in aged mice by regulating cell cycle inhibitor p16ink4a. Aging Cell (2013) 12:1000–11. doi: 10.1111/acel.12132
133. Ononye SN, Shi W, Wali VB, Aktas B, Jiang T, Hatzis C, et al. Metabolic isoenzyme shifts in cancer as potential novel therapeutic targets. Breast Cancer Res Treat. (2014) 148:477–88. doi: 10.1007/s10549-014-3194-1
134. Saltiel AR. New perspectives into the molecular pathogenesis and treatment of type 2 diabetes. Cell (2001) 104:517–29. doi: 10.1016/S0092-8674(01)00239-2
135. Brown MS, Goldstein JL. Selective versus total insulin resistance: a pathogenic paradox. Cell Metab. (2008) 7:95–6. doi: 10.1016/j.cmet.2007.12.009
136. Moon BC, Hernandez-Ono A, Stiles B, Wu H, Ginsberg HN. Apolipoprotein B secretion is regulated by hepatic triglyceride, and not insulin, in a model of increased hepatic insulin signaling. Arterioscler Thromb Vasc Biol. (2012) 32:236–46. doi: 10.1161/ATVBAHA.111.241356
137. Shearn CT, Mercer KE, Orlicky DJ, Hennings L, Smathers-McCullough RL, Stiles BL, et al. Short term feeding of a high fat diet exerts an additive effect on hepatocellular damage and steatosis in liver-specific PTEN knockout mice. PLoS ONE (2014) 9:e96553. doi: 10.1371/journal.pone.0096553
138. Zeng N, Li Y, He L, Xu X, Galicia V, Deng C, et al. Adaptive basal phosphorylation of eIF2alpha is responsible for resistance to cellular stress-induced cell death in Pten-null hepatocytes. Mol Cancer Res. (2011) 9:1708–17. doi: 10.1158/1541-7786.MCR-11-0299
139. Chen WT, Tseng CC, Pfaffenbach K, Kanel G, Luo B, Stiles BL, et al. Liver-specific knockout of GRP94 in mice disrupts cell adhesion, activates liver progenitor cells, and accelerates liver tumorigenesis. Hepatology (2014) 59:947–57. doi: 10.1002/hep.26711
140. He L, Gubbins J, Peng Z, Medina V, Fei F, Asahina K, et al. Activation of hepatic stellate cell in Pten null liver injury model. Fibrogenesis Tissue Repair (2016) 9:8. doi: 10.1186/s13069-016-0045-1
141. Shearn CT, Orlicky DJ, McCullough RL, Jiang H, Maclean KN, Mercer KE, et al. Liver-specific deletion of phosphatase and tensin homolog deleted on chromosome 10 significantly ameliorates chronic EtOH-induced increases in hepatocellular damage. PLoS ONE (2016) 11:e0154152. doi: 10.1371/journal.pone.0154152
Keywords: PTEN, PI3K, AKT, cancer, metabolism
Citation: Chen C-Y, Chen J, He L and Stiles BL (2018) PTEN: Tumor Suppressor and Metabolic Regulator. Front. Endocrinol. 9:338. doi: 10.3389/fendo.2018.00338
Received: 28 March 2018; Accepted: 05 June 2018;
Published: 09 July 2018.
Edited by:
Che-Pei Kung, School of Medicine, Washington University in St. Louis, United StatesReviewed by:
Roger Moorehead, University of Guelph, CanadaJean-Yves Scoazec, Institut Gustave Roussy, France
Copyright © 2018 Chen, Chen, He and Stiles. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bangyan L. Stiles, bstiles@usc.edu
†These authors have contributed equally to the work.