
Abstract
Epidermal growth factor receptor (EGFR) is an integral surface protein mediating cellular response to a number of growth factors. Its overexpression and increased activation due to mutations is one of the most common traits of many types of cancer. Development and clinical use of the agents, which block EGFR activation, became a prime example of the personalized targeted medicine. However, despite the obvious success in this area, cancer cure remains unattainable in most cases. Because of that, as well as the result of the search for possible ways to overcome the difficulties of treatment, a huge number of new treatment methods relying on the use of EGFR overexpression and its changes to destroy cancer cells. Modern data on the structure, functioning, and intracellular transport of EGFR, its natural ligands, as well as signaling cascades triggered by the EGFR activation, peculiarities of the EGFR expression and activation in oncological disorders, as well as applied therapeutic approaches aimed at blocking EGFR signaling pathway are summarized and analyzed in this review. Approaches to the targeted delivery of various chemotherapeutic agents, radionuclides, immunotoxins, photosensitizers, as well as the prospects for gene therapy aimed at cancer cells with EGFR overexpression are reviewed in detail. It should be noted that increasing attention is being paid nowadays to the development of multifunctional systems, either carrying several different active agents, or possessing several environment-dependent transport functions. Potentials of the systems based on receptor-mediated endocytosis of EGFR and their possible advantages and limitations are discussed.
Similar content being viewed by others

INTRODUCTION
Epidermal growth factor receptor (EGFR) is among one of the most studied proteins so that nearly 100,000 scientific publications are available in the PubMed database describing investigation of its structure and properties as well as functioning in norm and pathology. Despite that, multiple aspects of its involvement in regulation of the processes occurring in an organism remain poorly understood, whereas steadily increasing set of compounds affecting it as well as assessment of relevant impacts require regular systemic analysis allowing to choose the most promising research directions and develop novel pharmaceuticals. Being an integral surface receptor, EGFR mediates external signaling from entire family of regulatory signals, most renowned among which are epidermal growth factor (EGF) and transforming growth factor alpha (TGFα) [1]. Interaction of such extracellular signaling molecules with EGFR results in several activated intracellular cascades such as RAS/MAPK(ERK), PI3K/AKT/mTOR, and JAK/STAT, thereby promoting proliferation, apoptosis inhibition, and cell survival [2, 3]. Involvement of EGFR in carcinogenesis as well as possibility to use it as a target for treating oncological disorders attract special attention. EGFR activation represents one of the factors contributing to epithelial-mesenchymal transition resulting in emergence of cancer stem cell phenotype [4]. Mutations in this gene and its upregulated expression often account for emergence and progression of malignancies [5]. Increased amounts of EGFR have been detected in numerous oncological disorders (see “EGFR expression in oncological disorders” Section). Hence, EGFR obviously represents one of the most promising objects for targeted therapy. For instance, cetuximab, panitumumab, nimotuzumab and necitumumab being anti-EGFR monoclonal antibodies that prevent EGFR activation by natural ligands have been widely used in clinical practice. Another type of targeted therapy affecting EGFR is presented by the specific phosphorylation inhibitors including gefitinib, erlotinib, lapatinib, afatinib, etc. (see “Advances and limitations for targeted EGFR blockade” Section). Unfortunately, not all tumor types exhibiting upregulated expression of EGFR or gain-of-function mutations in the relevant gene and subsequent constitutively activated phosphorylation respond to the afore-mentioned antibodies and inhibitors due to the independent activation of the downstream signaling pathways (RAS BRAF kinase family), primarily KRAS and BRAF, thereby rendering EGFR targeting inefficient.
In this case, therapeutic inefficiency could be overcome by using EGFR-mediated endocytosis as a means for specific targeted delivery of diverse primarily cytotoxic agents into the cancer cells with upregulated EGFR expression allowing elimination of these cells regardless of activated downstream signaling pathways. For this purpose, cytotoxic agents conjugated with EGFR ligands (both natural and synthetic) as well as more complicated molecular and supramolecular constructs are being developed. The latter may include solid-core as well as liposomal, micellar, and similar type nanoparticles loaded with cytotoxic compounds. On the other hand, EGFR may be alternatively used to transport genes with the help of viral vectors or artificial delivery system that encode enzymes converting prodrugs into cytotoxic agents. Current approaches for the targeted delivery of cytotoxic agents into cancer cells exhibiting upregulated EGFR levels are presented in this review as well as their potentials and limitations.
EGFR AS A COMPONENT OF SIGNALING NETWORK
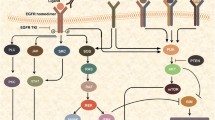
Activated EGFR initiates a set of biological reactions including cell proliferation, migration, and survival, which normally ensure epithelial integrity and regeneration. It is these properties that make the changes in the EGFR regulation important and often essential in the development of multiple types of oncological disorders. This receptor belongs to one of the tyrosine kinase families (ErbB or HER) that include four closely related membrane receptors: EGFR/ErbB1/HER1, Neu/ErbB2/HER2, ErbB3/HER3, ErbB4/HER4. All human ErbB (stands for avian erythroblastic leukemia viral oncogene B homolog – avian erythroblastic leukemia viral (v-erb-b) oncogene homolog) receptors are large transmembrane proteins containing cysteine-rich extracellular part, a single transmembrane-spanning segment, and intracellular cytosolic part consisting of a juxtamembrane domain, a tyrosine kinase domain, and a C-terminal domain [6]. Similar to other ErbB, extracellular EGFR part is subdivided into the four domains. Domains I and III of EGFR are required for ligand binding that initiates changes in the receptor conformation resulting in emergence of the protruding arm in the domain II of the extracellular region. This protruding arm is capable of interacting with the respective domain in another ErbB molecule. Normally, the emergence of this dimerization arm in inactive state is prevented by the interaction of domain II with domain IV of the receptor. Interaction of a ligand with the EGFR domains I and III releases the arm facilitating contact with the neighboring receptor [7]. Inactive EGFR molecules on the cell surface exist mainly as an equilibrium mixture of monomers, inactive dimers, and some oligomers [8]. EGFR activation is controlled by relevant external ligands and results in formation of its own homodimers and heterodimers with other ErbBs. Binding of an activating ligand leads to the equilibrium shift towards dimerization followed by formation of active dimers as well as activation of pre-existing inactive dimers. As a consequence, the tyrosine kinase domain becomes activated that is accompanied by specific phosphorylation of the tyrosine residues within the cytoplasmic region of the partner receptor in the dimer. Next, the motifs bearing phosphorylated tyrosine moieties bind the relevant intracellular signaling molecules initiating further activation of the inter-connected intracellular signaling cascades [9]. Thus, the signaling initiated by binding of the extracellular regulatory ligand to the EGFR is realized via phosphorylation of not only EGFR due to homodimerization, but also of its partner molecules, ErbB2 in particular, that lacks its own extracellular regulatory ligand [1]. To a much lesser extent, signal transduction from EGFR is conferred inside the cells by the remaining receptors of the same family called ErbB3 [10, 11] and ErbB4 [12], which are tissue-specific and expressed mainly in the nervous tissue. In addition, it was also demonstrated that apart from the related ErbB receptors, the plasma membrane EGFR may dimerize with other receptor tyrosine kinases. Interaction of active EGFR with platelet-derived growth factor beta receptor [13, 14], hepatocyte growth factor receptor / mesenchymal-epithelial transition factor receptor (MET) [15], and RON related to the latter [16], as well as insulin-like growth factor 1 receptor (IGF1R) [17] results in their subsequent phosphorylation followed by physiologically and therapeutically significant activation of the relevant downstream intracellular pathways (Fig. 1). It implies that cell response to EGFR ligand binding is determined not only by its surface expression and density, but also depends on composition and amount of the partner receptors. Moreover, cell response to external cues also depends on biological properties exerted by the growth factors interacting with EGFR (see “Natural EGFR ligands” Section).

Generalized scheme depicting major EGFR-activated signaling pathways. I-IV, EGFR extracellular domains; TK, tyrosine kinase domain; P, phosphorylated tyrosine residues; MET, mesenchymal-epithelial transition factor receptor; IGF1R, insulin-like growth factor 1 receptor. (Colored versions of Figs. 1 and 2 are available in online version of the article and can be accessed at: https://www.springer.com/journal/10541)
The additional level of signal transduction regulation provides the need to meet another receptor that depends on its diffusion properties and microdistribution within the plasma membrane. Some EGFR molecules exist as oligomers rendering that phosphorylation can occur in several receptors in response to binding of a single ligand molecule concentration of which is low [18]. Tracking individual EGFR molecules demonstrated that EGFR mobility alternated between several short-lived states: free, confined, and immobile states. In the immobile state, EGFR tends to form oligomers in the clathrin-coated pits. Mutual phosphorylation of receptors in the pits further enhances the transduced signal [19]. Apparently, oligomers could be coupled with other endocytosis-mediating structures [20].
Phosphorylation of EGFR and its dimerization partner ErbB2, which is frequently overexpressed in some cancer types, stimulates intracellular regulatory pathways such as RAS/RAF/MEK/ERK, PI3K/AKT/TOR, Src-kinases, and STAT transcription factors [3]. These pathways can be considered as components of more complex regulatory network containing large numbers of positive and negative feedback loops. The signaling network affected by EGFR contains over 300 components involved in more than 200 reactions [21]. EGFR signaling is fine-tuned intracellularly via multiple coordinated mechanisms including regulation mediated by phosphatases, feedback from the downstream components in the signaling pathway, endocytosis, and intracellular transport [21]. Specificity, direction, and magnitude of the cell response are determined by expression of the positive and negative regulators, activating ligand, components of the receptor dimers, as well as by the set of proteins interacting with phosphorylated tyrosine residues of the C-terminal domains of the receptor tyrosine kinases.
NATURAL EGFR LIGANDS
Normally, activation of the EGFR tyrosine kinase function is strictly regulated by quite extended set of natural external polypeptide ligands. This regulation is concentration-dependent so that EGF exhibits much higher affinity to the monomeric rather than dimeric receptor with one EGF molecule already bound, which can be interpreted as a negative cooperativity [22]. EGF is the most known EGFR ligand. Other natural ligands display structural similarity mainly represented by the three disulfide bridges accounting for rigid three-dimensional protein structure (EGF-like domain). This particular three-dimensional structure together with the specific amino acid sequences interacting with the extracellular domain I and III to a large extent determine interaction with the receptor binding site [23]. Majority of the ligands of the ErbB receptors are generated from large transmembrane precursor proteins via proteolytic cleavage resulting in production of soluble growth factors. Currently, more than a dozen of natural polypeptides able to interact with extracellular ERGR region with varying efficacy and impact have been identified. EGF, TGFα, and amphiregulin bind to EGFR, whereas betacellulin, heparin-binding epidermal growth factor, and epiregulin can also activate ErbB4 together with EGFR [9]. Recently it was shown that EGFR becomes activated following binding of angiogenin – one of the key regulators of angiogenesis. Angiogenin exhibits high affinity to EGFR (~40 nM) as well as a weak RNase activity that reflects its other name – RNase 5 [24, 25]. Two growth factors called neuregulin 2α and 2β commonly bind to their principle receptors ErbB3 and ErbB4, but can also bind and activate EGFR [26, 27]. In addition to agonists, EGFR antagonists have been also described. In particular, proteoglycan decorin binds to EGFR with nanomolar affinity [28] and lowers its cell surface exposure due to caveolae-mediated endocytosis and subsequent receptor degradation [29]. Decorin homologue – cleaved soluble ectodomain of LRIG1 (Leucine-rich repeats and immunoglobulin-like domains-1) domain – is also able to interact with EGFR with nanomolar affinity and compete with EGF for specific binding [30]. Macrophage migration inhibitory factor (MIF) known as regulator of innate immunity can block EGFR activation by binding to its ectodomain [31].
It became clear in recent years that various natural EGFR ligands stabilized structurally different dimer receptors causing diverse physiological outcomes [32]. Epiregulin and epigen, which could be considered as partial EGFR agonists, induce formation of less stable short-lived dimers than the ones formed following binding of EGF and TGFα. Nonetheless, such attenuated receptor dimerization results in more prolonged signal transduction causing breast cancer cell differentiation without cell proliferation contrary to the EGF effect [32]. Natural EGFR ligands can also differ in their potential to induce formation of dimers of the ErbB receptor family that can also trigger various biological effects upon activation of EGFR by different ligands in the same cell. Radioligand binding assay demonstrated higher affinity of EGF and TGFα to EGFR/ErbB2 heterodimers than to EGFR homodimers, whereas betacellulin and amphiregulin revealed similar affinity to both dimers. Amphiregulin acts as a partial agonist, which is manifested by the lower level of receptor phosphorylation as compared to the one initiated by EGF, TGFα, and betacellulin [33]. Compared to other ligands, amphiregulin demonstrates a two-phase kinetics in dimer formation allowing to assume that it could activate EGFR by binding both to monomers and pre-formed inactive dimers. In contrast, EGF, TGFα and betacellulin seem to mainly activate relevant receptors via binding to monomeric receptor and its dimerization [33]. Thus, different biological responses caused by binding diverse EGFR ligands could result from several processes: full or partial agonism with regard to dimer formation, differences in kinetic of generation of the dimers of active receptors, as well as in proportion of formed ErbB heterodimers versus homodimers.
In addition, affinity of EGFR ligands can be also controlled via methylation of its extracellular domain by arginine methyltransferase (PRMT1) that provides a three-fold increase of the ligand affinity towards EGFR [34]. PRMT1 knock-down or lack of relevant arginine residues (R198 and R200) decrease binding affinity of EGFR to EGF and TGFα [34] as well as angiogenin/RNase 5 [35]. Methylation of these arginine residues destabilizes interaction between the extracellular domains II/IV in EGFR, which, in turn, facilitates contacts between the domains I/III altering the receptor conformation and causing dimerization. Exogenous PRMT1 expression enhances EGFR ligand affinity and activates further receptor-mediated signaling events [34].
Currently accumulated evidence regarding binding of growth factors and other natural ligands to EGFR could hardly be described in full detail by the Langmuir adsorption isotherm with a single dissociation constant and single cell binding site, which represents an oversimplified model of the real process. Availability of negative cooperativity, dimer formation with partner receptors, known covalent modifications affecting binding, cell heterogeneity, as well as dynamic process of changing accessible receptors on the cell surface are clear manifestation of this. One can speak more confidently about the range where dissociation constants of the ligand–receptor complex lie. EGF binding constants range from tens of picomoles to several nanomoles per liter. For different objects it can reflect both the presence of negative cooperativity and the influence of another factor. The latter can be illustrated by complex formation with the partner receptors (e.g., with ErbB2), varying extent of arginine methylation at sites crucial for binding, as well as mutations found in some receptor genes. Furthermore, such variants can be also described by availability of subpopulations of receptor displaying different affinities.
ENDOCYTOSIS AND SUBSEQUENT INTRACELLULAR TRANSPORT
Similar to numerous cell surface receptors mediating response to external regulatory signals, EGFR uses endocytosis as a common means to reduce the response magnitude via negative feedback loop. Additional regulation is provided by sorting of internalized receptors, which may be directed back to the plasma membrane, for degradation inside the lysosomes, or towards some other intracellular compartments, wherein it may further regulate cellular processes [36]. In the absence of ligands EGFR is internalized very slowly: cultured cells expressing moderate amount of EGFR (<200,000 receptors per cell) display t½ (half-life) within the range of 6-10 h, whereas for the cells with aberrantly overexpressed EGFR level such as A431 human epidermoid carcinoma (~2,000,000 receptors per cell) the t½ value could be as long as 24 h or even more. Ligand binding and tyrosine kinase activation result in rapid endocytosis increasing EGFR endocytosis rate constant manifold [37]. Cytoplasmic regions of EGFR bear several internalization motifs unmasked upon ligand binding. Among them are two different motifs for recognizing subunits of the adaptor protein 2 (AP2) capable of binding membrane proteins to clathrin lattice [38]. Some phosphotyrosine residues located within the EGFR cytoplasmic tail after phosphorylation can also facilitate binding of E3 ubiquitin-ligase Cbl either directly [39] or via another adaptor protein Grb2 [40] that links phosphorylated receptor to E3 ubiquitin-ligase Cbl. It results in activation of the RAS/MAPK signaling cascade and EGFR endocytosis. Duplication of interaction between Cbl and EGFR is necessary for stable Cbl recruitment and efficient receptor ubiquitination [41]. It must be noted that multiple ubiquitination is not the only factor affecting EGFR endocytosis, because replacing even as many as 15 lysine residues in its kinase region that almost fully suppresses ubiquitination do not result in markedly suppressed receptor internalization [42]. Clathrin-dependent endocytosis of the ligand-activated EGFR is controlled by several mechanisms operating cooperatively providing exhaustive control, including ubiquitination of the receptor kinase domain and C-terminal lysine residues, as well as adaptor proteins AP-2 and Grb2 [43]. Complexity to EGFR endocytosis regulation is added by another mechanism of covalent receptor modification taking place along with ubiquitination. EGFR also undergoes modification by ubiquitin-like molecule Nedd8 (Neural Precursor Cell Expressed, Developmentally Down-Regulated 8) [44]. This modification is catalyzed by Cbl in complex with Nedd8-specific ligase E2 (Ubc12) and promotes ubiquitin binding with the help of Cbl and UbcH7 ligase. Such complexity and multiple duplications of mechanisms involved in internalization of the ligand-activated EGFR make this process resistant to perturbations (e.g., mutations or altered expression of endocytosis-related components) [38]. In particular, AP2-binding motifs, multiple ubiquitination, and EGFR acetylation should be simultaneously aborted (e.g., via mutations) to suppress the clathrin-dependent EGFR endocytosis significantly [43]. Stability of the EGFR endocytosis process can be likely explained by existence of clathrin-dependent and clathrin-independent processes. Cells are able to remove surface ligand-activated EGFR via several routes: clathrin-dependent, clathrin-independent, endophilin-dependent endocytosis and macropinocytosis [45]. All cell types expressing EGFR undergo internalization in response to added ligands via rapid clathrin-dependent endocytosis at all physiological EGF concentrations [36]. Significant proportion of EGFR that enter the cells via this way returns back to the plasma membrane [46], which can occur via two routes within varying time frame [47]. The clathrin-dependent endocytosis seems to act in a saturable manner that becomes more evident in the case of high EGFR expression, whereas the slower clathrin-independent endocytosis could be initiated in many cell types at higher concentration of added EGF (>2 nM) [36]. Moreover, endocytosis can also be affected by ligand saturation upon exposure to ligand at very high level exceeding several times dissociation constant for the ligand-receptor complex. Further increase of the ligand concentration lowers efficacy of its trafficking into cells via receptor-mediated endocytosis.
In addition to the clathrin-dependent endocytosis, there is also a rapid endophilin-dependent endocytosis that seems to be more active at the leading edges of migrating cells and required for spatially restricted EGF-signaling [45]. Such type of EGFR engulfment is mediated by CBL, adaptor protein CIN85, and controlled by dynamin activity similar to the clathrin-dependent endocytosis [45, 48].
The type of the EGFR endocytosis can depend on the receptor-bound ligand. For instance, in HeLa cells EGFR activated by EGF or TGFα binding is subjected solely to the clathrin-dependent endocytosis, whereas heparin-binding EGF-like growth factor (HB-EGF) and betacellulin stimulated both clathrin-dependent as well as clathrin-independent uptake [49]. EGFR internalization can be also mediated by macropinocytosis, which is observed after exposure of A431 cells to EGF [50, 51], or after formation of tubular-vesicular structures from the circular dorsal ruffles bearing dynamin and phosphatidylinositol 3-kinase (PI3) [51, 52]. It is worth-noting that EGFR ubiquitination is required for clathrin-independent endocytosis because mutations in the related motifs hindering ubiquitin binding also block endocytosis [41]. Such type of EGFR endocytosis depends on cholesterol-rich plasma membrane domains, but not caveolin [36].
Regardless of internalization route, EGFR enters early endosomes, wherein it undergoes sorting to regulate response to EGFR ligand binding. Routes of further EGFR trafficking substantially affect cellular regulatory events contributing to balanced response to input signals, which were summarized in detail in several reviews [36, 53]. It was noted above that destination to lysosomes for EGFR degradation resulting in down-modulated response to ligand-induced receptor stimulation and trafficking back to the plasma membrane to continue its signal response represented two major routes for transport of EGFR. However, small fraction of the receptors is delivered from endosomes into the endoplasmic reticulum (ER) via COPI transport vesicles [54]. In addition, alternative routes for EGFR nuclear and mitochondrial transport (see below) have been described. Mitochondrial transport of activated EGFR relies on c-Src and can result in phosphorylation of the cytochrome c-oxidase subunit COXII in the breast cancer cells, as shown after adding c-Src and EGFR to immunoprecipitated COXII or COXII-derived fragment [55]. Such EGFR trafficking facilitates resistance to apoptosis [56]. Furthermore, it was found that mitochondrial EGFR transport in non-small cell lung cancer cells resulted in enhanced invasiveness and metastasis spread [57, 58]. However, the proportion of EGFR found inside mitochondria is very low comprising as low as ~2% of phosphorylated EGFR [57].
NUCLEAR TRANSPORT OF EGFR
Intracellular juxtamembrane domain of EGFR contains three-part nuclear localization sequence (RRRHIVRKRTLRR) that mediates emergence of a small fraction of the full-sized ligand-activated receptor inside the cell nucleus [59]. Similar three-part nuclear localization signals are also found within the remaining ErbB molecules. EGFR transport from the plasma membrane towards the nucleus does not represent a common stable response to ligand receptor binding, but rather is observed in certain tissues or under certain circumstances [60]. Such trafficking occurs due to endocytosis and requires importin β1 implying that transport occurs through a nuclear pore complex [61]. Presence of intranuclear EGFR is a controlled process, which is assumed by the presence of active nuclear export signal within its structure [61]. EGFR is translocated into the nucleus being bound to EGF [62] and is found both inside the nucleoplasm as well as on the inner nuclear membrane [63]. EGFR ligands affect differently such trafficking: for instance, adding EGF, TGFα, HB-EGF and betacellulin led to an intranuclear dose-dependent EGFR accumulation in the human liver adenocarcinoma cells, whereas amphiregulin and epiregulin exerted no such effects [64]. Mechanisms resulting in EGFR nuclear accumulation remain poorly understood, it is likely that they are accomplished via several routes [60, 63, 65, 66]. In particular, EGFR can enter the ER from endosomes and exit into the cytosol due to a retrograde trafficking via Sec61translocon being involved both in transporting of de novo synthesized proteins into the ER as well as translocation of misfolded proteins back to the cytosol. Some studies demonstrated that EGFR interacted with Sec61β constituting one of the three subunits in the translocon [67, 68]. However, where exactly EGFR moves through the translocon, why receptor glycosylation does not interfere with it, as well as how it ends up inside the nucleus being bound to the ligand remains unclear. Moreover, it remains unclear what does provide EGFR solubility in nucleoplasm. Among the proposed nuclear trafficking routes exit of EGFR into the cytosol from the ER has been noted followed by movement through the nuclear pore complex and transport as an integral protein to the outer nuclear membrane and then through the peripheral channels of the nuclear pore complex. In addition, some endosomal fraction can also undergo nuclear trafficking followed by the fusion with nuclear envelope and transport through the nuclear pore complex to the inner nuclear membrane [69]. Because Sec61 was found at the inner nuclear membrane, the latter could allow EGFR to be directly translocated into the nucleus [67].
Intranuclear EGFR can interact with transcription factors STAT3, STAT5, E2F1, phosphorylate proliferating cell nuclear antigen (PCNA) ensuring accelerated proliferation and repair events as well as enhanced cell radioresistance. Owing to this, nuclear EGFR takes part in proliferation, tumorigenesis, metastasis spread, DNA repair as well as provides resistance to DNA-damaging radiation and alkylating anti-cancer agents [63].
EGFR EXPRESSION IN ONCOLOGICAL DISORDERS
EGFR activation upon malignant transformation can result from upregulated receptor expression, enhanced autocrine and paracrine production of activating growth factors, as well as altered intrinsic tyrosine kinase activity due to mutations. EGFR activation is often caused by combination of several factors, which complicates search for proper therapy.
One of the most common causes of EGFR activation is its upregulated expression, which could be due to not only gene amplification, but also enhanced transcription, translation, decreased receptor degradation, and altered expression of noncoding RNAs [70-72]. The data examining EGFR expression in oncological disorders presented in multiple studies vary a lot depending on the disease stage, applied therapeutic interventions, population, study methods (mRNA assay, immunohistochemistry), analyzed parameters, available antibodies, and duration of the sample storage. In some cases, quite strong increase of EGFR expression was observed. In particular, head and neck cancer samples contained EGFR mRNA amounts on average 69-fold higher than in normal tissues [73]. However, no direct unambiguous correlation between the mRNA and the relevant protein level was revealed, whereas immunohistochemistry-based expression was not strictly quantitative, and was additionally impacted by multiple factors not always taken into consideration [74]. In the case when several methods were used, immunohistochemical staining demonstrated lower prevalence of the overexpressed EGFR protein vs. mRNA level. In particular, the reported information revealed that in colorectal cancer overexpression of EGFR protein was detected in 61% cases examined with immunohistochemistry assays, whereas the relevant mRNA levels were increased in 79% cases, employing of either of the methods – detected EGFR overexpression in 82% cases [75]. Importantly, depending on the used commercially available antibodies detection of EGFR protein overexpression in non-small cell lung cancer samples varied within 5-56% range as was highlighted in the same study [76]. Nonetheless, it was precisely this type of assessing EGFR expression that allowed obtaining data on the receptor amount summarizing changes in its transcription, translation and protein degradation. Most often, investigators subdivide the data into several groups depending on intensity of the specimen staining (usually assigned to the four groups), assigning the enhanced expression to one or two groups with most prominent receptor expression. Analysis of a large number of conducted studies allows identifying oncological disorders with markedly increased EGFR expression (Table 1). All experimental studies cited in the table were conducted using immunohistochemistry staining. To provide more detained picture, we also describe the data from previous reviews, some of which cited studies based both on immunohistochemistry staining as well as other methods. It is also worth noting that such data are difficult to match with those obtained while dealing with cultured cells in terms of EGFR quantity per cell and correlate to the changes of the receptor level in the same tissue from healthy individuals. In some cases (bladder cancer, glioma, head and neck cancer, lung cancer) it provides the basis for developing local targeted therapeutic approaches employing radionuclides, photosensitizers, and nanoparticles reacting to various radiation types.
Overall, upregulated EGFR expression is one of the most common events recorded in oncological disorders. For instance, analysis of tumors with unknown primary site revealed EGFR expression in 55% cases, so that its amplified gene (17%) turned out to be the most prevalent among all the markers examined [77].
Heterogeneity of the changes triggering oncological disorders results in the situation when essentially distinct disease variants emerge from the tumors with the same localization. A thorough genetic profiling allows to elucidate cancer types within the same localization, which exhibit with high probability the same changes in the key regulatory proteins including EGFR. In particular, overexpressed EGFR was found in 97% cases of the classic glioblastoma multiforme [78].
ADVANCES AND LIMITATIONS OF TARGETED BLOCKING OF THE EGFR SIGNALING PATHWAY
EGFR serves as a crucial marker of malignant transformation as it controls one of the most common entry into in signaling pathway mediating regulation of cell proliferation. More importantly is that in many cases EGFR state or expression represent one of the essential steps in the cell malignant transformation. Altered EGFR activation often results in a continuous proliferative signaling so that the cell division becomes perpetual until the signal is interrupted. It should be noted that upregulated EGFR expression in solid tumors is often associated with increased production of its appropriate ligands that results in chronic receptor activation. Hence, it seems rational to interrupt EGFR activation that can be achieved either by blocking binding of agonist ligands or inhibiting receptor tyrosine kinase activity. The former is efficient in the case of upregulated EGFR level, whereas the latter – in the case of permanent receptor activation due to mutations especially while selecting inhibitors specific to the particular mutant.
Anti-EGFR monoclonal antibodies. A large number (currently reaching as many as ~40) of available diverse artificial polypeptides able to bind extracellular EGFR domains points out to the fact that interrupting of interaction between EGFR and its ligands seems to be in high demand [109]. In particular, they include chimeric antibodies, antibody fragments, single-domain antibodies, antibody mimetics engineered by using diverse scaffold proteins such as ankyrin-repeat-based DARPin, affibodies based on the protein A Z-domain, as well as adnectins based on the 10th domain of the type III fibronectin. A number of monoclonal antibodies such as cetuximab (Erbitux), panitumumab (Vectibix), nimotuzumab (Theraloc), necitumumab (Portrazza) have been successfully introduced into the common clinical practice (Table 2). These antibodies bind to the EGFR extracellular domain III preventing receptor activation by growth factors. Cetuximab is derived from the high-affinity murine monoclonal antibody (mAb) C225 fused to the human IgG1 constant region [110]. Importantly, IgG1 isotype in the structure of cetuximab also accounts for eliciting antibody-dependent cellular cytotoxicity by recruiting NK cells to eliminate cancer cells in contrast to panitumumab [111]. However, the latter is advantageous due to the fact that it is fully human monoclonal IgG antibody so that its use is less prone to trigger allergic reactions and anaphylaxis [112]. Since the mid-2000s these two first therapeutic monoclonal antibodies are widely used in clinical practice. In 2015, this panel of antibodies was supplemented with another human mAb called necitumumab [113] used to treat squamous cell non-small cell lung cancer. Compared to cetuximab and panitumumab, this antibody has much longer half-life (~2 weeks). Furthermore, nimotuzumab represents a humanized murine antibody characterized with less significant side effects compared to the rest of therapeutic antibodies [114] due to its EGFR affinity optimized for therapy that allows to exert effects mainly on cells with moderate-to-high receptor expression, but spare those with moderate surface level of the receptor [115, 116]. Moreover, there is a number of monoclonal antibodies targeting EGFR, which have passed several phases of clinical trials but received no approval for wide clinical use due to the lack of marked improvement in clinical outcomes compared to other therapeutic interventions. Among them are humanized IgG1 antibodies matuzumab and imgatuzumab, human zalutumumab and doligotuzumab that interact both with EGFR and ErbB3 [109].
The majority of currently used and developed blockers of EGFR binding display two mechanisms of action: inhibition of proliferative signaling and induction of antibody-dependent cellular cytotoxicity. However, cytotoxic component of their activity is unable to produce sufficient anti-cancer effect. Emergence of the gain-of-function mutations in the downstream elements of the signaling cascade such as well-known KRAS mutations usually nullifies blocking of the receptor-induced signal transduction. Tumors bearing wild type KRAS are sensitive to antibody blockade, whereas mutations in the codons 12 and 13 within exon 2 in the KRAS gene result in stabilization of the functionally active complex RAS-GTP and continuous signaling via the MAPK-pathway that renders such cancer cells insensitive to the blocking antibodies. This type of mutations is recorded in 40-45% and 15-30% cases of colorectal cancer and non-small cell lung cancer, respectively [117]. Gain-of-function mutations are not only restricted to the gene KRAS, and the cause of tumor resistance to EGFR blocking therapy can also be associated with the altered genes NRAS, BRAF, PIK3CA, PTEN, etc. [118] as well as expression of noncoding RNAs [119]. Moreover, a secondary resistance inevitably emerges after the initial therapeutic response to the EGFR blocking antibodies, which is likely caused by the mutated KRAS [120] and other genes [121].
EGFR tyrosine kinase inhibitors. Tremendous efforts have been made to develop and test EGFR phosphorylation inhibitors after elucidating the details of EGFR activation caused by binding to growth factors. Treatment of some oncological disorders with such inhibitory agents as gefitinib (Iressa), erlotinib (Tarceva), lapatinib (Tyverb), afatinib (Tovok), osimertinib (Tagrisso), brigatinib (Alunbrig), and dacomitinib (Vizimpro) [122] as well as icotinib (Conmana) (Table 3) was introduced into clinical practice [123]. Use of the first-generation peroral inhibitors (gefitinib and erlotinib) for treatment of non-small cell lung cancer (squamous cell carcinoma and adenocarcinoma) demonstrated much higher therapeutic efficacy than the standard chemotherapy [124]. Most common EGFR mutations are presented by deletion 746-750 in the exon 19 or L858R point mutation with leucine to arginine substitution [125]. Patients with gain-of-function EGFR mutations were found to be sensitive enough to the reversible first-generation inhibitors displaying manifold higher affinity to this mutant than to the wild type EGFR isoforms. Despite the fact that these inhibitors initially induced therapeutic effect, they did not function in the case of combined mutations and some other mutations, primarily T790M [126]. It stimulated introduction of the irreversible second-generation inhibitors with broader range of activity towards other ErbB receptors: afatinib and dacomitinib were supposed to overcome resistance of EGFR T790M mutation carriers to erlotinib and gefitinib. Such intervention did not demonstrate higher efficacy than the first-generation inhibitors due to inability to achieve sufficient therapeutic impact limited by toxicity for normal host tissues [127]. The use of more selective inhibitor osimertinib for treatment of non-small cell lung cancer was found to be more successful [128, 129]. Despite the noticeably prolonged progression-free survival period, the gradually developed drug resistance requires both the use of a combination of different inhibitors and the search for more effective interventions. Mutations emerging in the EGFR C797 locus – binding site of osimertinib – result in generation of the resistant tumor variants [130].
Most success in using EGFR tyrosine kinase inhibitors was reported for treatment of non-small cell lung cancer. Such lung cancer type is characterized by EGFR overexpression (around 50% cases) as well as mutations leading to its permanent activation [127]. Selection of tyrosine kinase inhibitors with varying sensitivity of the mutant EGFR isoforms to different inhibitors [131, 132] in combination with blocking antibodies could likely improve therapeutic efficacy [127]. Hence, development of the panel of EGFR inhibitor represents an exciting example of a personalized approach to cancer therapy relying on the structural data and mutations in one of the key components of carcinogenesis.
The necessity of permanent medication uses as well as insufficient eradication of cancer cells comprise common limitations of the EGFR inhibitor therapy. Prolonged use of inhibitors and presence of a large number of cancer cells results in emergence of therapy-insensitive variants and subsequent selection of most resistant clones [127]. Abolition of apoptosis suppression due to the permanently activated EGFR-triggered Ras/Raf/MEK/ERK signaling axis [133] turns out to be insufficient for tumor eradication. Along with the mutated EGFR gene, rising tumor resistance is also accounted for by other alterations in the signaling pathway controlling cell proliferation. Such disturbances include gain-of-function mutations in KRAS, BRAF, phosphatidylinositol-4,5-bisphosphate kinase catalytic subunit (PIK3CA), gene fusion affecting activation of RET, FGFR3, and BRAF, amplification of MET, FGFR, ErbB2, etc. [134-139], as well as expression of long noncoding RNAs, e.g., UCA1 (urothelial cancer-associated 1) [140]. In some cases, inhibitor treatment results in changing of the disease type, for example, transformation of the non-small cell lung cancer into the small cell lung cancer [141].
Thus, targeted therapy that is well illustrated by approaches targeting EGFR faces the same problem of emerging resistant cancer cells similar to the traditional chemotherapy. As in the aforementioned case, one of the ways to solve this issue could rely on selecting of a set of inhibitors blocking activated signaling pathway including not only EGFR inhibitors, but also those able to suppress downstream arms such as KRAS, BRAF, etc. Rapidly developing liquid biopsy techniques [142] and advanced sequencing methods such as the next generation sequencing (NGS) [143] make this problem technically solvable. Nonetheless, such approach does not guarantee an absolute success due to high heterogeneity, genetic instability, and clonal evolution of tumors [144-146]. Blocking proliferative signaling inhibits tumor growth, but does not cause its disappearance. Addition of toxic chemotherapeutic agents to the cancer treatment does not lead to tumor eradication in all cases. Hence, researches in many laboratories concentrate their attention on developing alternative approaches to address therapeutic resistance by applying a targeted intervention not to inhibit the pathologically altered steps in the normal regulatory processes, but to selectively eliminate cancer cells. Numerous variants of different oncological disorders exhibiting EGFR overexpression serve as promising candidates for developing such approaches.
THE USE OF EGFR AS A DELIVERY SYSTEM
Traditionally, chemotherapy is the most commonly used therapy of oncological disorders acting primarily on the rapidly dividing cells together with radiotherapy that mainly affects DNA and more effectively acting on the dividing cells. Both interventions are not selective towards exclusively cancer cells, which cause severe side effects complicating recovery. Apart from these most common methods, photodynamic therapy (PDT) and gene therapy have been developed and used in some cases. All these types of therapeutic interventions can be combined with the targeted therapy not only using simultaneous or sequential administration, but also for targeted delivery of some therapeutic agent [toxic chemotherapeutic substance, source of ionizing radiation, photosensitizer (PS), or key gene] into the target cancer cells. EGFR is considered as one of the most promising targets for creating targeted anticancer therapy due to its direct involvement in activating cell division, undergoing endocytosis, and frequently upregulated expression in many oncological disorders.
It was noted above (see “Endocytosis and subsequent intracellular transport” Section) that EGFR internalization increases by many orders of magnitude after stimulation with extracellular cognate ligands. This process depends on receptor dimerization and autophosphorylation, whereas anti-EGFR antibodies block it, but at least some of them are able to relatively effectively enter cells. In particular, variable region of the mAb 225 antibody ensures functionality of cetuximab and becomes endocytosed at the rate 6-7-fold lower than that one for EGF endocytosis [147]. Nevertheless, it markedly exceeds cell penetration rate of the ligand-free EGFR. It was shown experimentally that in A549 human adenocarcinoma cells moderately expressing EGFR (~105 receptors per cell) more than half of captured mAb and EGFR recirculate back to the cell surface within the first hour as compared to the EGFR-EGF complex, more than 70% of which is detained intracellularly to be further degraded in lysosomes. Interestingly, cetuximab enables trafficking of some EGFR into the ER, where it is found in complex with Sec61 and transported to the nucleus [148]. Such transport depended on interaction between the antibodies and the ligand binding site as well as on polyvalent antibody binding, because such trafficking was not observed for the blocking antibody targeting another receptor site and for the cetuximab Fab′-fragments [148]. Ligand–receptor complex clustering in glycosphingolipid domains is required for subsequent caveolin- and dynamin-2-dependent endocytosis of both cetuximab and nanoparticles coated with it [149]. Cetuximab-receptor complex clustering as an intermediate stage in the endocytosis initiation was also detected with the help of the single particle tracking-based reaction progress kinetic analysis [150]. The aforementioned data explains great interest to cetuximab as a ligand for delivery of cytotoxic agents into target cancer cells. Artificial antibody-like scaffold proteins allowing to spatially positioning of a set of amino acid residues ensuring interaction with a target molecule are considered as a promising type of ligands. Among them are small sized protein A-based affibodies [151]. Some of them effectively interact with EGFR, but also exhibit low endocytosis rate [152]. In addition, there is a short peptide GE11used for creating various delivery systems of anticancer agents, which is able to bind with EGFR facilitating internalization of nanoparticles coated with this peptide [153]. Along with antibodies and peptides utilized as artificial ligands capable of binding to EGFR, there are also aptamers. The RNA-aptamer E07 was demonstrated to be able to bind EGFR with nanomolar affinity, so that almost a quarter of the cell surface-bound aptamer could be internalized within 30 min [154].
Nanoparticles. Nanoparticles exhibit extremely heterogenous properties, and due to their size, they are capable to attach or to incorporate large amounts of low-molecular antitumor agents such as standard chemotherapeutic compounds. This combination alone could profoundly alter drug distribution and its pharmacokinetics. With respect to oncological disorders it is worth noting that attachment of antitumor agents to nanoparticles is performed in hope to achieve the enhanced permeability and retention effect (EPR) [155]. Hopes of successful use of nanoparticles in cancer treatment were based on the fact that at the sites of fast and chaotic tumor angiogenesis there is a significant number of large pores. However, clinical studies demonstrated that compared to murine experimental tumors the EPR level in human tumors was less pronounced and could differ substantially not only between the same type tumors in different patients, but also between various sites of metastasis in a single patient [156]. In attempts to overcome instability of nanoparticles it was suggested to make them functionally addressed by, for example, attaching EGFR-specific ligands [157, 158].
So far immunoliposomes loaded with doxorubicin as one of the most commonly used anticancer drugs is the only example of this most often used approach that reached clinical trial stage. To ensure their stability in blood such liposomes are coated with polyethylene glycol (PEG) moieties, whereas their specificity to cancer cells overexpressing EGFR is provided by the Fab′-fragment of cetuximab [159]. Such liposomes called anti-EGFR ILs-dox, on average, contain 4,000 doxorubicin molecules, elicit better tolerability compared to the free doxorubicin at a similar dose, and show no side effects associated with EGFR blockade. Currently, such kind of doxorubicin delivery into the cells with upregulated EGFR expression for the treatment of triple-negative breast cancer patients goes through the phase 2 clinical trial. Administration of such liposomes carrying Fab′-fragment of cetuximab to animals showed that they were able to enhance drug accumulation by 6-fold in MDA-MB-468 human breast cancer xenografts [160].
A whole set of similar delivery systems utilizing chemotherapeutic agents encapsulated into liposomes modified by EGFR ligands undergo preclinical trials (Fig. 2). Among them are immunoliposomes loaded with doxorubicin and cetuximab attached via folate binding protein [161], boron-containing anion for neutron-capture therapy, as well as cholesterol-cetuximab [162], cisplatin and nimotuzumab [163], 5-fluorouracil and cetuximab [164], liposomes containing zinc phthalocyanine PS and single-domain anti-EGFR antibody EGa1 [165], liposomes with oxaliplatin and EGF [166], cisplatin and anti-EGFR mAb [167], gemcitabine-loaded liposomes with attached PEG and anti-EGFR mAb [168]. It should be noted that such approaches with targeted delivery of anticancer agents showed an opportunity to overcome multi-drug resistance, which often poses problems for cancer therapy [160].

Schematic representation of major developed approaches for targeted EGFR-mediated delivery of therapeutic agents. 1 – Receptor binding; 2 – endocytosis: macropinocytosis, phagocytosis, caveolin, rapid clathrin-dependent, endophilin-dependent endocytosis; 3 – exit from endosomes; 4 – nuclear transport. Abbreviations: ITs, immunotoxins; MNTs, modular nanotransporters; NPs, nanoparticles; PS, photosensitizers.
All variants mentioned above could be beneficial for clinical practice, but the search for optimal drug, ligand, and route of attachment still continues. For instance, sortase that links the specific sequence LPETG at the C-terminus of the donor with the N-terminal pentaglycine was used to ensure homogeneous conjugation to liposomes [169]. Another approach for advancing anticancer agents relies on the use of several cytostatic compounds encapsulated in immunoliposomes. For example, immunoliposomes loaded with doxorubicin and vinorelbine together with the new single-domain scFv EGFR-s10 antibody were investigated [170]. In addition, a combined use can be illustrated by immunoliposomes loaded with cetuximab carrying doxorubicin and 188Re β-emitter as a potential theranostic agent [171]. Development of multifunctional delivery systems for pharmaceuticals comprises a general direction of search for most promising anticancer drugs. In case of the liposome-based delivery it is accomplished primarily by using functionalized liposomes, e.g., thermosensitive liposomes loaded with doxorubicin and EGFR peptide GE11 [172]. Such liposomes are stable at body temperature but become unstable upon temperature rising and release encapsulated drugs at 40°C, which can provide additional selectivity mediated by local heating. Moreover, sensitivity to environmental pH-value provides another type of modification for conferring additional functions to liposomes: immunoliposomes bearing anti-EGFR mAb could start releasing gemcitabine upon lowering pH [173], which can happen both in endosomes as well as within acidified areas of the extracellular space in some tumors.
Micelles represent another type of EGFR-targeted nanoparticles used for delivery of anticancer agents. For instance, pH-sensitive copolymer hydroxypropyl methacrylamide-lactate micelles loaded with doxorubicin were modified by nanoantibody EGa1 targeting EGFR. Such tailored micelles were able both to retard growth of head and neck cancer cell xenografts by blocking EGFR signaling as well as exhibit toxicity due to the delivered doxorubicin [174]. Similar micellar construct carrying paclitaxel was generated based on the Poly Lactic-co-Glycolic Acid (PLGA)-PEG copolymer loaded with cetuximab [175]. Another type of nanoparticles co-loaded with paclitaxel and quantum dots for theranostics were generated that contained phospholipid-conjugated PEG and conjugated with cetuximab or aptamer serving as ligand to EGFR. The micelles coupled with EGFR ligands suppressed growth of pancreatic cancer xenografts to a significantly higher degree than the non-targeted variants of nanoparticles [176]. Moreover, other micellar constructs delivering several therapeutic agents have been designed. Such constructs can be exemplified by cationic micelles decorated with cetuximab and co-loaded with cytostatic agent gemcitabine and microRNA miR-205 low level of which is associated with cancer stem cells. This approach aimed at treating advanced pancreatic cancer demonstrated enhanced therapeutic efficacy in pancreatic cancer xenografts compared to micelle-mediated delivery of every single component separately [177]. Another variant of micelles – tocopheryl-PEG conjugated to cetuximab and loaded with paclitaxel – was successfully used for therapy of triple-negative breast cancer xenografts [178].
A number of studies on targeted delivery of cytotoxic agents is based on using several ligands binding to several cancer cell-specific receptors. Most promising approaches simultaneously targeted EGFR (because this receptor promotes epithelial-mesenchymal transition [4]) and cancer stem cell markers. In particular, dual specificity PLGA copolymer nanoparticles conjugated to aptamers against EGFR and CD133 and loaded with antibiotic salinomycin effective against cancer stem cells were generated [179]. The similar functionality was also conferred to hyaluronic acid-based nanoparticles with dual specificity: EGFR (peptide GE11) and CD44 to which hyaluronic acid could be bound with high specificity. These particles carried granzyme B serine protease as a cytotoxic agent [180].
By now, a large number of EGFR-targeting nanoparticles loaded with diverse therapeutic agents has been generated and tested in animal tumor models. In particular, hybrid lipid–PLGA nanoparticles conjugated to PEG and Fab′-fragment of anti-EGFR antibody and loaded with adrianomycin suppressed growth of the hepatocellular carcinoma xenograft [181]. The pluronic- and PEG-stabilized self-assembled lipid nanoparticles loaded with paclitaxel and decorated with EGFR-specific mAb also demonstrated enhanced efficacy against ovarian cancer cells both in vitro and in vivo [182]. In contrast, the PLGA nanoparticles conjugated to cetuximab and loaded with the prodrug paclitaxel acetate were tested for therapy of the A549 lung cancer cell xenografts and showed no enhanced intra-tumor accumulation. Nonetheless, the cetuximab-conjugated immuno-nanoparticles demonstrated better therapeutic effect in comparison with the initial nanoparticles [183]. Delivery of paclitaxel to gastric cancer cells in vitro and in vivo was provided by the cetuximab-conjugated poly(γ-glutamic acid)-chitosan nanoparticles [184]. In addition, cetuximab-conjugated paclitaxel-loaded PLGA nanoparticles also demonstrated better efficacy in treating lung cancer xenografts [185]. The cetuximab-conjugated doxorubicin-loaded silica nanoparticles were also able to improve therapeutic effect against hepatocellular carcinoma xenografts [186].
Many promising types of drugs are based on using substances, which have not been sufficiently investigated so far. In this case, a transfer from the data obtained on cultured cells and animal models to clinical practice will require substantial time and expenses. The attempts to create a construct containing mostly components already approved for therapy can be illustrated by the EGF-conjugated gemcitabine-loaded lecithin-based nanoparticles modified with PEG [187].
Intrinsic properties of nanoparticle material can be also used for EGFR targeted delivery. For instance, cetuximab-conjugated iron oxide nanoparticles were applied not only for magnetic resonance imaging, but also for enhancing effects of radiotherapy. When inside the cells these nanoparticles were able to trigger production of reactive oxygen species (ROS) that enhanced radiosensitivity in of the irradiated glioblastoma xenografts [188].
Cetuximab-conjugated gemcitabine-loaded gold nanoparticles exerted a therapeutic effect against pancreatic adenocarcinoma xenografts [189]. Moreover, one should not forget mentioning an opportunity of using anti-EGFR antibody-conjugated gold nanoparticles for photo-thermal destruction of cancer cells [190]. In this case specificity of the nanoparticle internalization accounted for their long-term intra-tumor retention.
The presented examples show that there is a wide field of activities for advancing traditional anti-cancer chemotherapeutic agents via targeted delivery supplementing them with favorable pharmacokinetic, intrinsic trafficking, and cytotoxic properties. The common feature of these approaches is the necessity to include elements preventing capture of such constructs by the monocyte-macrophage system, which typically rely on using PEG.
Radionuclide therapy. α-, β-Emitting radioisotopes, γ-radiation, and Auger electrons (AEs) are used as cytotoxic agents in the targeted radionuclide therapy. Intracellular location of emitters of β-particles is not necessary for damaging cancer cells, and endocytosis may only improve the intra-tumor retention of a β-particle emitter attached to a drug. The effect of α-particles is profoundly more prominent when they are located inside the cell nucleus. Effects exerted by AEs usually require their intranuclear location. Radionuclides often used for therapy and imaging emit several types of radiation. AEs emitted due to electron capture or internal conversion are of top interest from the point of view of delivery into the target cells. They possess short-range in tissues (normally within nanometer range) and exhibit high linear energy transfer (9-26 keV/µm). Upon decay in the immediate vicinity to nuclear DNA, AE emitters are considered as promising agents for selective elimination of target cell with minimal damage to adjacent normal cells [191]. Diverse types of EGFR-targeted AE emitter delivery of varying complexity have been developed by taking into consideration EGFR overexpression typical in numerous cancer types as well as ability of EGFR to undergo nuclear transport. Monotherapy with intravenously administered [125I]-labeled mouse anti-EGFR mAb 425 was found to markedly extend (from 7 to 16 months) overall survival in patients with aggressive glioblastoma multiforme [192]. Experiments with glioma cell lines showed that 20-40% of cell-surface-bound antibodies underwent internalization [193]. A chelating agent DTPA used for binding of one of the most promising for clinical purposes AE emitter 111In conjugated to EGF is the simple and most common delivery systems investigated. First efficacy studies of [111In]-DTPA-EGF were published 20 years ago [191], which demonstrated that [111In]-DTPA-EGF penetrated into the EGFR-positive breast cancer cells with 15% of internalized radioactivity deposited in the nucleus. Incubation with [111In]-DTPA-EGF markedly retarded growth of target cells. Further studies demonstrated efficacy and safety of such approach in vivo in mouse breast cancer model [194] as well as assessed potential opportunity to replace natural EGF for its truncated isoform able to bind but not activate EGFR [195]. Moreover, phase I clinical trial demonstrated lack of high toxicity and immunogenicity of [111In]-DTPA-EGF after a single administration [196]. In addition, efficacy of such intervention can be improved by introducing a nuclear localization sequence (NLS) into the EGFR-recognizing constructs for enhancing their subsequent nuclear import. For instance, introducing NLS into the anti-EGFR antibody nimotuzumab-based delivery system for 111In significantly increased amount of the intranuclear radioactivity deposited into the breast cancer cells with EGFR overexpression, thereby conferring higher cytotoxicity compared to the NLS-free control construct. Moreover, nuclear localization sequence was also successfully utilized in the 111In dual-targeted delivery system, wherein EGF accounted for target cell recognition, whereas anti-γ-H2AX antibody allowed radionuclide delivery to the DNA damage repair signaling protein called histone γ-H2AX [197]. Interaction with the nuclear import-promoting factors occurred in the cytosol, however after binding EGFR-targeted construct underwent internalization into the closed intracellular vesicles. Trafficking of the intrinsic cell surface receptor including EGFR into the nucleus is low (see “Nuclear EGFR transport” Section) and is directed mainly to lysosomes and plasma membrane. At the same time, cells have extended inter-organelle trafficking system that may be utilized for controlling transport of the designed pharmaceuticals [198]. Sobolev et al. developed recombinant modular constructs called modular nanotransporters (MNTs) for delivering cytotoxic agents into the cell nucleus [199-201] consisting of the following components: EGF, binding of which to EGFR on the surface of the target cells provides selectivity and triggers internalization; translocation domain of the diphtheria toxin (DTox) responsible for release of transporters into cytosol; nuclear localization sequence derived from SV-40 large T-antigen; and bacterial hemoglobin-like protein (HMP) as a carrier [199]. It was shown that each module constituting MNT preserved functional activity [202], and that MNTs could reach the nucleus in the EGFR-positive cancer cells in vitro [202] and in vivo [203]. Initial data regarding application of MNTs to deliver AE emitters into the nucleus of EGFR-positive cancer cells were obtained in vitro by using two radionuclides: 125I [204] and 67Ga [205]. It was found that in both cases 55-60% of internalized MNT-delivered radionuclides were deposited in the nuclei of the target cells, and manifold increased cytotoxicity was observed when compared with the control constructs. To develop efficient MNT-mediated 111In delivery, there was first designed a method for attachment of this AE emitter to proteins allowing to obtain a labeled product with high specific activity [206]. As a result, the level of damage caused by 111In attached to MNTs was markedly increased both in the EGFR-expressing cultured cells [206-208] and in the in vivo animal model after a single local administration into the EGFR-expressing tumor that resulted in a dose-dependent therapeutic effect causing tumor eradication in 40% of animals treated with the highest dose of the preparation [207].
Another option for overexpressed EGFR-based radionuclide therapy is to use liposomes coupled to 186Re (emitting β-particles and AEs) or 188Re (solely β-particles) administered intracavitary after surgery in rats with orthotopic human breast cancer. This approach is aimed at improving current chemotherapeutic and radiotherapeutic clinical protocols [209].
High linear energy transfer (50-230 keV/µm) and relatively short range (50-100 µm) of α-particles make emitting them radionuclides more cytotoxic in the case of intracellular decay [200]. Targeted delivery of α-particle emitters into cancer cells expressing EGFR was successfully performed in some studies. Generally, mAb cetuximab targeting EGFR, which was used for a long time in clinical practice, is applied as the delivery vehicle. For instance, the α-emitter 213Bi conjugated to cetuximab demonstrated high cytotoxicity in vitro against the triple-negative breast cancer cells expressing EGFR [210]. Moreover, 213Bi attached to anti-EGFR antibody matuzumab revealed high efficacy in vivo against orthotopic human bladder cancer by remarkably improving overall survival as compared to control animals [211, 212]. A pilot study on efficacy of topically administered 213Bi conjugated to cetuximab for treatment of bladder carcinoma in situ demonstrated potential feasibility of this approach: three out 12 patients were observed to have full remission after a single or double instillation [213].
Furthermore, high anticancer efficiency was shown for another α-emitter – 212Pb conjugated to cetuximab in the murine LS-174T (human colon carcinoma cell line) disseminated intraperitoneal tumor model demonstrating increase of the median survival of animals to 84 days vs. 34 days for the case of 212Pb conjugated to the non-specific antibody [214]. Efficacy of the 212Pb–cetuximab therapy can be further improved both by gemcitabine pre-treatment as well as by conjugating 212Pb to the anti-HER-2 antibody trastuzumab.
Delivery of α-particle emitting radionuclides into the target cell nuclei allows both enhancing the damaging effect from α-particle itself and enabling cytotoxic potential of the recoil nuclei emitted upon α-decay [215]. In particular, 211At-attached to MNTs, was able to specifically enhance its intrinsic cytotoxicity up to 18-fold in several cancer cell lines overexpressing EGFR [216].
Immunotoxins. Catalytic subunits of bacterial toxins exerting toxic activity represent another type of antitumor preparations. Such substances consist of toxins with the ligand portion ensuring cell binding replaced with an antibody or its fragment or any other ligand specific to the internalizable receptor on the target cell. Generally, such substances are called immunotoxins, which is not very appropriate because these anticancer agents also include toxin fragments chemically conjugated or genetically engineered to be fused with some natural ligands. By now, there has been designed and tested more than 15 constructs consisting of EGFR ligands coupled to various toxins fragments mainly derived from Pseudomonas exotoxin, diphtheria toxin, and saporin [217-219]. Natural toxins exert some intrinsic mechanism for entering cell cytosol along with their own toxic activity. For instance, owing to its translocation domain the diphtheria toxin is able under condition of low acidity to insert its catalytic subunit into the endosome membrane and transfer it into the cytosol, whereas Pseudomonas exotoxin can be transferred via vesicular trafficking system through the Golgi apparatus into the ER and then into the cytosol by retrograde transport. ADP-ribosylation subunits of such toxins exhibit extremely high cytotoxicity, which often rises a question as to whether they exhibit therapeutic range adequate for clinical practice [220]. Nonetheless, the three immunotoxins have been already approved for clinical application in treatment of several hematological cancers: IL-2 fused to diphtheria toxin fragment (Ontak) for treatment of T-cell lymphoma; anti-CD22 antibody linked to a Pseudomonas exotoxin (Lumoxiti) for treatment of hairy-cell leukemia, and IL-3 coupled to a portion of diphtheria toxin (Elzonris) for treatment of blastic plasmacytoid dendritic cell neoplasm [220].
In contrast, using the same strategy for designing drugs effective against solid tumors is a more complicated task. Nonetheless, one of such compounds called TP-38 consisting of EGFR ligand TGFα fused to Pseudomonas exotoxin fragment truncated to exert solely toxic ADP-ribosylation activity, but unable to cell binding demonstrated promising results in preclinical studies. Such immunotoxin was administered for assessing its potential in treatment of glioblastoma upon local intracerebral inoculation via convection enhanced delivery. Glioblastoma is characterized by frequent EGFR overexpression (see “EGFR expression in oncology diseases” Section). It was found that such intraparenchymal drug delivery in several patients produced significant response [221]. Similar immunotoxin (D2C7-(scdsFv)-PE38KDEL) [222] was created by using antibody variable fragment able to bind both wild type EGFR and EGFRvIII (gain-of-function mutant with deleted extracellular domain) [223] most often observed in glioblastoma. Such construct demonstrated markedly extended overall survival in immunocompromised mice after treatment of orthotopic human glioma [224]. Combination of the D2C7-(scdsFv)-PE38KDEL therapy of the orthotopic human brain tumor in mice with the checkpoint inhibitors αCTLA-4/αPD-1/αPD-L1 allowed to eradicate experimental glioma in some animals [225].
Photosensitizer delivery. PDT represents an interesting and promising therapeutic approach for treatment of some diseases primarily oncological disorders [226]. Such type of therapy is based on selective intra-tumor accumulation of PS and subsequent tumor illumination with light with wavelength corresponding to the maximum of PS absorption. This triggers photodynamic reactions resulting in generation of ROS that exert powerful damaging effect. A distance the most reactive ROS (singlet oxygen, hydroxyl radical) can travel is limited by several dozens of nanometers, which makes them most efficient in the case they enter the nucleus of target cells [227, 228]. MNTs including those targeting EGFR have been examined as delivery vehicles of PS into the nuclei of target cells. It was shown that PS conjugated to MNTs preserve their potential to generate ROS upon illumination [202]. PS conjugated to MNTs vs. free PS exerted phototoxicity by three orders of magnitude higher in cancer cells with overexpressed EGFR [202, 229]. Substantially increased efficacy of MNT-conjugated PS vs. free PS photodynamic action was shown in vivo in nude mice inoculated with A431 human epidermoid carcinoma xenografts characterized with overexpression of EGFR. In particular, PDT mediated by the PS-MNTs construct resulted in 75% overall animal survival rate vs. 20% in the case when free PS was used [203].
The preparation RM-1929 – an immunoconjugate of the PS dyeIRDye700DX with anti-EGFR antibody cetuximab – is currently in the later phase of clinical trials for treating recurrent head and neck squamous cell carcinoma with PDT [230]. A large number of immunoconjugates between photosensitizers and mAb or their fragments currently undergoes preclinical studies [231].
Gene therapy. Several combined approaches have been proposed to transfer genetic material, genes, and miRNAs via EGFR. For this purpose, transfection with adenovirus redirected towards cells overexpressing EGFR by using chimeric protein composed of EGFR coupled to extracellular domain of the adenovirus receptor hCAR (human coxsackie B and adenovirus receptor) was used [232]. Modified adenovirus delivering human herpes simplex virus thymidine kinase gene ensured elimination of ovarian cancer cells, which delayed xenograft growth [232]. Another version of the combined approach is the system delivering sodium-iodide symporter gene that mediates iodide uptake by the cell. Polyethyleneimine-based GE11-conjugated nanoparticles delivered this gene into the colon cancer xenografts, which enabled bioimaging as well as suppressed tumor growth [233]. The liposomes loaded with miRNAs to inactivate survivin that carried two ligands – GE11 targeting EGFR and hyaluronic acid targeting CD44 – inhibited growth of the hepatocellular carcinoma xenografts [234]. It must be also emphasized that EGFR especially its mutant variant EGFRvIII that forms a unique epitope, have been extensively examined including in several clinical trials for their potential use in CAR T cell therapy [235].
CONCLUDING REMARKS: CURRENT TRENDS AND PERSPECTIVES
Analysis of current trends in the studies of EGFR, its significance in oncological disorders, and in the development of interventions for its therapeutic application demonstrates that as in the past most attention is paid to the ways of blocking EGFR-mediated signaling. However, it has been increasingly evident that approaches combining EGFR overexpression and its alteration with the agents affecting cancer cells primarily by damaging their activity deserved much attention [93, 236, 237]. Along with the traditional widely used approaches involving concurrent use of several therapeutic agents, multifunctional systems have been developed. These systems combine delivery with several simultaneously or sequentially applied addresses followed by eliciting lethal impact inside the altered cells. Together with the targeted cytotoxic effect, the receptor-mediated delivery to target cell can be also used for interrupting intracellular regulation, e.g., by delivering antibodies or transcription factors blocking regulatory processes [238-240]. EGFR as one of the most examined entry into the signaling network externally regulating cell proliferation is often used for testing new delivery systems. A necessity to take into account potential of the host body to neutralize foreign substances and particles represents another challenge on the path to practical use of such approaches, which, however, does not seem to be unsolvable in the near future.
Abbreviations
- AEs:
-
Auger electrons
- EGF:
-
epidermal growth factor
- EGFR:
-
epidermal growth factor receptor
- ER:
-
endoplasmic reticulum
- ErbB:
-
avian erythroblastic leukemia viral (v-erb-b) oncogene homolog (synonym: EGFR/HER)
- mAb:
-
monoclonal antibody
- MNTs:
-
modular nanotransporters
- PDT:
-
photodynamic therapy
- PEG:
-
polyethylene glycol
- PLGA:
-
poly lactic-co-glycolic acid
- PRMT1:
-
arginine methyltransferase 1
- PS:
-
photosensitizer
- ROS:
-
reactive oxygen species
- TGFα:
-
transforming growth factor alpha
References
Jorissen, R. N., Walker, F., Pouliot, N., Garrett, T. P., Ward, C. W., and Burgess, A. W. (2003) Epidermal growth factor receptor: mechanisms of activation and signalling, Exp. Cell Res., 284, 31-53, doi: https://doi.org/10.1016/s0014-4827(02)00098-8 .
Yarden, Y., and Pines, G. (2012) The ERBB network: at last, cancer therapy meets systems biology, Nat. Rev. Cancer, 12, 553-563, doi: https://doi.org/10.1038/nrc3309 .
Roskoski, R., Jr. (2014) The ErbB/HER family of protein-tyrosine kinases and cancer, Pharmacol. Res., 79, 34-74, doi: https://doi.org/10.1016/j.phrs.2013.11.002 .
Lamouille, S., Xu, J., and Derynck, R. (2014) Molecular mechanisms of epithelial-mesenchymal transition, Nat. Rev. Mol. Cell Biol., 15, 178-196, doi: https://doi.org/10.1038/nrm3758 .
Sigismund, S., Avanzato, D., and Lanzetti, L. (2018) Emerging functions of the EGFR in cancer, Mol. Oncol., 12, 3-20, doi: https://doi.org/10.1002/1878-0261.12155 .
Lemmon, M. A., Schlessinger, J., and Ferguson, K. M. (2014) The EGFR family: not so prototypical receptor tyrosine kinases, Cold Spring Harb. Perspect. Biol., 6, a020768, doi: https://doi.org/10.1101/cshperspect.a020768 .
Arkhipov, A., Shan, Y., Das, R., Endres, N. F., Eastwood, M. P., Wemmer, D. E., Kuriyan, J., and Shaw, D. E. (2013) Architecture and membrane interactions of the EGF receptor, Cell, 152, 557-569, doi: https://doi.org/10.1016/j.cell.2012.12.030 .
Purba, E. R., Saita, E. I., and Maruyama, I. N. (2017) Activation of the EGF receptor by ligand binding and oncogenic mutations: the “rotation model”, Cells, 6, 13, doi: https://doi.org/10.3390/cells6020013 .
Holbro, T., and Hynes, N. E. (2004) ErbB receptors: directing key signaling networks throughout life, Annu. Rev. Pharmacol. Toxicol., 44, 195-217, doi: https://doi.org/10.1146/annurev.pharmtox.44.101802.121440 .
Frolov, A., Schuller, K., Tzeng, C. W., Cannon, E. E., Ku, B. C., Howard, J. H., Vickers, S. M., Heslin, M. J., Buchsbaum, D. J., and Arnoletti, J. P. (2007) ErbB3 expression and dimerization with EGFR influence pancreatic cancer cell sensitivity to erlotinib, Cancer Biol. Ther., 6, 548-554, doi: https://doi.org/10.4161/cbt.6.4.3849 .
Zhu, S., Belkhiri, A., and El-Rifai, W. (2011) DARPP-32 increases interactions between epidermal growth factor receptor and ERBB3 to promote tumor resistance to gefitinib, Gastroenterology, 141, 1738-1748, doi: https://doi.org/10.1053/j.gastro.2011.06.070 .
Huang, Z., Wang, Y., Nayak, P. S., Dammann, C. E., and Sanchez-Esteban, J. (2012) Stretch-induced fetal type II cell differentiation is mediated via ErbB1-ErbB4 interactions, J. Biol. Chem., 287, 18091-18102, doi: https://doi.org/10.1074/jbc.M111.313163 .
Saito, Y., Haendeler, J., Hojo, Y., Yamamoto, K., and Berk, B. C. (2001) Receptor heterodimerization: essential mechanism for platelet-derived growth factor-induced epidermal growth factor receptor transactivation, Mol. Cell Biol., 21, 6387-6394, doi: https://doi.org/10.1128/mcb.21.19.6387-6394.2001 .
Black, P. C., Brown, G. A., Dinney, C. P., Kassouf, W., Inamoto, T., Arora, A., Gallagher, D., Munsell, M. F., Bar-Eli, M., McConkey, D. J., and Adam, L. (2011) Receptor heterodimerization: a new mechanism for platelet-derived growth factor induced resistance to anti-epidermal growth factor receptor therapy for bladder cancer, J. Urol., 185, 693-700, doi: https://doi.org/10.1016/j.juro.2010.09.082 .
Tanizaki, J., Okamoto, I., Sakai, K., and Nakagawa, K. (2011) Differential roles of trans-phosphorylated EGFR, HER2, HER3, and RET as heterodimerisation partners of MET in lung cancer with MET amplification, Br. J. Cancer, 105, 807-813, doi: https://doi.org/10.1038/bjc.2011.322 .
Peace, B. E., Hill, K. J., Degen, S. J., and Waltz, S. E. (2003) Cross-talk between the receptor tyrosine kinases Ron and epidermal growth factor receptor, Exp. Cell Res., 289, 317-325, doi: https://doi.org/10.1016/s0014-4827(03)00280-5 .
Morgillo, F., Woo, J. K., Kim, E. S., Hong, W. K., and Lee, H. Y. (2006) Heterodimerization of insulin-like growth factor receptor/epidermal growth factor receptor and induction of survivin expression counteract the antitumor action of erlotinib, Cancer Res., 66, 10100-10111, doi: https://doi.org/10.1158/0008-5472.CAN-06-1684 .
Needham, S. R., Roberts, S. K., Arkhipov, A., Mysore, V. P., Tynan, C. J., Zanetti-Domingues, L. C., Kim, E. T., Losasso, V., Korovesis, D., Hirsch, M., Rolfe, D. J., Clarke, D. T., Winn, M. D., Lajevardipour, A., Clayton, A. H., Pike, L. J., Perani, M., Parker, P. J., Shan, Y., Shaw, D. E., and Martin-Fernandez, M. L. (2016) EGFR oligomerization organizes kinase-active dimers into competent signalling platforms, Nat. Commun., 7, 13307, doi: https://doi.org/10.1038/ncomms13307 .
Ibach, J., Radon, Y., Gelleri, M., Sonntag, M. H., Brunsveld, L., Bastiaens, P. I., and Verveer, P. J. (2015) Single particle tracking reveals that EGFR signaling activity is amplified in clathrin-coated pits, PLoS One, 10, e0143162, doi: https://doi.org/10.1371/journal.pone.0143162 .
Clarke, D. T., and Martin-Fernandez, M. L. (2019) A brief history of single-particle tracking of the epidermal growth factor receptor, Methods Protoc., 2, 12, doi: https://doi.org/10.3390/mps2010012 .
Lemmon, M. A., and Schlessinger, J. (2010) Cell signaling by receptor tyrosine kinases, Cell, 141, 1117-1134, doi: https://doi.org/10.1016/j.cell.2010.06.011 .
Macdonald, J. L., and Pike, L. J. (2008) Heterogeneity in EGF-binding affinities arises from negative cooperativity in an aggregating system, Proc. Natl. Acad. Sci. USA, 105, 112-117, doi: https://doi.org/10.1073/pnas.0707080105 .
Riese, D. J., and Stern, D. F. (1998) Specificity within the EGF family/ErbB receptor family signaling network, Bioessays, 20, 41-48, doi: https://doi.org/10.1002/(SICI)1521-1878(199801)20:1<41::AID-BIES7>3.0.CO;2-V .
Wang, Y. N., Lee, H. H., Chou, C. K., Yang, W. H., Wei, Y., Chen, C. T., Yao, J., Hsu, J. L., Zhu, C., Ying, H., Ye, Y., Wang, W. J., Lim, S. O., Xia, W., Ko, H. W., Liu, X., Liu, C. G., Wu, X., Wang, H., Li, D., Prakash, L. R., Katz, M. H., Kang, Y., Kim, M., Fleming, J. B., Fogelman, D., Javle, M., Maitra, A., and Hung, M. C. (2018) Angiogenin/ribonuclease 5 is an EGFR ligand and a serum biomarker for erlotinib sensitivity in pancreatic cancer, Cancer Cell, 33, 752-769, doi: https://doi.org/10.1016/j.ccell.2018.02.012 .
Wang, Y. N., Lee, H. H., and Hung, M. C. (2018) A novel ligand-receptor relationship between families of ribonucleases and receptor tyrosine kinases, J. Biomed. Sci., 25, 83, doi: https://doi.org/10.1186/s12929-018-0484-7 .
Pinkas-Kramarski, R., Shelly, M., Guarino, B. C., Wang, L. M., Lyass, L., Alroy, I., Alimandi, M., Kuo, A., Moyer, J. D., Lavi, S., Eisenstein, M., Ratzkin, B. J., Seger, R., Bacus, S. S., Pierce, J. H., Andrews, G. C., and Yarden, Y. (1998) ErbB tyrosine kinases and the two neuregulin families constitute a ligand-receptor network, Mol. Cell Biol., 18, 6090-6101, doi: https://doi.org/10.1128/mcb.18.10.6090 .
Gilmore, J. L., Gallo, R. M., and Riese, D. J. (2006) The epidermal growth factor receptor (EGFR)-S442F mutant displays increased affinity for neuregulin-2beta and agonist-independent coupling with downstream signalling events, Biochem. J., 396, 79-88, doi: https://doi.org/10.1042/BJ20051687 .
Santra, M., Reed, C. C., and Iozzo, R. V. (2002) Decorin binds to a narrow region of the epidermal growth factor (EGF) receptor, partially overlapping but distinct from the EGF-binding epitope, J. Biol. Chem., 277, 35671-35681, doi: https://doi.org/10.1074/jbc.M205317200 .
Zhu, J. X., Goldoni, S., Bix, G., Owens, R. T., McQuillan, D. J., Reed, C. C., and Iozzo, R. V. (2005) Decorin evokes protracted internalization and degradation of the epidermal growth factor receptor via caveolar endocytosis, J. Biol. Chem., 280, 32468-32479, doi: https://doi.org/10.1074/jbc.M503833200 .
Goldoni, S., Iozzo, R. A., Kay, P., Campbell, S., McQuillan, A., Agnew, C., Zhu, J. X., Keene, D. R., Reed, C. C., and Iozzo, R. V. (2007) A soluble ectodomain of LRIG1 inhibits cancer cell growth by attenuating basal and ligand-dependent EGFR activity, Oncogene, 26, 368-381, doi: https://doi.org/10.1038/sj.onc.1209803 .
Zheng, Y., Li, X., Qian, X., Wang, Y., Lee, J. H., Xia, Y., Hawke, D. H., Zhang, G., Lyu, J., and Lu, Z. (2015) Secreted and O-GlcNAcylated MIF binds to the human EGF receptor and inhibits its activation, Nat. Cell Biol., 17, 1348-1355, doi: https://doi.org/10.1038/ncb3222 .
Freed, D. M., Bessman, N. J., Kiyatkin, A., Salazar-Cavazos, E., Byrne, P. O., Moore, J. O., Valley, C. C., Ferguson, K. M., Leahy, D. J., Lidke, D. S., and Lemmon, M. A. (2017) EGFR ligands differentially stabilize receptor dimers to specify signaling kinetics, Cell, 171, 683-695, doi: https://doi.org/10.1016/j.cell.2017.09.017 .
Macdonald-Obermann, J. L., and Pike, L. J. (2014) Different epidermal growth factor (EGF) receptor ligands show distinct kinetics and biased or partial agonism for homodimer and heterodimer formation, J. Biol. Chem., 289, 26178-26188, doi: https://doi.org/10.1074/jbc.M114.586826 .
Liao, H. W., Hsu, J. M., Xia, W., Wang, H. L., Wang, Y. N., Chang, W. C., Arold, S. T., Chou, C. K., Tsou, P. H., Yamaguchi, H., Fang, Y. F., Lee, H. J., Lee, H. H., Tai, S. K., Yang, M. H., Morelli, M. P., Sen, M., Ladbury, J. E., Chen, C. H., Grandis, J. R., Kopetz, S., and Hung, M. C. (2015) PRMT1-mediated methylation of the EGF receptor regulates signaling and cetuximab response, J. Clin. Invest., 125, 4529-4543, doi: https://doi.org/10.1172/JCI82826 .
Wang, W. J., Hsu, J. M., Wang, Y. N., Lee, H. H., Yamaguchi, H., Liao, H. W., and Hung, M. C. (2019) An essential role of PRMT1-mediated EGFR methylation in EGFR activation by ribonuclease 5, Am. J. Cancer Res., 9, 180-185.
Caldieri, G., Malabarba, M. G., Di Fiore, P. P., and Sigismund, S. (2018) EGFR trafficking in physiology and cancer, Prog. Mol. Subcell. Biol., 57, 235-272, doi: https://doi.org/10.1007/978-3-319-96704-2_9 .
Sorkin, A., and Goh, L. K. (2008) Endocytosis and intracellular trafficking of ErbBs, Exp. Cell Res., 315, 683-696, doi: https://doi.org/10.1016/j.yexcr.2008.07.029 .
Goh, L. K., and Sorkin, A. (2013) Endocytosis of receptor tyrosine kinases, Cold Spring Harb. Perspect. Biol., 5, a017459, doi: https://doi.org/10.1101/cshperspect.a017459 .
Waterman, H., Levkowitz, G., Alroy, I., and Yarden, Y. (1999) The RING finger of c-Cbl mediates desensitization of the epidermal growth factor receptor, J. Biol. Chem., 274, 22151-22154, doi: https://doi.org/10.1074/jbc.274.32.22151 .
Jiang, X., Huang, F., Marusyk, A., and Sorkin, A. (2003) Grb2 regulates internalization of EGF receptors through clathrin-coated pits, Mol. Biol. Cell, 14, 858-870, doi: https://doi.org/10.1091/mbc.e02-08-0532 .
Sigismund, S., Algisi, V., Nappo, G., Conte, A., Pascolutti, R., Cuomo, A., Bonaldi, T., Argenzio, E., Verhoef, L. G., Maspero, E., Bianchi, F., Capuani, F., Ciliberto, A., Polo, S., and Di Fiore, P. P. (2013) Threshold-controlled ubiquitination of the EGFR directs receptor fate, EMBO J., 32, 2140-2157, doi: https://doi.org/10.1038/emboj.2013.149 .
Huang, F., Goh, L. K., and Sorkin, A. (2007) EGF receptor ubiquitination is not necessary for its internalization, Proc. Natl. Acad. Sci. USA, 104, 16904-16909.
Goh, L. K., Huang, F., Kim, W., Gygi, S., and Sorkin, A. (2010) Multiple mechanisms collectively regulate clathrin-mediated endocytosis of the epidermal growth factor receptor, J. Cell Biol., 189, 871-883, doi: https://doi.org/10.1083/jcb.201001008 .
Oved, S., Mosesson, Y., Zwang, Y., Santonico, E., Shtiegman, K., Marmor, M. D., Kochupurakkal, B. S., Katz, M., Lavi, S., Cesareni, G., and Yarden, Y. (2006) Conjugation to Nedd8 instigates ubiquitylation and down-regulation of activated receptor tyrosine kinases, J. Biol. Chem., 281, 21640-21651, doi: https://doi.org/10.1074/jbc.M513034200 .
Boucrot, E., Ferreira, A. P., Almeida-Souza, L., Debard, S., Vallis, Y., Howard, G., Bertot, L., Sauvonnet, N., and McMahon, H. T. (2015) Endophilin marks and controls a clathrin-independent endocytic pathway, Nature, 517, 460-465, doi: https://doi.org/10.1038/nature14067 .
Sigismund, S., Argenzio, E., Tosoni, D., Cavallaro, E., Polo, S., and Di Fiore, P. P. (2008) Clathrin-mediated internalization is essential for sustained EGFR signaling but dispensable for degradation, Dev. Cell, 15, 209-219, doi: https://doi.org/10.1016/j.devcel.2008.06.012 .
Sorkin, A., Krolenko, S., Kudrjavtceva, N., Lazebnik, J., Teslenko, L., Soderquist, A. M., and Nikolsky, N. (1991) Recycling of epidermal growth factor-receptor complexes in A431 cells: identification of dual pathways, J. Cell Biol., 112, 55-63, doi: https://doi.org/10.1016/0167-4889(89)90083-9 .
Soubeyran, P., Kowanetz, K., Szymkiewicz, I., Langdon, W. Y., and Dikic, I. (2002) Cbl-CIN85-endophilin complex mediates ligand-induced downregulation of EGF receptors, Nature, 416, 183-187, doi: https://doi.org/10.1038/416183a .
Henriksen, L., Grandal, M. V., Knudsen, S. L. J., van Deurs, B., and Grovdal, L. M. (2013) Internalization mechanisms of the epidermal growth factor receptor after activation with different ligands, PLoS One, 8, e58148, doi: https://doi.org/10.1371/journal.pone.0058148 .
West, M. A., Bretscher, M. S., and Watts, C. (1989) Distinct endocytotic pathways in epidermal growth factor-stimulated human carcinoma A431 cells, J. Cell Biol., 109, 2731-2739, doi: https://doi.org/10.1083/jcb.109.6.2731 .
Yamazaki, T., Zaal, K., Hailey, D., Presley, J., Lippincott-Schwartz, J., and Samelson, L. E. (2002) Role of Grb2 in EGF-stimulated EGFR internalization, J. Cell Sci., 115, 1791-1802.
Orth, J. D., Krueger, E. W., Weller, S. G., and McNiven, M. A. (2006) A novel endocytic mechanism of epidermal growth factor receptor sequestration and internalization, Cancer Res., 66, 3603-3610, doi: https://doi.org/10.1158/0008-5472.CAN-05-2916 .
Tomas, A., Futter, C. E., and Eden, E. R. (2014) EGF receptor trafficking: consequences for signaling and cancer, Trends Cell Biol., 24, 26-34.
Wang, Y. N., Wang, H., Yamaguchi, H., Lee, H. J., Lee, H. H., and Hung, M. C. (2010) COPI-mediated retrograde trafficking from the Golgi to the ER regulates EGFR nuclear transport, Biochem. Biophys. Res. Commun., 399, 498-504, doi: https://doi.org/10.1016/j.bbrc.2010.07.096 .
Demory, M. L., Boerner, J. L., Davidson, R., Faust, W., Miyake, T., Lee, I., Huttemann, M., Douglas, R., Haddad, G., and Parsons, S. J. (2009) Epidermal growth factor receptor translocation to the mitochondria: regulation and effect, J. Biol. Chem., 284, 36592-36604, doi: https://doi.org/10.1074/jbc.M109.000760 .
Cao, X., Zhu, H., Ali-Osman, F., and Lo, H. W. (2011) EGFR and EGFRvIII undergo stress- and EGFR kinase inhibitor-induced mitochondrial translocalization: a potential mechanism of EGFR-driven antagonism of apoptosis, Mol. Cancer, 10, 26, doi: https://doi.org/10.1186/1476-4598-10-26 .
Wang, T. H., Lin, Y. H., Yang, S. C., Chang, P. C., Wang, T. C., and Chen, C. Y. (2017) Tid1-S regulates the mitochondrial localization of EGFR in non-small cell lung carcinoma, Oncogenesis, 6, e361, doi: https://doi.org/10.1038/oncsis.2017.62 .
Che, T. F., Lin, C. W., Wu, Y. Y., Chen, Y. J., Han, C. L., Chang, Y. L., Wu, C. T., Hsiao, T. H., Hong, T. M., and Yang, P. C. (2015) Mitochondrial translocation of EGFR regulates mitochondria dynamics and promotes metastasis in NSCLC, Oncotarget, 6, 37349-37366, doi: https://doi.org/10.18632/oncotarget.5736 .
Hsu, S. C., and Hung, M. C. (2007) Characterization of a novel tripartite nuclear localization sequence in the EGFR family, J. Biol. Chem., 282, 10432-10440, doi: https://doi.org/10.1074/jbc.M610014200 .
Shah, P., Chaumet, A., Royle, S. J., and Bard, F. A. (2019) The NAE pathway: autobahn to the nucleus for cell surface receptors, Cells, 8, doi: https://doi.org/10.3390/cells8080915 .
Lo, H. W., Ali-Seyed, M., Wu, Y., Bartholomeusz, G., Hsu, S. C., and Hung, M. C. (2006) Nuclear-cytoplasmic transport of EGFR involves receptor endocytosis, importin beta1 and CRM1, J. Cell Biochem., 98, 1570-1583, doi: https://doi.org/10.1002/jcb.20876 .
De Angelis Campos, A. C., Rodrigues, M. A., de Andrade, C., de Goes, A. M., Nathanson, M. H., and Gomes, D. A. (2011) Epidermal growth factor receptors destined for the nucleus are internalized via a clathrin-dependent pathway, Biochem. Biophys. Res. Commun., 412, 341-346, doi: https://doi.org/10.1016/j.bbrc.2011.07.100 .
Lo, H. W. (2010) Nuclear mode of the EGFR signaling network: biology, prognostic value, and therapeutic implications, Discov. Med., 10, 44-51.
Faria, J. A. Q. A., de Andrade, C., Goes, A. M., Rodrigues, M. A., and Gomes, D. A. (2016) Effects of different ligands on epidermal growth factor receptor (EGFR) nuclear translocation, Biochem. Biophys. Res. Commun., 478, 39-45, doi: https://doi.org/10.1016/j.bbrc.2016.07.097 .
Han, W., and Lo, H. W. (2012) Landscape of EGFR signaling network in human cancers: biology and therapeutic response in relation to receptor subcellular locations, Cancer Lett., 318, 124-134, doi: https://doi.org/10.1016/j.canlet.2012.01.011 .
Wang, Y. N., Yamaguchi, H., Hsu, J. M., and Hung, M. C. (2010) Nuclear trafficking of the epidermal growth factor receptor family membrane proteins, Oncogene, 29, 3997-4006, doi: https://doi.org/10.1038/onc.2010.157 .
Wang, Y. N., Yamaguchi, H., Huo, L., Du, Y., Lee, H. J., Lee, H. H., Wang, H., Hsu, J. M., and Hung, M. C. (2010) The translocon Sec61beta localized in the inner nuclear membrane transports membrane-embedded EGF receptor to the nucleus, J. Biol. Chem., 285, 38720-38729, doi: https://doi.org/10.1074/jbc.M110.158659 .
Liao, H. J., and Carpenter, G. (2007) Role of the Sec61 translocon in EGF receptor trafficking to the nucleus and gene expression, Mol. Biol. Cell, 18, 1064-1072, doi: https://doi.org/10.1091/mbc.e06-09-0802 .
Chaumet, A., Wright, G. D., Seet, S. H., Tham, K. M., Gounko, N. V., and Bard, F. (2015) Nuclear envelope-associated endosomes deliver surface proteins to the nucleus, Nat. Commun., 6, 8218, doi: https://doi.org/10.1038/ncomms9218 .
Chia, P. L., Scott, A. M., and John, T. (2019) Epidermal growth factor receptor (EGFR)-targeted therapies in mesothelioma, Expert Opin. Drug Deliv., 16, 441-451, doi: https://doi.org/10.1080/17425247.2019.1598374 .
Franovic, A., Gunaratnam, L., Smith, K., Robert, I., Patten, D., and Lee, S. (2007) Translational up-regulation of the EGFR by tumor hypoxia provides a nonmutational explanation for its overexpression in human cancer, Proc. Natl. Acad. Sci. USA, 104, 13092-13097, doi: https://doi.org/10.1073/pnas.0702387104 .
Liu, X., Wang, P., Zhang, C., and Ma, Z. (2017) Epidermal growth factor receptor (EGFR): A rising star in the era of precision medicine of lung cancer, Oncotarget, 8, 50209-50220, doi: https://doi.org/10.18632/oncotarget.16854 .
Grandis, J. R., and Tweardy, D. J. (1993) Elevated levels of transforming growth factor alpha and epidermal growth factor receptor messenger RNA are early markers of carcinogenesis in head and neck cancer, Cancer Res., 53, 3579-3584.
Atkins, D., Reiffen, K. A., Tegtmeier, C. L., Winther, H., Bonato, M. S., and Storkel, S. (2004) Immunohistochemical detection of EGFR in paraffin-embedded tumor tissues: variation in staining intensity due to choice of fixative and storage time of tissue sections, J. Histochem. Cytochem., 52, 893-901, doi: https://doi.org/10.1369/jhc.3A6195.2004 .
Yen, L. C., Uen, Y. H., Wu, D. C., Lu, C. Y., Yu, F. J., Wu, I. C., Lin, S. R., and Wang, J. Y. (2010) Activating KRAS mutations and overexpression of epidermal growth factor receptor as independent predictors in metastatic colorectal cancer patients treated with cetuximab, Ann. Surg., 251, 254-260, doi: https://doi.org/10.1097/SLA.0b013e3181bc9d96 .
Lee, H. J., Xu, X., Choe, G., Chung, D. H., Seo, J. W., Lee, J. H., Lee, C. T., Jheon, S., Sung, S. W., and Chung, J. H. (2010) Protein overexpression and gene amplification of epidermal growth factor receptor in nonsmall cell lung carcinomas: comparison of four commercially available antibodies by immunohistochemistry and fluorescence in situ hybridization study, Lung Cancer, 68, 375-382, doi: https://doi.org/10.1016/j.lungcan.2009.07.014 .
Gatalica, Z., Millis, S. Z., Vranic, S., Bender, R., Basu, G. D., Voss, A., and Von Hoff, D. D. (2014) Comprehensive tumor profiling identifies numerous biomarkers of drug response in cancers of unknown primary site: analysis of 1806 cases, Oncotarget, 5, 12440-12447, doi: https://doi.org/10.18632/oncotarget.2574 .
Verhaak, R. G., Hoadley, K. A., Purdom, E., Wang, V., Qi, Y., et al. (2010) Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1, Cancer Cell, 17, 98-110.
Herbst, R. S., and Shin, D. M. (2002) Monoclonal antibodies to target epidermal growth factor receptor-positive tumors: a new paradigm for cancer therapy, Cancer, 94, 1593-1611, doi: https://doi.org/10.1002/cncr.10372 .
Kalyankrishna, S., and Grandis, J. R. (2006) Epidermal growth factor receptor biology in head and neck cancer, J. Clin. Oncol., 24, 2666-2672, doi: https://doi.org/10.1200/JCO.2005.04.8306 .
Chua, D. T., Nicholls, J. M., Sham, J. S., and Au, G. K. (2004) Prognostic value of epidermal growth factor receptor expression in patients with advanced stage nasopharyngeal carcinoma treated with induction chemotherapy and radiotherapy, Int. J. Radiat. Oncol. Biol. Phys., 59, 11-20, doi: https://doi.org/10.1016/j.ijrobp.2003.10.038 .
Wei, Q., Chen, L., Sheng, L., Nordgren, H., Wester, K., and Carlsson, J. (2007) EGFR, HER2 and HER3 expression in esophageal primary tumours and corresponding metastases, Int. J. Oncol., 31, 493-499.
Grobe, A., Eichhorn, W., Fraederich, M., Kluwe, L., Vashist, Y., Wikner, J., Smeets, R., Simon, R., Sauter, G., Heiland, M., and Blessmann, M. (2014) Immunohistochemical and FISH analysis of EGFR and its prognostic value in patients with oral squamous cell carcinoma, J. Oral. Pathol. Med., 43, 205-210, doi: https://doi.org/10.1111/jop.12111 .
Wang, X., Niu, H., Fan, Q., Lu, P., Ma, C., Liu, W., Liu, Y., Li, W., Hu, S., Ling, Y., Guo, L., Ying, J., and Huang, J. (2016) Predictive value of EGFR overexpression and gene amplification on icotinib efficacy in patients with advanced esophageal squamous cell carcinoma, Oncotarget, 7, 24744-24751, doi: https://doi.org/10.18632/oncotarget.8271 .
Politi, A., Tsiambas, E., Mastronikolis, N. S., Peschos, D., Asproudis, I., Kyrodimos, E., Armata, I. E., Chrysovergis, A., Asimakopoulos, A., Papanikolaou, V. S., Batistatou, A., and Ragos, V. (2019) Combined EGFR/ALK expression analysis in laryngeal squamous cell carcinoma, In vivo, 33, 815-819, doi: https://doi.org/10.21873/invivo.11544 .
Schrevel, M., Gorter, A., Kolkman-Uljee, S. M., Trimbos, J. B., Fleuren, G. J., and Jordanova, E. S. (2011) Molecular mechanisms of epidermal growth factor receptor overexpression in patients with cervical cancer, Mod. Pathol., 24, 720-728, doi: https://doi.org/10.1038/modpathol.2010.239 .
Gui, T., and Shen, K. (2012) The epidermal growth factor receptor as a therapeutic target in epithelial ovarian cancer, Cancer Epidemiol., 36, 490-496, doi: https://doi.org/10.1016/j.canep.2012.06.005 .
Tian, W. J., Huang, M. L., Qin, Q. F., Chen, Q., Fang, K., and Wang, P. L. (2016) Prognostic impact of epidermal growth factor receptor overexpression in patients with cervical cancer: a meta-analysis, PLoS One, 11, e0158787, doi: https://doi.org/10.1371/journal.pone.0158787 .
Li, Q., Tang, Y., Cheng, X., Ji, J., Zhang, J., and Zhou, X. (2014) EGFR protein expression and gene amplification in squamous intraepithelial lesions and squamous cell carcinomas of the cervix, Int. J. Clin. Exp. Pathol., 7, 733-741.
Rokita, M., Stec, R., Bodnar, L., Charkiewicz, R., Korniluk, J., Smoter, M., Cichowicz, M., Chyczewski, L., Niklinski, J., Kozlowski, W., and Szczylik, C. (2013) Overexpression of epidermal growth factor receptor as a prognostic factor in colorectal cancer on the basis of the Allred scoring system, Onco Targets Ther., 6, 967-976, doi: https://doi.org/10.2147/OTT.S42446 .
Yun, S., Kwak, Y., Nam, S. K., Seo, A. N., Oh, H. K., Kim, D. W., Kang, S. B., and Lee, H. S. (2018) Ligand-independent epidermal growth factor receptor overexpression correlates with poor prognosis in colorectal cancer, Cancer Res. Treat., 50, 1351-1361, doi: https://doi.org/10.4143/crt.2017.487 .
Huang, C. W., Chen, Y. T., Tsai, H. L., Yeh, Y. S., Su, W. C., Ma, C. J., Tsai, T. N., and Wang, J. Y. (2017) EGFR expression in patients with stage III colorectal cancer after adjuvant chemotherapy and on cancer cell function, Oncotarget, 8, 114663-114676, doi: https://doi.org/10.18632/oncotarget.23072 .
Grapa, C. M., Mocan, T., Gonciar, D., Zdrehus, C., Mosteanu, O., Pop, T., and Mocan, L. (2019) Epidermal growth factor receptor and its role in pancreatic cancer treatment mediated by nanoparticles, Int. J. Nanomedicine, 14, 9693-9706, doi: https://doi.org/10.2147/IJN.S226628 .
Cook, N., Frese, K. K., and Moore, M. (2014) Assessing the role of the EGF receptor in the development and progression of pancreatic cancer, Gastrointest. Cancer Targets Therapy, 4, 23-37.
Karandish, F., and Mallik, S. (2016) Biomarkers and targeted therapy in pancreatic cancer, Biomarkers Cancer, 8, BIC-S34414.
Park, S. J., Gu, M. J., Lee, D. S., Yun, S. S., Kim, H. J., and Choi, J. H. (2015) EGFR expression in pancreatic intraepithelial neoplasia and ductal adenocarcinoma, Int. J. Clin. Exp. Pathol., 8, 8298-8304.
Gonzalez-Conchas, G. A., Rodriguez-Romo, L., Hernandez-Barajas, D., Gonzalez-Guerrero, J. F., Rodriguez-Fernandez, I. A., Verdines-Perez, A., Templeton, A. J., Ocana, A., Seruga, B., Tannock, I. F., Amir, E., and Vera-Badillo, F. E. (2018) Epidermal growth factor receptor overexpression and outcomes in early breast cancer: a systematic review and a meta-analysis, Cancer Treat. Rev., 62, 1-8, doi: https://doi.org/10.1016/j.ctrv.2017.10.008 .
Sheng, Q., and Liu, J. (2011) The therapeutic potential of targeting the EGFR family in epithelial ovarian cancer, Br. J. Cancer, 104, 1241-1245, doi: https://doi.org/10.1038/bjc.2011.62 .
Wang, K., Li, D., and Sun, L. (2016) High levels of EGFR expression in tumor stroma are associated with aggressive clinical features in epithelial ovarian cancer, Onco Targets Ther., 9, 377-386, doi: https://doi.org/10.2147/OTT.S96309 .
Perren, T. J. (2016) Mucinous epithelial ovarian carcinoma, Ann. Oncol., 27 Suppl. 1, i53-i57, doi: https://doi.org/10.1093/annonc/mdw087 .
Shinojima, N., Tada, K., Shiraishi, S., Kamiryo, T., Kochi, M., Nakamura, H., Makino, K., Saya, H., Hirano, H., Kuratsu, J., Oka, K., Ishimaru, Y., and Ushio, Y. (2003) Prognostic value of epidermal growth factor receptor in patients with glioblastoma multiforme, Cancer Res., 63, 6962-6970.
Zhao, L. L., Xu, K. L., Wang, S. W., Hu, B. L., and Chen, L. R. (2012) Pathological significance of epidermal growth factor receptor expression and amplification in human gliomas, Histopathology, 61, 726-736, doi: https://doi.org/10.1111/j.1365-2559.2012.04354.x .
Wang, X., Zhang, S., MacLennan, G. T., Eble, J. N., Lopez-Beltran, A., Yang, X. J., Pan, C. X., Zhou, H., Montironi, R., and Cheng, L. (2007) Epidermal growth factor receptor protein expression and gene amplification in small cell carcinoma of the urinary bladder, Clin. Cancer Res., 13, 953-957, doi: https://doi.org/10.1158/1078-0432.CCR-06-2167 .
Chaux, A., Cohen, J. S., Schultz, L., Albadine, R., Jadallah, S., Murphy, K. M., Sharma, R., Schoenberg, M. P., and Netto, G. J. (2012) High epidermal growth factor receptor immunohistochemical expression in urothelial carcinoma of the bladder is not associated with EGFR mutations in exons 19 and 21: a study using formalin-fixed, paraffin-embedded archival tissues, Hum. Pathol., 43, 1590-1595, doi: https://doi.org/10.1016/j.humpath.2011.11.016 .
Carlsson, J., Wester, K., De La Torre, M., Malmstrom, P. U., and Gardmark, T. (2015) EGFR-expression in primary urinary bladder cancer and corresponding metastases and the relation to HER2-expression. On the possibility to target these receptors with radionuclides, Radiol. Oncol., 49, 50-58, doi: https://doi.org/10.2478/raon-2014-0015 .
Girard, N. (2010) Thymic tumors: relevant molecular data in the clinic, J. Thorac. Oncol., 5, S291-S295, doi: https://doi.org/10.1097/JTO.0b013e3181f209b9 .
Rusch, V. W., Klimstra, D. S., and Venkatraman, E. S. (1996) Molecular markers help characterize neuroendocrine lung tumors, Ann. Thorac. Surg., 62, 798-809, doi: https://doi.org/10.1016/s0003-4975(96)00435-3 .
Srirajaskanthan, R., Shah, T., Watkins, J., Marelli, L., Khan, K., and Caplin, M. E. (2010) Expression of the HER-1-4 family of receptor tyrosine kinases in neuroendocrine tumours, Oncol. Rep., 23, 909-915, doi: https://doi.org/10.3892/or_00000714 .
London, M., and Gallo, E. (2020) Epidermal growth factor receptor (EGFR) involvement in epithelial-derived cancers and its current antibody-based immunotherapies, Cell Biol. Int., 44, 1267-1282, doi: https://doi.org/10.1002/cbin.11340 .
Cunningham, D., Humblet, Y., Siena, S., Khayat, D., Bleiberg, H., Santoro, A., Bets, D., Mueser, M., Harstrick, A., Verslype, C., Chau, I., and Van Cutsem, E. (2004) Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer, N. Engl. J. Med., 351, 337-345, doi: https://doi.org/10.1056/NEJMoa033025 .
Trivedi, S., Srivastava, R. M., Concha-Benavente, F., Ferrone, S., Garcia-Bates, T. M., Li, J., and Ferris, R. L. (2016) Anti-EGFR targeted monoclonal antibody isotype influences antitumor cellular immunity in head and neck cancer patients, Clin. Cancer Res., 22, 5229-5237, doi: https://doi.org/10.1158/1078-0432.CCR-15-2971 .
Cohenuram, M., and Saif, M. W. (2007) Panitumumab the first fully human monoclonal antibody: from the bench to the clinic, Anticancer Drugs, 18, 7-15, doi: https://doi.org/10.1097/CAD.0b013e32800feecb .
Thatcher, N., Hirsch, F. R., Luft, A. V., Szczesna, A., Ciuleanu, T. E., Dediu, M., Ramlau, R., Galiulin, R. K., Balint, B., Losonczy, G., Kazarnowicz, A., Park, K., Schumann, C., Reck, M., Depenbrock, H., Nanda, S., Kruljac-Letunic, A., Kurek, R., Paz-Ares, L., and Socinski, M. A. (2015) Necitumumab plus gemcitabine and cisplatin versus gemcitabine and cisplatin alone as first-line therapy in patients with stage IV squamous non-small-cell lung cancer (SQUIRE): an open-label, randomised, controlled phase 3 trial, Lancet Oncol., 16, 763-774, doi: https://doi.org/10.1016/S1470-2045(15)00021-2 .
Li, J., and Yan, H. (2018) Skin toxicity with anti-EGFR monoclonal antibody in cancer patients: a meta-analysis of 65 randomized controlled trials, Cancer Chemother. Pharmacol., 82, 571-583, doi: https://doi.org/10.1007/s00280-018-3644-2 .
Boland, W. K., and Bebb, G. (2009) Nimotuzumab: a novel anti-EGFR monoclonal antibody that retains anti-EGFR activity while minimizing skin toxicity, Expert Opin. Biol. Ther., 9, 1199-1206, doi: https://doi.org/10.1517/14712590903110709 .
Garrido, G., Tikhomirov, I. A., Rabasa, A., Yang, E., Gracia, E., Iznaga, N., Fernandez, L. E., Crombet, T., Kerbel, R. S., and Perez, R. (2011) Bivalent binding by intermediate affinity of nimotuzumab: a contribution to explain antibody clinical profile, Cancer Biol. Ther., 11, 373-382, doi: https://doi.org/10.4161/cbt.11.4.14097 .
Ciardiello, F., and Tortora, G. (2008) EGFR antagonists in cancer treatment, N. Engl. J. Med., 358, 1160-1174, doi: https://doi.org/10.1056/NEJMra0707704 .
Therkildsen, C., Bergmann, T. K., Henrichsen-Schnack, T., Ladelund, S., and Nilbert, M. (2014) The predictive value of KRAS, NRAS, BRAF, PIK3CA and PTEN for anti-EGFR treatment in metastatic colorectal cancer: a systematic review and meta-analysis, Acta Oncol., 53, 852-864, doi: https://doi.org/10.3109/0284186X.2014.895036 .
Lu, Y., Zhao, X., Liu, Q., Li, C., Graves-Deal, R., Cao, Z., Singh, B., Franklin, J. L., Wang, J., Hu, H., Wei, T., Yang, M., Yeatman, T. J., Lee, E., Saito-Diaz, K., Hinger, S., Patton, J. G., Chung, C. H., Emmrich, S., Klusmann, J. H., Fan, D., and Coffey, R. J. (2017) lncRNA MIR100HG-derived miR-100 and miR-125b mediate cetuximab resistance via Wnt/beta-catenin signaling, Nat. Med., 23, 1331-1341, doi: https://doi.org/10.1038/nm.4424 .
Misale, S., Yaeger, R., Hobor, S., Scala, E., Janakiraman, M., et al. (2012) Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer, Nature, 486, 532-536, doi: https://doi.org/10.1038/nature11156 .
Siravegna, G., Mussolin, B., Buscarino, M., Corti, G., Cassingena, A., et al. (2015) Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients, Nat. Med., 21, 795-801, doi: https://doi.org/10.1038/nm.3870 .
Roskoski, R., Jr. (2020) Properties of FDA-approved small molecule protein kinase inhibitors: a 2020 update, Pharmacol. Res., 152, 104609, doi: https://doi.org/10.1016/j.phrs.2019.104609 .
Yang, J. J., Zhou, C., Huang, Y., Feng, J., Lu, S., Song, Y., Huang, C., Wu, G., Zhang, L., Cheng, Y., Hu, C., Chen, G., Zhang, L., Liu, X., Yan, H. H., Tan, F. L., Zhong, W., and Wu, Y. L. (2017) Icotinib versus whole-brain irradiation in patients with EGFR-mutant non-small-cell lung cancer and multiple brain metastases (BRAIN): a multicentre, phase 3, open-label, parallel, randomised controlled trial, Lancet Respir. Med., 5, 707-716, doi: https://doi.org/10.1016/S2213-2600(17)30262-X .
Mok, T. S., Wu, Y. L., Thongprasert, S., Yang, C. H., Chu, D. T., Saijo, N., Sunpaweravong, P., Han, B., Margono, B., Ichinose, Y., Nishiwaki, Y., Ohe, Y., Yang, J. J., Chewaskulyong, B., Jiang, H., Duffield, E. L., Watkins, C. L., Armour, A. A., and Fukuoka, M. (2009) Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma, N. Engl. J. Med., 361, 947-957, doi: https://doi.org/10.1056/NEJMoa0810699 .
Sequist, L. V., Waltman, B. A., Dias-Santagata, D., Digumarthy, S., Turke, A. B., Fidias, P., Bergethon, K., Shaw, A. T., Gettinger, S., Cosper, A. K., Akhavanfard, S., Heist, R. S., Temel, J., Christensen, J. G., Wain, J. C., Lynch, T. J., Vernovsky, K., Mark, E. J., Lanuti, M., Iafrate, A. J., Mino-Kenudson, M., and Engelman, J. A. (2011) Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors, Sci. Transl. Med., 3, 75ra26, doi: https://doi.org/10.1126/scitranslmed.3002003 .
Pao, W., Miller, V. A., Politi, K. A., Riely, G. J., Somwar, R., Zakowski, M. F., Kris, M. G., and Varmus, H. (2005) Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain, PLoS Med., 2, e73, doi: https://doi.org/10.1371/journal.pmed.0020073 .
Murtuza, A., Bulbul, A., Shen, J. P., Keshavarzian, P., Woodward, B. D., Lopez-Diaz, F. J., Lippman, S. M., and Husain, H. (2019) Novel third-generation EGFR tyrosine kinase inhibitors and strategies to overcome therapeutic resistance in lung cancer, Cancer Res., 79, 689-698, doi: https://doi.org/10.1158/0008-5472.CAN-18-1281 .
Soria, J. C., Ohe, Y., Vansteenkiste, J., Reungwetwattana, T., Chewaskulyong, B., Lee, K. H., Dechaphunkul, A., Imamura, F., Nogami, N., Kurata, T., Okamoto, I., Zhou, C., Cho, B. C., Cheng, Y., Cho, E. K., Voon, P. J., Planchard, D., Su, W. C., Gray, J. E., Lee, S. M., Hodge, R., Marotti, M., Rukazenkov, Y., and Ramalingam, S. S. (2018) Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer, N. Engl. J. Med., 378, 113-125, doi: https://doi.org/10.1056/NEJMoa1713137 .
Ramalingam, S. S., Vansteenkiste, J., Planchard, D., Cho, B. C., Gray, J. E., Ohe, Y., Zhou, C., Reungwetwattana, T., Cheng, Y., Chewaskulyong, B., Shah, R., Cobo, M., Lee, K. H., Cheema, P., Tiseo, M., John, T., Lin, M. C., Imamura, F., Kurata, T., Todd, A., Hodge, R., Saggese, M., Rukazenkov, Y., and Soria, J. C. (2020) Overall survival with osimertinib in untreated, EGFR-mutated advanced NSCLC, N. Engl. J. Med., 382, 41-50, doi: https://doi.org/10.1056/NEJMoa1913662 .
Nagano, T., Tachihara, M., and Nishimura, Y. (2018) Mechanism of resistance to epidermal growth factor receptor-tyrosine kinase inhibitors and a potential treatment strategy, Cells, 7, 212, doi: https://doi.org/10.3390/cells7110212 .
Li, R., Zhou, X., Yao, H., and Li, L. (2020) Four generations of EGFR TKIs associated with different pathogenic mutations in non-small cell lung carcinoma, J. Drug Target, 1-12, doi: https://doi.org/10.1080/1061186X.2020.1737934 .
Vyse, S., and Huang, P. H. (2019) Targeting EGFR exon 20 insertion mutations in non-small cell lung cancer, Signal. Transduct. Target Ther., 4, 5, doi: https://doi.org/10.1038/s41392-019-0038-9 .
Sordella, R., Bell, D. W., Haber, D. A., and Settleman, J. (2004) Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways, Science, 305, 1163-1167, doi: https://doi.org/10.1126/science.1101637 .
Oxnard, G. R., Hu, Y., Mileham, K. F., Husain, H., Costa, D. B., Tracy, P., Feeney, N., Sholl, L. M., Dahlberg, S. E., Redig, A. J., Kwiatkowski, D. J., Rabin, M. S., Paweletz, C. P., Thress, K. S., and Janne, P. A. (2018) Assessment of resistance mechanisms and clinical implications in patients with EGFR T790M-positive lung cancer and acquired resistance to osimertinib, JAMA Oncol., 4, 1527-1534, doi: https://doi.org/10.1001/jamaoncol.2018.2969 .
Piotrowska, Z., Isozaki, H., Lennerz, J. K., Gainor, J. F., Lennes, I. T., Zhu, V. W., Marcoux, N., Banwait, M. K., Digumarthy, S. R., Su, W., Yoda, S., Riley, A. K., Nangia, V., Lin, J. J., Nagy, R. J., Lanman, R. B., Dias-Santagata, D., Mino-Kenudson, M., Iafrate, A. J., Heist, R. S., Shaw, A. T., Evans, E. K., Clifford, C., Ou, S. I., Wolf, B., Hata, A. N., and Sequist, L. V. (2018) Landscape of acquired resistance to osimertinib in EGFR-mutant NSCLC and clinical validation of combined EGFR and RET inhibition with osimertinib and BLU-667 for acquired RET fusion, Cancer Discov., 8, 1529-1539, doi: https://doi.org/10.1158/2159-8290.CD-18-1022 .
Wang, Q., Yang, S., Wang, K., and Sun, S. Y. (2019) MET inhibitors for targeted therapy of EGFR TKI-resistant lung cancer, J. Hematol. Oncol., 12, 63, doi: https://doi.org/10.1186/s13045-019-0759-9 .
Terai, H., Soejima, K., Yasuda, H., Nakayama, S., Hamamoto, J., Arai, D., Ishioka, K., Ohgino, K., Ikemura, S., Sato, T., Yoda, S., Satomi, R., Naoki, K., and Betsuyaku, T. (2013) Activation of the FGF2-FGFR1 autocrine pathway: a novel mechanism of acquired resistance to gefitinib in NSCLC, Mol. Cancer Res., 11, 759-767, doi: https://doi.org/10.1158/1541-7786.MCR-12-0652 .
Schoenfeld, A. J., Chan, J. M., Kubota, D., Sato, H., Rizvi, H., Daneshbod, Y., Chang, J. C., Paik, P. K., Offin, M., Arcila, M. E., Davare, M. A., Shinde, U., Pe’er, D., Rekhtman, N., Kris, M. G., Somwar, R., Riely, G. J., Ladanyi, M., and Yu, H. A. (2020) Tumor analyses reveal squamous transformation and off-target alterations as early resistance mechanisms to first-line osimertinib in EGFR-mutant lung cancer, Clin. Cancer Res., 26, 2654-2663, doi: https://doi.org/10.1158/1078-0432.CCR-19-3563 .
Zhu, V. W., Klempner, S. J., and Ou, S. I. (2019) Receptor tyrosine kinase fusions as an actionable resistance mechanism to EGFR TKIs in EGFR-mutant non-small-cell lung cancer, Trends Cancer, 5, 677-692, doi: https://doi.org/10.1016/j.trecan.2019.09.008 .
Cheng, N., Cai, W., Ren, S., Li, X., Wang, Q., Pan, H., Zhao, M., Li, J., Zhang, Y., Zhao, C., Chen, X., Fei, K., Zhou, C., and Hirsch, F. R. (2015) Long non-coding RNA UCA1 induces non-T790M acquired resistance to EGFR-TKIs by activating the AKT/mTOR pathway in EGFR-mutant non-small cell lung cancer, Oncotarget, 6, 23582-23593, doi: https://doi.org/10.18632/oncotarget.4361 .
Lee, J. K., Lee, J., Kim, S., Kim, S., Youk, J., Park, S., An, Y., Keam, B., Kim, D. W., Heo, D. S., Kim, Y. T., Kim, J. S., Kim, S. H., Lee, J. S., Lee, S. H., Park, K., Ku, J. L., Jeon, Y. K., Chung, D. H., Park, P. J., Kim, J., Kim, T. M., and Ju, Y. S. (2017) Clonal history and genetic predictors of transformation into small-cell carcinomas from lung adenocarcinomas, J. Clin. Oncol., 35, 3065-3074, doi: https://doi.org/10.1200/JCO.2016.71.9096 .
Heitzer, E., Ulz, P., and Geigl, J. B. (2015) Circulating tumor DNA as a liquid biopsy for cancer, Clin. Chem., 61, 112-123, doi: https://doi.org/10.1373/clinchem.2014.222679 .
Yohe, S., and Thyagarajan, B. (2017) Review of clinical next-generation sequencing, Arch. Pathol. Lab. Med., 141, 1544-1557, doi: https://doi.org/10.5858/arpa.2016-0501-RA .
Greaves, M., and Maley, C. C. (2012) Clonal evolution in cancer, Nature, 481, 306-313, doi: https://doi.org/10.1038/nature10762 .
McGranahan, N., and Swanton, C. (2017) Clonal heterogeneity and tumor evolution: past, present, and the future, Cell, 168, 613-628, doi: https://doi.org/10.1016/j.cell.2017.01.018 .
Negrini, S., Gorgoulis, V. G., and Halazonetis, T. D. (2010) Genomic instability – an evolving hallmark of cancer, Nat. Rev. Mol. Cell Biol., 11, 220-228, doi: https://doi.org/10.1038/nrm2858 .
Jaramillo, M. L., Leon, Z., Grothe, S., Paul-Roc, B., Abulrob, A., and O’Connor, M. M. (2006) Effect of the anti-receptor ligand-blocking 225 monoclonal antibody on EGF receptor endocytosis and sorting, Exp. Cell Res., 312, 2778-2790, doi: https://doi.org/10.1016/j.yexcr.2006.05.008 .
Liao, H. J., and Carpenter, G. (2009) Cetuximab/C225-induced intracellular trafficking of epidermal growth factor receptor, Cancer Res., 69, 6179-6183, doi: https://doi.org/10.1158/0008-5472.CAN-09-0049 .
Bhattacharyya, S., Singh, R. D., Pagano, R., Robertson, J. D., Bhattacharya, R., and Mukherjee, P. (2012) Switching the targeting pathways of a therapeutic antibody by nanodesign, Angew. Chem. Int. Ed Engl., 51, 1563-1567, doi: https://doi.org/10.1002/anie.201105432 .
Kim, D. H., Kim, D. K., Zhou, K., Park, S., Kwon, Y., Jeong, M. G., Lee, N. K., and Ryu, S. H. (2017) Single particle tracking-based reaction progress kinetic analysis reveals a series of molecular mechanisms of cetuximab-induced EGFR processes in a single living cell, Chem. Sci., 8, 4823-4832, doi: https://doi.org/10.1039/c7sc01159h .
Tolmachev, V., and Orlova, A. (2020) Affibody molecules as targeting vectors for PET imaging, Cancers (Basel), 12, doi: https://doi.org/10.3390/cancers12030651 .
Gostring, L., Chew, M. T., Orlova, A., Hoiden-Guthenberg, I., Wennborg, A., Carlsson, J., and Frejd, F. Y. (2010) Quantification of internalization of EGFR-binding Affibody molecules: methodological aspects, Int. J. Oncol., 36, 757-763, doi: https://doi.org/10.3892/ijo_00000551 .
Genta, I., Chiesa, E., Colzani, B., Modena, T., Conti, B., and Dorati, R. (2017) GE11 peptide as an active targeting agent in antitumor therapy: a minireview, Pharmaceutics, 10, doi: https://doi.org/10.3390/pharmaceutics10010002 .
Li, N., Nguyen, H. H., Byrom, M., and Ellington, A. D. (2011) Inhibition of cell proliferation by an anti-EGFR aptamer, PLoS One, 6, e20299, doi: https://doi.org/10.1371/journal.pone.0020299 .
Maeda, H., Wu, J., Sawa, T., Matsumura, Y., and Hori, K. (2000) Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review, J. Control. Release, 65, 271-284, doi: https://doi.org/10.1016/s0168-3659(99)00248-5 .
Danhier, F. (2016) To exploit the tumor microenvironment: since the EPR effect fails in the clinic, what is the future of nanomedicine? J. Control. Release, 244, 108-121, doi: https://doi.org/10.1016/j.jconrel.2016.11.015 .
Golombek, S. K., May, J. N., Theek, B., Appold, L., Drude, N., Kiessling, F., and Lammers, T. (2018) Tumor targeting via EPR: strategies to enhance patient responses, Adv. Drug Deliv. Rev., 130, 17-38, doi: https://doi.org/10.1016/j.addr.2018.07.007 .
Park, J., Choi, Y., Chang, H., Um, W., Ryu, J. H., and Kwon, I. C. (2019) Alliance with EPR effect: combined strategies to improve the EPR effect in the tumor microenvironment, Theranostics, 9, 8073-8090, doi: https://doi.org/10.7150/thno.37198 .
Mamot, C., Ritschard, R., Wicki, A., Stehle, G., Dieterle, T., Bubendorf, L., Hilker, C., Deuster, S., Herrmann, R., and Rochlitz, C. (2012) Tolerability, safety, pharmacokinetics, and efficacy of doxorubicin-loaded anti-EGFR immunoliposomes in advanced solid tumours: a phase 1 dose-escalation study, Lancet Oncol., 13, 1234-1241, doi: https://doi.org/10.1016/S1470-2045(12)70476-X .
Mamot, C., Drummond, D. C., Noble, C. O., Kallab, V., Guo, Z., Hong, K., Kirpotin, D. B., and Park, J. W. (2005) Epidermal growth factor receptor-targeted immunoliposomes significantly enhance the efficacy of multiple anticancer drugs in vivo, Cancer Res., 65, 11631-11638, doi: https://doi.org/10.1158/0008-5472.CAN-05-1093 .
Pan, X., and Lee, R. J. (2007) Construction of anti-EGFR immunoliposomes via folate-folate binding protein affinity, Int. J. Pharm., 336, 276-283, doi: https://doi.org/10.1016/j.ijpharm.2006.12.007 .
Pan, X., Wu, G., Yang, W., Barth, R. F., Tjarks, W., and Lee, R. J. (2007) Synthesis of cetuximab-immunoliposomes via a cholesterol-based membrane anchor for targeting of EGFR, Bioconjug. Chem., 18, 101-108, doi: https://doi.org/10.1021/bc060174r .
Vazquez-Becerra, H., Perez-Cardenas, E., Muniz-Hernandez, S., Izquierdo-Sanchez, V., and Medina, L. A. (2017) Characterization and in vitro evaluation of nimotuzumab conjugated with cisplatin-loaded liposomes, J. Liposome Res., 27, 274-282, doi: https://doi.org/10.1080/08982104.2016.1207665 .
Petrilli, R., Eloy, J. O., Saggioro, F. P., Chesca, D. L., de Souza, M. C., Dias, M. V. S., daSilva, L. L. P., Lee, R. J., and Lopez, R. F. V. (2018) Skin cancer treatment effectiveness is improved by iontophoresis of EGFR-targeted liposomes containing 5-FU compared with subcutaneous injection, J. Control. Release, 283, 151-162, doi: https://doi.org/10.1016/j.jconrel.2018.05.038 .
Broekgaarden, M., van Vught, R., Oliveira, S., Roovers, R. C., van Bergen en Henegouwen, P. M. P., Pieters, R. J., Van Gulik, T. M., Breukink, E., and Heger, M. (2016) Site-specific conjugation of single domain antibodies to liposomes enhances photosensitizer uptake and photodynamic therapy efficacy, Nanoscale, 8, 6490-6494, doi: https://doi.org/10.1039/c6nr00014b .
Zalba, S., Contreras, A. M., Merino, M., Navarro, I., de Ilarduya, C. T., Troconiz, I. F., Koning, G., and Garrido, M. J. (2016) EGF-liposomes promote efficient EGFR targeting in xenograft colocarcinoma model, Nanomedicine (Lond.), 11, 465-477, doi: https://doi.org/10.2217/nnm.15.208 .
Jung, J., Jeong, S. Y., Park, S. S., Shin, S. H., Ju, E. J., Choi, J., Park, J., Lee, J. H., Kim, I., Suh, Y. A., Hwang, J. J., Kuroda, S., Lee, J. S., Song, S. Y., and Choi, E. K. (2015) A cisplatinincorporated liposome that targets the epidermal growth factor receptor enhances radiotherapeutic efficacy without nephrotoxicity, Int. J. Oncol., 46, 1268-1274, doi: https://doi.org/10.3892/ijo.2014.2806 .
Aggarwal, S., Gupta, S., Pabla, D., and Murthy, R. S. (2013) Gemcitabine-loaded PLGA-PEG immunonanoparticles for targeted chemotherapy of pancreatic cancer, Cancer Nanotechnol., 4, 145-157, doi: https://doi.org/10.1007/s12645-013-0046-3 .
Wöll, S., Bachran, C., Schiller, S., Schröder, M., Conrad, L., Swee, L. K., and Scherliess, R. (2018) Sortaggable liposomes: evaluation of reaction conditions for single-domain antibody conjugation by Sortase-A and targeting of CD11b(+) myeloid cells, Eur. J. Pharm. Biopharm., 133, 138-150, doi: https://doi.org/10.1016/j.ejpb.2018.09.017 .
Wang, Y. P., Liu, I. J., Chung, M. J., and Wu, H. C. (2020) Novel anti-EGFR scFv human antibody-conjugated immunoliposomes enhance chemotherapeutic efficacy in squamous cell carcinoma of head and neck, Oral Oncol., 106, 104689, doi: https://doi.org/10.1016/j.oraloncology.2020.104689 .
Hsu, W. C., Cheng, C. N., Lee, T. W., and Hwang, J. J. (2015) Cytotoxic effects of PEGylated anti-EGFR immunoliposomes combined with doxorubicin and Rhenium-188 against cancer cells, Anticancer Res., 35, 4777-4788.
Haeri, A., Zalba, S., Ten Hagen, T. L., Dadashzadeh, S., and Koning, G. A. (2016) EGFR targeted thermosensitive liposomes: a novel multifunctional platform for simultaneous tumor targeted and stimulus responsive drug delivery, Colloids Surf. B Biointerfaces, 146, 657-669, doi: https://doi.org/10.1016/j.colsurfb.2016.06.012 .
Kim, I. Y., Kang, Y. S., Lee, D. S., Park, H. J., Choi, E. K., Oh, Y. K., Son, H. J., and Kim, J. S. (2009) Antitumor activity of EGFR targeted pH-sensitive immunoliposomes encapsulating gemcitabine in A549 xenograft nude mice, J. Control. Release, 140, 55-60, doi: https://doi.org/10.1016/j.jconrel.2009.07.005 .
Talelli, M., Oliveira, S., Rijcken, C. J., Pieters, E. H., Etrych, T., Ulbrich, K., van Nostrum, R. C., Storm, G., Hennink, W. E., and Lammers, T. (2013) Intrinsically active nanobody-modified polymeric micelles for tumor-targeted combination therapy, Biomaterials, 34, 1255-1260, doi: https://doi.org/10.1016/j.biomaterials.2012.09.064 .
Gener, P., Gouveia, L. P., Sabat, G. R., de Sousa Rafael, D. F., Fort, N. B., Arranja, A., Fernandez, Y., Prieto, R. M., Ortega, J. S., Arango, D., Abasolo, I., Videira, M., and Schwartz, S., Jr. (2015) Fluorescent CSC models evidence that targeted nanomedicines improve treatment sensitivity of breast and colon cancer stem cells, Nanomedicine, 11, 1883-1892.
Kang, S. J., Jeong, H. Y., Kim, M. W., Jeong, I. H., Choi, M. J., You, Y. M., Im, C. S., Song, I. H., Lee, T. S., and Park, Y. S. (2018) Anti-EGFR lipid micellar nanoparticles co-encapsulating quantum dots and paclitaxel for tumor-targeted theranosis, Nanoscale, 10, 19338-19350, doi: https://doi.org/10.1039/c8nr05099f .
Mondal, G., Almawash, S., Chaudhary, A. K., and Mahato, R. I. (2017) EGFR-targeted cationic polymeric mixed micelles for codelivery of gemcitabine and miR-205 for treating advanced pancreatic cancer, Mol. Pharm., 14, 3121-3133, doi: https://doi.org/10.1021/acs.molpharmaceut.7b00355 .
Kutty, R. V., Chia, S. L., Setyawati, M. I., Muthu, M. S., Feng, S. S., and Leong, D. T. (2015) In vivo and ex vivo proofs of concept that cetuximab conjugated vitamin E TPGS micelles increases efficacy of delivered docetaxel against triple negative breast cancer, Biomaterials, 63, 58-69, doi: https://doi.org/10.1016/j.biomaterials.2015.06.005 .
Jiang, J., Chen, H., Yu, C., Zhang, Y., Chen, M., Tian, S., and Sun, C. (2015) The promotion of salinomycin delivery to hepatocellular carcinoma cells through EGFR and CD133 aptamers conjugation by PLGA nanoparticles, Nanomedicine (Lond.), 10, 1863-1879, doi: https://doi.org/10.2217/nnm.15.43 .
Chen, J., Ouyang, J., Chen, Q., Deng, C., Meng, F., Zhang, J., Cheng, R., Lan, Q., and Zhong, Z. (2017) EGFR and CD44 dual-targeted multifunctional hyaluronic acid Nanogels boost protein delivery to ovarian and breast cancers in vitro and in vivo, ACS Appl. Mater. Interfaces, 9, 24140-24147, doi: https://doi.org/10.1021/acsami.7b06879 .
Gao, J., Xia, Y., Chen, H., Yu, Y., Song, J., Li, W., Qian, W., Wang, H., Dai, J., and Guo, Y. (2014) Polymer-lipid hybrid nanoparticles conjugated with anti-EGF receptor antibody for targeted drug delivery to hepatocellular carcinoma, Nanomedicine (Lond.), 9, 279-293, doi: https://doi.org/10.2217/nnm.13.20 .
Zhai, J., Luwor, R. B., Ahmed, N., Escalona, R., Tan, F. H., Fong, C., Ratcliffe, J., Scoble, J. A., Drummond, C. J., and Tran, N. (2018) Paclitaxel-loaded self-assembled lipid nanoparticles as targeted drug delivery systems for the treatment of aggressive ovarian cancer, ACS Appl. Mater. Interfaces, 10, 25174-25185, doi: https://doi.org/10.1021/acsami.8b08125 .
Karra, N., Nassar, T., Ripin, A. N., Schwob, O., Borlak, J., and Benita, S. (2013) Antibody conjugated PLGA nanoparticles for targeted delivery of paclitaxel palmitate: efficacy and biofate in a lung cancer mouse model, Small, 9, 4221-4236, doi: https://doi.org/10.1002/smll.201301417 .
Sreeranganathan, M., Uthaman, S., Sarmento, B., Mohan, C. G., Park, I. K., and Jayakumar, R. (2017) In vivo evaluation of cetuximab-conjugated poly(gamma-glutamic acid)-docetaxel nanomedicines in EGFR-overexpressing gastric cancer xenografts, Int. J. Nanomedicine, 12, 7165-7182, doi: https://doi.org/10.2147/IJN.S143529 .
Patel, J., Amrutiya, J., Bhatt, P., Javia, A., Jain, M., and Misra, A. (2018) Targeted delivery of monoclonal antibody conjugated docetaxel loaded PLGA nanoparticles into EGFR overexpressed lung tumour cells, J. Microencapsul., 35, 204-217, doi: https://doi.org/10.1080/02652048.2018.1453560 .
Wang, J. K., Zhou, Y. Y., Guo, S. J., Wang, Y. Y., Nie, C. J., Wang, H. L., Wang, J. L., Zhao, Y., Li, X. Y., and Chen, X. J. (2017) Cetuximab conjugated and doxorubicin loaded silica nanoparticles for tumor-targeting and tumor microenvironment responsive binary drug delivery of liver cancer therapy, Mater. Sci. Eng. C Mater. Biol. Appl., 76, 944-950, doi: https://doi.org/10.1016/j.msec.2017.03.131 .
Sandoval, M. A., Sloat, B. R., Lansakara, P., Kumar, A., Rodriguez, B. L., Kiguchi, K., Digiovanni, J., and Cui, Z. (2012) EGFR-targeted stearoyl gemcitabine nanoparticles show enhanced anti-tumor activity, J. Control. Release, 157, 287-296, doi: https://doi.org/10.1016/j.jconrel.2011.08.015 .
Bouras, A., Kaluzova, M., and Hadjipanayis, C. G. (2015) Radiosensitivity enhancement of radioresistant glioblastoma by epidermal growth factor receptor antibody-conjugated iron-oxide nanoparticles, J. Neurooncol., 124, 13-22, doi: https://doi.org/10.1007/s11060-015-1807-0 .
Chen, C. H., Wu, Y. J., and Chen, J. J. (2016) Photo-thermal therapy of bladder cancer with Anti-EGFR antibody conjugated gold nanoparticles, Front. Biosci. (Landmark Ed.), 21, 1211-1221, doi: https://doi.org/10.2741/4451 .
El-Sayed, I. H., Huang, X., and El-Sayed, M. A. (2006) Selective laser photo-thermal therapy of epithelial carcinoma using anti-EGFR antibody conjugated gold nanoparticles, Cancer Lett., 239, 129-135, doi: https://doi.org/10.1016/j.canlet.2005.07.035 .
Rosenkranz, A. A., Slastnikova, T. A., Georgiev, G. P., Zalutsky, M. R., and Sobolev, A. S. (2020) Delivery systems exploiting natural cell transport processes of macromolecules for intracellular targeting of Auger electron emitters, Nucl. Med. Biol., 80-81, 45-56, doi: https://doi.org/10.1016/j.nucmedbio.2019.11.005 .
Li, L., Quang, T. S., Gracely, E. J., Kim, J. H., Emrich, J. G., Yaeger, T. E., Jenrette, J. M., Cohen, S. C., Black, P., and Brady, L. W. (2010) A phase II study of anti-epidermal growth factor receptor radioimmunotherapy in the treatment of glioblastoma multiforme, J. Neurosurg., 113, 192-198, doi: https://doi.org/10.3171/2010.2.JNS091211 .
Emrich, J. G., Bender, H., Class, R., Eshleman, J., Miyamoto, C., and Brady, L. W. (1996) In vitro evaluation of iodine-125-labeled monoclonal antibody (MAb 425) in human high-grade glioma cells, Am. J. Clin. Oncol., 19, 601-608, doi: https://doi.org/10.1097/00000421-199612000-00015 .
Reilly, R. M., Chen, P., Wang, J., Scollard, D., Cameron, R., and Vallis, K. A. (2006) Preclinical pharmacokinetic, biodistribution, toxicology, and dosimetry studies of 111In-DTPA-human epidermal growth factor: an Auger electron-emitting radiotherapeutic agent for epidermal growth factor receptor-positive breast cancer, J. Nucl. Med., 47, 1023-1031.
Panosa, C., Fonge, H., Ferrer-Batalle, M., Menendez, J. A., Massaguer, A., De Llorens, R., and Reilly, R. M. (2015) A comparison of non-biologically active truncated EGF (EGFt) and full-length hEGF for delivery of Auger electron-emitting 111In to EGFR-positive breast cancer cells and tumor xenografts in athymic mice, Nucl. Med. Biol., 42, 931-938, doi: https://doi.org/10.1016/j.nucmedbio.2015.08.003 .
Vallis, K. A., Reilly, R. M., Scollard, D., Merante, P., Brade, A., Velauthapillai, S., Caldwell, C., Chan, I., Freeman, M., Lockwood, G., Miller, N. A., Cornelissen, B., Petronis, J., and Sabate, K. (2014) Phase I trial to evaluate the tumor and normal tissue uptake, radiation dosimetry and safety of (111)In-DTPA-human epidermal growth factor in patients with metastatic EGFR-positive breast cancer, Am. J. Nucl. Med. Mol. Imaging, 4, 181-192.
Cornelissen, B., Waller, A., Able, S., and Vallis, K. A. (2013) Molecular radiotherapy using cleavable radioimmunoconjugates that target EGFR and gammaH2AX, Mol. Cancer Ther., 12, 2472-2482, doi: https://doi.org/10.1158/1535-7163.MCT-13-0369 .
Rosenkranz, A. A., Ulasov, A. V., Slastnikova, T. A., Khramtsov, Y. V., and Sobolev, A. S. (2014) Use of intracellular transport processes for targeted drug delivery into a specified cellular compartment, Biochemistry (Moscow), 79, 928-946, doi: https://doi.org/10.1134/S0006297914090090 .
Sobolev, A. S. (2009) Modular nanotransporters of anticancer drugs conferring cell specificity and higher efficiency, Biochemistry (Moscow), 74, 1567-1574, doi: https://doi.org/10.1134/s0006297909130094 .
Sobolev, A. S., Aliev, R. A., and Kalmykov, S. N. (2016) Radionuclides emitting short-range particles and modular nanotransporters for their delivery to target cancer cells, Rus. Chem. Rev., 85, 1011-1032.
Sobolev, A. S. (2018) Modular nanotransporters for nuclear-targeted delivery of Auger electron emitters, Front. Pharmacol., 9, 952, doi: https://doi.org/10.3389/fphar.2018.00952 .
Gilyazova, D. G., Rosenkranz, A. A., Gulak, P. V., Lunin, V. G., Sergienko, O. V., Khramtsov, Y. V., Timofeyev, K. N., Grin, M. A., Mironov, A. F., Rubin, A. B., Georgiev, G. P., and Sobolev, A. S. (2006) Targeting cancer cells by novel engineered modular transporters, Cancer Res., 66, 10534-10540, doi: https://doi.org/10.1158/0008-5472.CAN-06-2393 .
Slastnikova, T. A., Rosenkranz, A. A., Gulak, P. V., Schiffelers, R. M., Lupanova, T. N., Khramtsov, Y. V., Zalutsky, M. R., and Sobolev, A. S. (2012) Modular nanotransporters: a multipurpose in vivo working platform for targeted drug delivery, Int. J. Nanomed., 7, 467-482, doi: https://doi.org/10.2147/IJN.S28249 .
Slastnikova, T. A., Koumarianou, E., Rosenkranz, A. A., Vaidyanathan, G., Lupanova, T. N., Sobolev, A. S., and Zalutsky, M. R. (2012) Modular nanotransporters: a versatile approach for enhancing nuclear delivery and cytotoxicity of Auger electron-emitting 125I, EJNMMI Res., 2, 59, doi: https://doi.org/10.1186/2191-219X-2-59 .
Koumarianou, E., Slastnikova, T. A., Pruszynski, M., Rosenkranz, A. A., Vaidyanathan, G., Sobolev, A. S., and Zalutsky, M. R. (2014) Radiolabeling and in vitro evaluation of (67)Ga-NOTA-modular nanotransporter – a potential Auger electron emitting EGFR-targeted radiotherapeutic, Nucl. Med. Biol., 41, 441-449, doi: https://doi.org/10.1016/j.nucmedbio.2014.03.026 .
Slastnikova, T. A., Rosenkranz, A. A., Morozova, N. B., Vorontsova, M. S., Petriev, V. M., Lupanova, T. N., Ulasov, A. V., Zalutsky, M. R., Yakubovskaya, R. I., and Sobolev, A. S. (2017) Preparation, cytotoxicity, and in vivo antitumor efficacy of (111)In-labeled modular nanotransporters, Int. J. Nanomed., 12, 395-410, doi: https://doi.org/10.2147/IJN.S125359 .
Rosenkranz, A. A., Slastnikova, T. A., Karmakova, T. A., Vorontsova, M. S., Morozova, N. B., Petriev, V. M., Abrosimov, A. S., Khramtsov, Y. V., Lupanova, T. N., Ulasov, A. V., Yakubovskaya, R. I., Georgiev, G. P., and Sobolev, A. S. (2018) Antitumor activity of Auger electron emitter 111In delivered by modular nanotransporter for treatment of bladder cancer with EGFR overexpression, Front. Pharmacol., 9, 1331, doi: https://doi.org/10.3389/fphar.2018.01331 .
Karyagina, T. S., Ulasov, A. V., Slastnikova, T. A., Rosenkranz, A. A., Lupanova, T. N., Khramtsov, Y. V., Georgiev, G. P., and Sobolev, A. S. (2020) Targeted delivery of 111In into the nuclei of EGFR overexpressing cells via modular nanotransporters with anti-EGFR affibody, Front. Pharmacol., 11, 176, doi: https://doi.org/10.3389/fphar.2020.00176 .
Li, S., Goins, B., Hrycushko, B. A., Phillips, W. T., and Bao, A. (2012) Feasibility of eradication of breast cancer cells remaining in postlumpectomy cavity and draining lymph nodes following intracavitary injection of radioactive immunoliposomes, Mol. Pharm., 9, 2513-2522, doi: https://doi.org/10.1021/mp300132f .
Song, H., Hedayati, M., Hobbs, R. F., Shao, C., Bruchertseifer, F., Morgenstern, A., Deweese, T. L., and Sgouros, G. (2013) Targeting aberrant DNA double-strand break repair in triple-negative breast cancer with alpha-particle emitter radiolabeled anti-EGFR antibody, Mol. Cancer Ther., 12, 2043-2054, doi: https://doi.org/10.1158/1535-7163.MCT-13-0108 .
Pfost, B., Seidl, C., Autenrieth, M., Saur, D., Bruchertseifer, F., Morgenstern, A., Schwaiger, M., and Senekowitsch-Schmidtke, R. (2009) Intravesical alpha-radioimmunotherapy with 213Bi-anti-EGFR-mAb defeats human bladder carcinoma in xenografted nude mice, J. Nucl. Med., 50, 1700-1708, doi: https://doi.org/10.2967/jnumed.109.065961 .
Fazel, J., Rotzer, S., Seidl, C., Feuerecker, B., Autenrieth, M., Weirich, G., Bruchertseifer, F., Morgenstern, A., and Senekowitsch-Schmidtke, R. (2015) Fractionated intravesical radioimmunotherapy with 213Bi-anti-EGFR-MAb is effective without toxic side-effects in a nude mouse model of advanced human bladder carcinoma, Cancer Biol. Ther., 16, 1526-1534, doi: https://doi.org/10.1080/15384047.2015.1071735 .
Autenrieth, M. E., Seidl, C., Bruchertseifer, F., Horn, T., Kurtz, F., Feuerecker, B., D’Alessandria, C., Pfob, C., Nekolla, S., Apostolidis, C., Mirzadeh, S., Gschwend, J. E., Schwaiger, M., Scheidhauer, K., and Morgenstern, A. (2018) Treatment of carcinoma in situ of the urinary bladder with an alpha-emitter immunoconjugate targeting the epidermal growth factor receptor: a pilot study, Eur. J. Nucl. Med. Mol. Imaging, 45, 1364-1371, doi: https://doi.org/10.1007/s00259-018-4003-6 .
Milenic, D. E., Baidoo, K. E., Kim, Y. S., and Brechbiel, M. W. (2015) Evaluation of cetuximab as a candidate for targeted alpha-particle radiation therapy of HER1-positive disseminated intraperitoneal disease, MAbs, 7, 255-264, doi: https://doi.org/10.4161/19420862.2014.985160 .
Zidenberg-Cherr, S., Parks, N. J., and Keen, C. L. (1987) Tissue and subcellular distribution of bismuth radiotracer in the rat: considerations of cytotoxicity and microdosimetry for bismuth radiopharmaceuticals, Radiat. Res., 111, 119-129.
Rosenkranz, A. A., Vaidyanathan, G., Pozzi, O. R., Lunin, V. G., Zalutsky, M. R., and Sobolev, A. S. (2008) Engineered modular recombinant transporters: application of new platform for targeted radiotherapeutic agents to alpha-particle emitting 211 At, Int. J. Radiat. Oncol. Biol. Phys., 72, 193-200, doi: https://doi.org/10.1016/j.ijrobp.2008.05.055 .
Simon, N., and FitzGerald, D. (2016) Immunotoxin therapies for the treatment of epidermal growth factor receptor-dependent cancers, Toxins (Basel), 8, doi: https://doi.org/10.3390/toxins8050137 .
Yang, Y., Tian, Z., Ding, Y., Li, X., Zhang, Z., Yang, L., Zhao, F., Ren, F., and Guo, R. (2018) EGFR-targeted immunotoxin exerts antitumor effects on esophageal cancers by increasing ROS accumulation and inducing apoptosis via inhibition of the Nrf2-Keap1 pathway, J. Immunol. Res., 2018, 1090287, doi: https://doi.org/10.1155/2018/1090287 .
Deng, C., Xiong, J., Gu, X., Chen, X., Wu, S., Wang, Z., Wang, D., Tu, J., and Xie, J. (2017) Novel recombinant immunotoxin of EGFR specific nanobody fused with cucurmosin, construction and antitumor efficiency in vitro, Oncotarget, 8, 38568-38580, doi: https://doi.org/10.18632/oncotarget.16930 .
Kim, J. S., Jun, S. Y., and Kim, Y. S. (2020) Critical issues in the development of immunotoxins for anticancer therapy, J. Pharm. Sci., 109, 104-115, doi: https://doi.org/10.1016/j.xphs.2019.10.037 .
Sampson, J. H., Akabani, G., Archer, G. E., Berger, M. S., Coleman, R. E., Friedman, A. H., Friedman, H. S., Greer, K., Herndon, J. E., Kunwar, S., McLendon, R. E., Paolino, A., Petry, N. A., Provenzale, J. M., Reardon, D. A., Wong, T. Z., Zalutsky, M. R., Pastan, I., and Bigner, D. D. (2008) Intracerebral infusion of an EGFR-targeted toxin in recurrent malignant brain tumors, Neuro. Oncol., 10, 320-329, doi: https://doi.org/10.1215/15228517-2008-012 .
Chandramohan, V., Pegram, C. N., Piao, H., Szafranski, S. E., Kuan, C. T., Pastan, I. H., and Bigner, D. D. (2017) Production and quality control assessment of a GLP-grade immunotoxin, D2C7-(scdsFv)-PE38KDEL, for a phase I/II clinical trial, Appl. Microbiol. Biotechnol., 101, 2747-2766, doi: https://doi.org/10.1007/s00253-016-8063-x .
Zalutsky, M. R., Boskovitz, A., Kuan, C. T., Pegram, C. N., Ayriss, J., Wikstrand, C. J., Buckley, A. F., Lipp, E. S., Herndon, J. E., McLendon, R. E., and Bigner, D. D. (2012) Radioimmunotargeting of malignant glioma by monoclonal antibody D2C7 reactive against both wild-type and variant III mutant epidermal growth factor receptors, Nucl. Med. Biol., 39, 23-34, doi: https://doi.org/10.1016/j.nucmedbio.2011.06.005 .
Chandramohan, V., Bao, X., Keir, S. T., Pegram, C. N., Szafranski, S. E., Piao, H., Wikstrand, C. J., McLendon, R. E., Kuan, C. T., Pastan, I. H., and Bigner, D. D. (2013) Construction of an immunotoxin, D2C7-(scdsFv)-PE38KDEL, targeting EGFRwt and EGFRvIII for brain tumor therapy, Clin. Cancer Res., 19, 4717-4727, doi: https://doi.org/10.1158/1078-0432.CCR-12-3891 .
Chandramohan, V., Bao, X., Yu, X., Parker, S., McDowall, C., Yu, Y. R., Healy, P., Desjardins, A., Gunn, M. D., Gromeier, M., Nair, S. K., Pastan, I. H., and Bigner, D. D. (2019) Improved efficacy against malignant brain tumors with EGFRwt/EGFRvIII targeting immunotoxin and checkpoint inhibitor combinations, J. Immunother. Cancer, 7, 142, doi: https://doi.org/10.1186/s40425-019-0614-0 .
Agostinis, P., Berg, K., Cengel, K. A., Foster, T. H., Girotti, A. W., Gollnick, S. O., Hahn, S. M., Hamblin, M. R., Juzeniene, A., Kessel, D., Korbelik, M., Moan, J., Mroz, P., Nowis, D., Piette, J., Wilson, B. C., and Golab, J. (2011) Photodynamic therapy of cancer: an update, CA Cancer J. Clin., 61, 250-281, doi: https://doi.org/10.3322/caac.20114 .
Liang, H., Shin, D. S., Lee, Y. E., Nguyen, D. C., Trang, T. C., Pan, A. H., Huang, S. L., Chong, D. H., and Berns, M. W. (1998) Subcellular phototoxicity of 5-aminolaevulinic acid (ALA), Lasers Surg. Med., 22, 14-24, doi: https://doi.org/10.1002/(sici)1096-9101(1998)22:1<14::aid-lsm6>3.0.co;2-# .
Rosenkranz, A. A., Jans, D. A., and Sobolev, A. S. (2000) Targeted intracellular delivery of photosensitizers to enhance photodynamic efficiency, Immunol. Cell Biol., 78, 452-464, doi: https://doi.org/10.1046/j.1440-1711.2000.00925.x .
Gilyazova, D. G., Rosenkranz, A. A., Gulak, P. V., Lunin, V. G., Sergienko, O. V., Grin, M. A., Mironov, A. F., Rubin, A. B., and Sobolev, A. S. (2006) Recombinant modular transporters on the basis of epidermal growth factor for targeted intracellular delivery of photosensitizers, Cur. Res. Laser Use Oncol.: 2000-2004, 5973, 59730E, doi: https://doi.org/10.1117/12.640049 .
Gillenwater, A. M., Johnson, J. M., Curry, J. M., Kochuparambil, S. T., McDonald, D., Fidler, M., Stenson, K. M., Vasan, N. R., Razaq, M. A., and Campana, J. (2020) Survival following photoimmunotherapy in patients (Pts) with recurrent Head and neck squamous cell carcinoma (rHNSCC), Int. J. Radiat. Oncol. Biol. Phys., 106, 1180-1180.
Fernandes, S. R. G., Fernandes, R., Sarmento, B., Pereira, P. M. R., and Tome, J. P. C. (2019) Photoimmunoconjugates: novel synthetic strategies to target and treat cancer by photodynamic therapy, Org. Biomol. Chem., 17, 2579-2593, doi: https://doi.org/10.1039/c8ob02902d .
Harvey, T. J., Burdon, D., Steele, L., Ingram, N., Hall, G. D., Selby, P. J., Vile, R. G., Cooper, P. A., Shnyder, S. D., and Chester, J. D. (2010) Retargeted adenoviral cancer gene therapy for tumour cells overexpressing epidermal growth factor receptor or urokinase-type plasminogen activator receptor, Gene Ther., 17, 1000-1010, doi: https://doi.org/10.1038/gt.2010.45 .
Urnauer, S., Muller, A. M., Schug, C., Schmohl, K. A., Tutter, M., Schwenk, N., Rodl, W., Morys, S., Ingrisch, M., Bertram, J., Bartenstein, P., Clevert, D. A., Wagner, E., and Spitzweg, C. (2017) EGFR-targeted nonviral NIS gene transfer for bioimaging and therapy of disseminated colon cancer metastases, Oncotarget, 8, 92195-92208, doi: https://doi.org/10.18632/oncotarget.21028 .
Liang, Y., Peng, J., Li, N., Yu-Wai-Man, C., Wang, Q., Xu, Y., Wang, H., Tagalakis, A. D., and Du, Z. (2019) Smart nanoparticles assembled by endogenous molecules for siRNA delivery and cancer therapy via CD44 and EGFR dual-targeting, Nanomedicine, 15, 208-217, doi: https://doi.org/10.1016/j.nano.2018.09.018 .
Bagley, S. J., and O’Rourke, D. M. (2020) Clinical investigation of CAR T cells for solid tumors: lessons learned and future directions, Pharmacol. Ther., 205, 107419, doi: https://doi.org/10.1016/j.pharmthera.2019.107419 .
Westphal, M., Maire, C. L., and Lamszus, K. (2017) EGFR as a target for glioblastoma treatment: an unfulfilled promise, CNS Drugs, 31, 723-735, doi: https://doi.org/10.1007/s40263-017-0456-6 .
Xu, M. J., Johnson, D. E., and Grandis, J. R. (2017) EGFR-targeted therapies in the post-genomic era, Cancer Metastasis Rev., 36, 463-473, doi: https://doi.org/10.1007/s10555-017-9687-8 .
Slastnikova, T. A., Ulasov, A. V., Rosenkranz, A. A., and Sobolev, A. S. (2018) Targeted intracellular delivery of antibodies: the state of the art, Front. Pharmacol., 9, 1208, doi: https://doi.org/10.3389/fphar.2018.01208 .
Ulasov, A. V., Rosenkranz, A. A., and Sobolev, A. S. (2018) Transcription factors: time to deliver, J. Control. Release, 269, 24-35, doi: https://doi.org/10.1016/j.jconrel.2017.11.004 .
Jin, J. O., Kim, G., Hwang, J., Han, K. H., Kwak, M., and Lee, P. C. W. (2020) Nucleic acid nanotechnology for cancer treatment, Biochim. Biophys. Acta Rev. Cancer, 1874, 188377, doi: https://doi.org/10.1016/j.bbcan.2020.188377 .
Funding
This work was financially supported by the Russian Foundation for Basic Research (project no. 19-14-50385\19).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare no conflict of interest in financial or any other sphere. This article does not contain any studies with human participants or animals performed by any of the authors.
Rights and permissions
Open access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License ( http://creativecommons.org/licenses/by/4.0/ ), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Rosenkranz, A.A., Slastnikova, T.A. Epidermal Growth Factor Receptor: Key to Selective Intracellular Delivery. Biochemistry Moscow 85, 967–993 (2020). https://doi.org/10.1134/S0006297920090011
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0006297920090011