
Abstract
The p53 tumor-suppressor plays a critical role in the prevention of human cancer. In the absence of cellular stress, the p53 protein is maintained at low steady-state levels and exerts very little, if any, effect on cell fate. However, in response to various types of stress, p53 becomes activated; this is reflected in elevated protein levels, as well as augmented biochemical capabilities. As a consequence of p53 activation, cells can undergo marked phenotypic changes, ranging from increased DNA repair to senescence and apoptosis. This review deals with the mechanisms that underlie the apoptotic activities of p53, as well as the complex interactions between p53 and central regulatory signaling networks. In p53-mediated apoptosis, the major role is played by the ability of p53 to transactivate specific target genes. The choice of particular subsets of target genes, dictated by covalent p53 modifications and protein–protein interactions, can make the difference between life and apoptotic death of a cell. In addition, transcriptional repression of antiapoptotic genes, as well as transcription-independent activities of p53, can also contribute to the apoptotic effects of p53. Regarding the crosstalk between p53 and signaling networks, this review focuses on the interplay between p53 and two pivotal regulatory proteins: β-catenin and Akt/PKB. Both proteins can regulate p53 as well as be regulated by it. In addition, p53 interacts with the GSK-3β kinase, which serves as a link between Akt and β-catenin. This review discusses how the functional balance between these different interactions might dictate the likelihood of a given cell to become cancerous or be eliminated from the replicative pool, resulting in suppression of cancer.
Similar content being viewed by others

Introduction
Over a decade ago, it was appreciated that the p53 protein is the product of a pivotal tumor-suppressor gene, whose inactivation is implicated in a high percentage of individual cases of human cancer.1 Since then, p53 has become the focus of intensive research, basic as well as clinical. This has led to an unprecedented wealth of knowledge about p53, its biochemistry, its biological roles and its relevance to cancer. This knowledge is summarized in many excellent review articles; for some recent reviews on p53, its regulation and its role in apoptosis see Alarcon-Vargas and Ronai,2 Ashcroft et al.,3 Bargonetti and Manfredi,4 Hickman et al.,5 Michael and Oren,6 Sears and Nevins,7 Shen and White,8 Vogelstein et al.,9 and Woods Vousden.10
In a nutshell, p53 is primarily a sequence-specific transcriptional activator. It binds to cognate p53 responsive elements within the genome and activates the transcription of genes residing in the vicinity of these binding sites. The proteins encoded by the p53 target genes, whose number is probably in the hundreds, contribute in multiple ways to the biological effects of p53. The biological outcomes of p53 activity include apoptosis, inhibition of cell cycle progression, senescence, differentiation and accelerated DNA repair. However, these effects of p53 are not exerted indiscriminately. Rather, p53 is believed to reside in a biologically latent state in the absence of cellular stress, although the exact biochemical nature of this latency remains under debate.11 When cells experience a variety of stress conditions, p53 becomes activated. Activation involves a marked increase in the cellular abundance of p53 molecules, as well as qualitative changes that endow each p53 molecule with improved capabilities to modulate gene expression and alter the cell phenotype. The types of stress that promote p53 activation include many conditions associated with cancer initiation and progression, such as direct DNA damage, chromosomal aberrations, illegitimate activation of oncogenes, hypoxia, telomere shortening and more. The practical implication of these facts is that when a cell undergoes alterations that predispose it to become cancerous, p53 is activated to trigger a response that either takes care of the damage (by augmented DNA repair) or else eliminates the affected cell from the replicative pool, thereby preventing its expansion into a large population of malignant progeny. The diversity of cancer-related signals that trigger a protective p53 response probably accounts for its being such a central tumor-suppressor, and explains why its inactivation is so frequently selected for in almost all types of cancer.
This review will address some of the basic rules that govern the activity of p53, and describe in some detail several examples illustrating the complexity of p53 regulation as it pertains to apoptosis and to cancer.
Mdm2 and p53: caught in a loop
In the absence of stress, p53 is maintained at very low steady-state levels, and is thus prevented from exerting profound effects on the cell phenotype. Furthermore, the relatively few p53 molecules that do exist under such conditions appear to be rather ineffective as transcriptional activators, although they do contribute to the maintenance of basal levels of at least several p53 target genes. Multiple lines of evidence indicate that the lion's share of the negative regulation of p53, under nonstressed conditions, is performed by the Mdm2 protein (for recent reviews on Mdm2 see Michael and Oren,6 Daujat et al.,12 Deb13 and Momand et al.14) Mdm2 is a product of a proto-oncogene, amplified or otherwise overexpressed in a significant number of human tumors. Its main claim to fame is that it binds tightly to p53 and renders it inactive. This inactivation is achieved through at least two distinct molecular mechanisms (Figure 1). On the one hand, Mdm2 interferes with the transcriptional activity of p53, by virtue of its binding to the N-terminal transactivation domain of p53; this blocks critical interactions with other proteins necessary for p53-dependent regulation of gene expression. On the other hand, Mdm2 plays a cardinal role in the ongoing degradation of p53 under nonstressed conditions. This is achieved through the ability of Mdm2 to act as a p53-specific E3 ubiquitin ligase: upon binding to p53, it attaches to it ubiquitin moieties, which target p53 for rapid degradation by the 26S proteasome. Thus, elevated levels of Mdm2 will interfere with the activity of p53, even under conditions where p53 is normally expected to be functional. Parenthetically, it should be mentioned that there is evidence for additional, Mdm2-independent mechanisms for p53 degradation, whose relative contribution to the maintenance of low steady-state p53 levels may vary under different conditions.15,16,17,18

p53–Mdm2 autoregulatory loop. Mdm2 protein binds to p53 and inactivates it through at least two distinct mechanisms: physical blockage of the transcriptional activities of p53, and promotion of p53 ubiquitination and subsequent proteasomal degradation. In parallel, p53 can bind to p53 binding sites within the promoter of the mdm2 gene, and positively regulate Mdm2 expression
There exists, however, a second tier in the p53-Mdm2 interplay. The mdm2 gene contains two adjacent p53 binding sites (p53BS) within its first intron, rendering it a favorite target of p53. In its capacity as a sequence-specific transcriptional activator, p53 can bind to these p53BS and trigger the expression of Mdm2 (Figure 1). This thus establishes a negative autoregulatory feedback loop, wherein p53 stimulates Mdm2 synthesis, which in turn shuts off p53 activity (Figure 1). This loop serves as the receiving end for a plethora of incoming signals, which collectively dictate the state of p53 activity within a given cell under a given set of conditions.6 Simply put, signals that render p53 immune to Mdm2 will drive p53 activation, whereas signals that boost Mdm2 and allow it to overpower p53 will repress p53 activity. As will be illustrated later, this simple equation appears to generally hold true, although the actual outcome depends on many more factors than just p53 and Mdm2.
Life and death choices of p53
Once p53 wins over Mdm2 and becomes activated, it is potentially free to realize its capacity for driving dramatic changes in cell fate. However, this potential is not always fully realized. In particular, normal cells often seem to be more refractory to the effects of p53 than their tumor-derived counterparts. Moreover, rapidly proliferating cells appear to be more sensitive to p53 activation than resting or slowly proliferating cells. One encouraging outcome of this knowledge is that excessive activation of p53, considered by many drug hunters as an appealing option for novel cancer therapies, is expected to target preferentially tumor cells while relatively sparing unaffected normal tissue.
When cells do respond adequately to p53-activating signals, the actual biological outcome may vary greatly. A question that has received great attention, in part owing to its paramount relevance to the successful application of cancer chemotherapy, is how p53 chooses between induction of apoptosis versus induction of a viable growth arrest. As it appears now, much of the choice is not in the hands of p53; rather, it is the cellular context, as defined by the balance of intracellular and extracellular signaling events, which dictates whether p53 activation will spare the cell or lead to its apoptotic demise. A key component in this ‘cellular context’ input is the availability of survival signals, in the form of secreted molecules as well as those emanating from cell–cell and cell–matrix interactions. Such survival signals will typically render a cell more resistant to apoptosis, including p53-mediated apoptosis. When survival signals are available, p53 activation will more likely result in cessation of cell cycle progression, either transient – perhaps pending on successful alleviation of the stress signal that triggered the p53 response – or irreversible, in which case it may bear the features of replicative senescence. In the absence of adequate survival factors, p53 will be more likely to drive apoptosis, although, as discussed below, the ability of p53 to do so relies also to a great extent on its likelihood to turn on particular molecular events necessary for p53-mediated apoptosis.
Beside the presence of survival factors and other inputs from the cellular microenvironment, the particular genotype of the cell can also have a major impact on the performance of p53. This is of particular relevance to cancer cells, which often acquire genetic alterations affecting directly or indirectly the functional status of p53. The simplest and most common event is of course direct mutational activation of the p53 gene itself, which will eliminate all wild-type p53 (wt p53) from the affected cells. Another easily understood mechanism is aberrant constitutive overexpression of Mdm2, because of mdm2 gene amplification or to other, less well-defined mechanisms.6,12,13,14 The p53 protein can also be inactivated directly by viral oncoproteins, such as the E6 protein of high-risk human papillomaviruses. However, many additional genetic alterations can also modulate the extent and consequences of p53 activation under stress conditions. To name just a few examples, the JunD protein, component of the AP-1 transcription factor, was shown to restrain the activity of p53; cells lacking JunD are more prone to undergo p53 activation with consequent apoptosis.19 Similarly, the transcription factor NF-κB can also function as an antagonist of p53, acting at least in part through upregulation of Mdm2 expression.20 This finding provides an appealing explanation for the ability of NF-κB to inhibit p53-mediated apoptosis in many cell types. One should be cautioned, however, that the relation between NF-κB and p53-mediated apoptosis is not that simple; in fact, there are instances where NF-κB appears to perform the opposite role, enabling rather than inhibiting the apoptotic effects of p53.21 Any genetic alterations that impact on the competence of these and many other proteins associated with apoptosis, cell cycle control and DNA damage repair, are expected to be capable of modulating the likelihood that p53 will be activated in response to particular types of stress, as well as the biological outcome of such activation.
Selective target gene activation: the sure road to the gallows?
So how does p53 trigger apoptosis when it is allowed to do so? Work done over the years has indicated that p53 has more than a single way of achieving this goal. However, it is fair to say that the function of p53 as a positive regulator of gene expression is the major contributor to its apoptotic activity; at least under physiological conditions, the transactivation function appears to be indispensable for the ability of p53 to elicit cell death in response to stress.22,23 It thus follows that proteins encoded by one or more p53 target genes play a critical role in bringing about p53-mediated apoptosis. This notion has prompted a search for proapoptotic genes that are subject to transcriptional activation by p53. This search turned out to be extremely fruitful, and has rounded up an impressive array of strong suspects, many of them with a well-proven deadly record.
Unlike the cell cycle inhibitory capacity of p53, which appears to be primarily mediated by a handful of genes dominated by p21Waf1, the death-promoting arm of p53 may have tens of genes employed directly in its service (Figure 2). p53 seems to hold shares in more than one apoptotic operation, being capable of transactivating genes encoding death receptors, for example, Fas/CD95/Apo-1 and Killer/DR5, as well as those encoding a multitude of mitochondrial proteins, for example, Bax, Noxa, Puma and p53AIP1, along with others having known or speculated roles in different steps of the apoptotic program. Several genes may be recruited simultaneously by p53 within the same cell, probably acting additively or synergistically, whereas others may be more cell type-restricted with regard to their requirement for p53-mediated apoptosis. Either way, Figure 2 suggests that the choice of p53 between life and death may be dictated by its ability to switch on preferentially particular subsets of genes. More specifically, one may propose that conditions that favor the activation of proapoptotic genes by p53 will end up in cell death, whereas conditions that favor the activation of cell cycle-inhibitory genes will leave the cell growth-arrested, but alive. This attractive conjecture gained initial support from experiments where particular tumor-derived p53 mutants, proficient in induction of a growth arrest, but deficient in apoptosis, were found to be capable of activating normally the p21 promoter but not the Bax promoter.24,25 On the assumption that such mutants may be ‘frozen’ in a state that is optional for the wt p53 protein, one could thus speculate that wt p53 can normally alternate between a mode where it favors growth inhibitory genes and one where it transactivates effectively proapoptotic target genes, the outcome being life or death, respectively.

Target genes subject to positive transcriptional regulation by p53 mediate its different biological outcomes. See text for further details
Since those early observations, additional support was gained for the existence and relevance of preferential promoter activation by wt p53 as it pertains to apoptosis (see below). It is fair to state, however, that at present it is too early to conclude that this is the main mode of regulation of the death/life outcome of p53 activation, rather than being just one of several contributors.
The molecular basis for the differential activation of particular sets of target genes by p53 is not fully understood. Most certainly, multiple molecular mechanisms contribute to p53 target gene selectivity. There is now ample evidence that covalent modifications on p53 may play a critical role in its target gene preference. The most widely studied type of p53 modification is phosphorylation, shown long ago to impact on the promoter selectivity of p53.26 A more recent striking example involves phosphorylation of serine 46, which is specifically required for the efficient transactivation of the proapoptotic p53AIP1 gene,27 and perhaps a small subset of additional death-promoting genes. Thus, agents that induce phosphorylation of p53 on serine 46 will bias the transcriptional repertoire of p53 towards proapoptotic genes, thereby favoring death. An additional twist is introduced by the finding that PPM1D/Wip1, a phosphatase that inhibits serine 46 phosphorylation, is a positive transcriptional target of p53.28 Hence, differential induction of PPM1D/Wip1 by p53 might also dictate the ability of a particular type of stress to elicit apoptosis in a particular cell type. Another interesting case has to do with the Chk2 kinase. Chk2 was identified as responsible for phosphorylation of p53 on serine 20, a site implicated in p53 activation and stabilization following DNA damage (Chehab et al.29; reviewed in Alarcon-Vargas and Ronai2 and Woods and Vousden10), It now appears that Chk2 is selectively required for the apoptotic outcome of p53 activation, but not for p53-mediated growth arrest induced by DNA damage.30 It remains to be elucidated whether this relates to a specific contribution of serine 20 to apoptosis, or to a role of Chk2 in modifying another site(s) on p53 or perhaps even another protein. Other covalent modifications of p53 may also play a role in dictating target gene specificity. It is noteworthy that acetylation of p73, a p53 family member, directs it towards proapoptotic genes; 31 given that p53 is also acetylated, particularly in response to genotoxic stress,2,10 it seems feasible that this modification may also affect the promoter selectivity of p53.
How do covalent modifications impact on the ability of p53 to choose among distinct transcriptional targets? The simplest possibility is that the modified p53 undergoes some conformational change, which alters directly its DNA-binding specificity. Such conjecture may be supported by the fact that the p53 DNA binding consensus sequence is rather degenerate, and there exist substantial differences in the precise composition of the p53BS among different p53 target genes. The modified protein is thus proposed to gain an elevated affinity for some variants of the p53BS and a reduced affinity for others, thereby directing it preferentially to the former. Indeed, phosphorylation on specific residues has been shown long ago to alter the DNA binding preference of p53 in vitro.32 However, the picture may not be as simple as that; in fact, the in vivo relevance of the conformational model for selective activation of p53 binding has been called into question recently, on the basis of chromatin immunoprecipitation analysis.11
Beside covalent modifications of p53 itself, the choice of particular target genes appears to be dictated also by the interaction of p53 with a variety of other proteins (Figure 2). Such modifiers of p53 selectivity include members of the ASPP family33 and the p300-binding protein JMY,34 which favor the transactivation of proapoptotic genes by p53 and thereby shift the p53 response towards apoptosis. Additional proteins, such as RB18A35 and the putative tumor-suppressor Zac-136 may exert similar effects. The need for additional p53 partners may be of particular importance in the case of genes harboring low-affinity p53BS, which may fail to be engaged effectively by the limited concentrations of p53 attained under physiological conditions.37 It is noteworthy that many, albeit not all, proapoptotic p53 target genes harbor p53BS of rather low binding affinity. Consequently, this subclass of genes may rely more heavily on the availability of cooperating proteins of the type described above, whereas cell cycle inhibitory genes may be turned on by p53 as a default option. This makes good intuitive sense in view of the irreversibility of the cell death. Thus, the greater dependence of proapoptotic p53 target genes on auxiliary proteins and additional modifications may serve well the need to call on the apoptotic option sparingly, only under conditions where death is the best solution, rather than any time that a cell is exposed to a p53-activating stress. Curiously, a major apoptotic target gene of p53, PIG3, was recently found to utilize a polymorphic microsatellite rather than its putative consensus p53BS for transcriptional activation by p53.38 It remains to be explored whether this is common to additional proapoptotic p53 target genes, and whether the binding of p53 to this microsatellite is regulated by mechanisms similar to those governing its interaction with consensus p53BS.
An important contribution to the regulation of p53 activity is made by E2F1, a transcription factor often implicated in apoptosis. E2F1 cooperates with p53 in the induction of apoptosis.39 Some of this cooperation is likely to rely on direct association of p53 with E2F1,40 which may impact on the biochemical properties of p53. Moreover, E2F1 was recently shown to induce phosphorylation of p53 on multiple residues, in a manner strikingly similar to that elicited by genotoxic stress.41 In addition, E2F1 and p53 can synergize to activate particular proapoptotic genes containing adjacent binding sites for each of these two transcription factors, as perhaps best illustrated for the Apaf1 gene.42 This is probably an example of a broader generic mechanism, where the availability of a particular rate-limiting transcription factor is expected to affect selectively the ability of p53 to trigger transcription from genes containing responsive elements for both p53 and that factor.
A rather unexpected insight into p53 promoter selectivity was provided by experiments involving mice rendered null for the p53 homologues p63 and p73.43 Surprisingly, these experiments revealed that both p63 and p73 enable the activation of proapoptotic genes such as Bax and PERP by p53. Apparently, p63 and/or p73 are constitutively associated with these genes within the chromatin, and are required for recruitment of p53 to those sites once p53 is induced in response to appropriate stress.
While the proteins discussed above instruct p53 to favor apoptotic target genes, it is conceivable that other proteins exist, which direct p53 selectively to growth inhibitory, DNA repair or antiapoptotic genes (see below), resulting in growth arrest and survival rather than apoptosis. An early example was the WT1 tumor-suppressor protein, shown to stabilize p53 in a mode conducive to growth inhibition, but prohibitory to apoptosis.44 More recently another tumor-suppressor protein, BRCA1, was found to exert a similar effect;45 moreover, BRCA1 was shown to selectively instruct p53 to transactivate target genes involved in cell cycle arrest and DNA repair, but not apoptosis.
The effects of covalent modifications and protein–protein interactions are not necessarily separable. In fact, particular covalent modifications may regulate the interaction of p53 with given proteins, in a manner that will impact on transcription rates from p53 target genes. A case in point is phosphorylation of serine 15 of p53, required for efficient recruitment of the transcriptional coactivators p300/CBP.46,47 Differential recruitment of p300/CBP is expected to lead to nonidentical rates of target-gene transactivation, independent of the extent of specific DNA binding. This may hold equally for another coactivator, hADA3, whose recruitment to p53 following genotoxic stress also requires phosphorylation of p53 on N-terminal residues, enabling more efficient p53-mediated apoptosis.48 Other covalent modifications of p53 may similarly anchor distinct subsets of proteins to p53, all eventually contributing to preferential target-gene activation and to the choice between survival and apoptosis.
Transcriptional repression by p53: a role in apoptosis?
In addition to its well-documented and extensively studied capabilities as a positive transcriptional activator, p53 can also repress transcription from a broad array of promoters.49 The molecular basis for p53-mediated transcriptional repression has been elaborated only partially. In general, unlike positive promoter activation by p53, repression does not rely on identifiable p53BS; rather, it involves recruitment of p53 into a relatively large repressor complex,50 whose exact mode of tethering to particular promoters remains to be worked out.
Irrespective of the mechanism of p53-mediated transrepression, numerous studies indicate that it can contribute substantially to the apoptotic capabilities of p53.51,52,53,54 The most sensible assumption is that p53 can selectively repress one or more antiapoptotic genes, with an outcome essentially similar to transactivation of proapoptotic genes. Indeed, several such repression targets have been identified. The first antiapoptotic protein whose expression was reported to be transcriptionally blocked by p53 is Bcl-2.55,56 Subsequently, p53 was shown to repress the Bcl-X promoter,57 and the latest to join this list is survivin.58,59 Most certainly, additional candidates will emerge with time. p53 thus appears to be a highly sophisticated executioner, simultaneously upregulating death-promoting genes and turning off protective genes. It remains to be found out whether, as in the case of transactivation, p53 can also be instructed to repress selec-tively either survival genes or proliferation-promoting ones, so as to orchestrate apoptosis or growth arrest, respectively.
Transcription-independent activities of p53 in apoptosis
While the crucial role of the transcriptional activities of p53 for its apoptotic action is unquestionable, transcription-independent activities of p53 may also contribute. Transcription-independent proapoptotic functions of p53 have been proposed many years ago,60,61,62 and were shown more recently to facilitate cell killing by genotoxic agents.63,64 However, only limited insight into the nature of these functions has been obtained.
A reasonable prediction is that transcription-independent proapoptotic activities of p53 may result from an ability of p53 to perturb or modulate the function of proteins implicated in the apoptotic machinery. One provocative candidate is the Fas/CD95 death receptor, whose translocation from cytoplasmic stores to the plasma membrane was shown to be promoted by p53;65 it remains to be determined how exactly p53 promotes this translocation, and whether this mechanism pertains also to other death receptors. Furthermore, p53 may direct the translocation of the Bax protein from the cytoplasm into mitochondria, with consequent cytochrome C release.66 Activation of caspase 8 was also shown to be necessary for p53-dependent, transcription-independent apoptosis.67 Particularly illuminating is the observation that p53 itself can become associated with mitochondria under apoptotic conditions, in a manner consistent with a possible direct involvement of p53 in the apoptotic machinery.68 This mechanism may also underlie the reported ability of a transactivation-incompetent p53 mutant to trigger apoptosis via the mitochondrial death pathway.69
Much more work remains to be done in order to figure out the precise biochemical nature of the transcription-independent apoptotic activities of p53, and their relative contribution to apoptosis. However, in view of compelling in vivo evidence,22,23 it appears likely that the main ‘apoptotic load’ is carried by the transcriptional activities, whereas the transcription-independent functions may serve mainly to augment the apoptotic response, particularly under conditions that entail accumulation of very high cellular p53 concentrations.
Antiapoptotic effects of p53
The proapoptotic activities of p53 are well documented and highly appreciated. Yet, there exists a second, relatively little unexplored facet to this story. As surprising as it may appear at first, p53 has in fact been observed to possess antiapoptotic capabilities under a variety of conditions. The common denominator of all these observations is that cells lacking p53 function are actually sometimes more sensitive to apoptosis than their p53-proficient counterparts. Whereas some of the early studies relied on artificially exaggerated wt p53 function,70,71 subsequent work has extended this surprising correlation also to scenarios implicating the endogenous wt p53 protein in protection against cell death.72
How does p53 inhibit or delay apoptosis? The simplest explanation, at least in the case of genotoxic stress, invokes the contribution of p53 to more effective DNA repair. A commitment to apoptosis in response to DNA damage may require that the damage persist beyond a critical time window, which somehow defines it as irreparable and turns on the death machine. In the absence of p53, the likelihood that this will happen may be greater, owing to the less efficient execution of at least some types of DNA repair. This idea gains support from experiments suggesting that p53 protects cells against UV- and cisplatin-induced apoptosis in a manner dependent on transcription-coupled DNA repair.73
In some cases, the antiapoptotic effect of p53 may be an indirect consequence of its impact on cell cycle progression. Rapidly proliferating cells are often more prone to apoptosis, while an extended growth arrest may sometimes provide an antiapoptotic ‘shelter’. Hence, cells lacking functional p53 may become more vulnerable to some apoptotic inducers owing to their failure to arrest properly in response to such agents (Figure 3). In this antiapoptotic capacity of p53, there is good evidence for a critical role of the p21 protein—a major transcriptional target of p53- in conferring increased survival in the face of various DNA-damaging agents and other apoptotic triggers.74,75,76,77,78,79,80 In extreme cases, this capacity of p21 may even lead to oncogenic outcomes.81 Of note, the c-jun proto-oncogene was shown to interfere selectively with the induction of p21 expression by p53, the consequence being augmented apoptosis in response to UV radiation.82 Interestingly, the Akt kinase, whose activation restrains p53-mediated apoptosis (see below), stabilizes the p21 protein, and this was shown to contribute to Akt's prosurvival effects.83,84 Furthermore, p53 can sometimes exert a direct cytoprotective effect, through the transcriptional activation of several target genes with overt antiapoptotic action. Instructive examples include the decoy receptor DcR2, which counteracts the activity of functional death receptors,85 as well as HB-EGF, a potent survival factor.86 It is highly likely that additional examples will follow.

Differential activation of different subsets of target genes by p53 can dictate the biological outcome of p53 activation. Arrows denote stimulatory interactions, whereas horizontal bars instead of arrowheads indicate inhibitory inputs
The ability of p53 to turn on at least several antiapoptotic genes, in addition to its many proapoptotic targets, raises the attractive possibility that the decision whether p53 will dictate death or life relies not only on its ability to turn on the latter group, as discussed above, but also on its ability to upregulate the expression of the former group (Figure 3). It will be of great interest to determine to what extent this rather obscure capacity of p53 plays a role in distinct physiological and pathological conditions, and whether there exist defined factors that direct p53 selectively towards its antiapoptotic targets, thereby favoring survival over death. In any event, the complexity of the relation between p53 function and apoptotic propensity may explain why it has been so difficult to establish an unequivocal, generally applicable correlation between p53 status and therapy response in many types of human cancer, despite extensive clinical studies and compelling evidence from genetically manipulated animal models.
Beta catenin, ARF, p53 and cancer
The following sections of this review will attempt to illustrate how the actual cellular outcome of p53-activating stress is determined, and particularly how the interplay between p53 and other key regulatory proteins and signaling pathways impacts on cell death and on cancer.
The first example focuses on the β-catenin protein. Normally, a structural component of cell–cell adhesive interactions, β-catenin can also double as a potent signaling molecule. In this capacity, β-catenin enters the cell nucleus and turns on the expression of a panel of target genes, a role it performs normally during developmental processes. However, when β-catenin becomes aberrantly and constitutively hyperactive, this can bear profound consequences on the cellular phenotype, as often happens in several types of human cancer.87,88,89 In such cancers, including colorectal carcinoma, hepatocellular carcinoma, melanoma and others, β-catenin behaves as a bona fide oncogene and plays an important role in cancer initiation and progression. Interestingly, deregulated hyperactive β-catenin, in addition to its oncogenic impact, can also cause activation of p53.90 Although this may seem counterintuitive, the concomitant activation of p53 is indeed a well- recognized feature of several oncogenes.91,92 Most certainly, this coupling between aberrant oncogene activation and the triggering of a p53 response is a protective mechanism aimed at alerting p53 to the imminent tumorigenic threat. The ability of p53 to orchestrate an inhibitory response to such threat, either by evoking apoptosis or by halting cell proliferation, is probably one of the keys to its power as a tumor-suppressor.
As is the case with some oncogenes,91,92 albeit not all,93 the induction of p53 by deregulated β-catenin is strictly dependent on the ARF protein, product of another important tumor-suppressor.94 Deregulated β-catenin elevates the production of ARF mRNA. The resultant ARF protein binds Mdm2, blocking its ability to promote the ubiquitination and degradation of p53 (Figure 4). This block probably relies on multiple mechanisms, including direct inhibition of Mdm2's E3 ubiquitin ligase activity, physical sequestration of Mdm2 in the cell nucleolus away from p53, and interference with a post-ubiquitination step required for Mdm2-mediated p53 degradation in the proteasome.2,6,10 When this happens in response to β-catenin deregulation, the cell phenotype can be altered in a p53-dependent manner, resulting in an antiproliferative effect.94 On the other hand, in the absence of a functional p53 pathway, β-catenin is rendered free to exert its oncogenic effects on the affected cell and drive it down the precipitous road to cancer.94 In actual human tumors, this is often achieved through mutation or deletion of the p53 gene, effectively eliminating wt p53 protein from the emerging cancer cells (Figure 4). However, an essentially similar outcome, at least with regard to uncoupling β-catenin deregulation from p53 induction, can potentially be attained by ARF inactivation. Indeed, methylation of the ARF promoter, leading to its silencing and prevention of ARF expression, is a frequent event in colorectal cancer.95 Moreover, ARF inactivation occurs at an early stage of tumor progression, closely following the initiating events involving aberrant activation of β-catenin.95 This strengthens the likelihood that at least one, if not the main, purpose for the early inactivation of ARF is to override the potential anticancer effects of the induction of p53 by deregulated β-catenin. It should be noted, however, that the role of ARF in the activation of p53 by oncogenes is not universal. Whereas in some cell types and with some oncogenes ARF is obligatory, as illustrated for cultured mouse fibroblasts,94 it may be entirely dispensable in other cell types, as shown for choroid plexus-derived tumors.96 This predicts that ARF inactivation will be of selective tumorigenic advantage only in some, but not all, types of cancer. It will be interesting to determine whether the types of tumors that exhibit frequent ARF silencing are also those where, in the normal cellular context, ARF is implicated in linking oncogene activation with p53 induction.

p53–β-catenin crossregulatory network. Symbols are as in Figure 3. See text for further details
p53 mutations do occur eventually in the vast majority of colorectal cancers harboring silenced ARF genes.95 A likely explanation is that, later in the course of tumor progression, additional alterations take place that can trigger a p53 response in an ARF-independent manner. For instance, acquisition of irreparable genomic damage, characteristic of advanced cancer, is expected to exert a strong stress signal that will feed into p53 irrespective of ARF (Figure 4). Other alterations, for example, augmented E2F activity owing to inactivation of the RB pathway, can also have a similar impact.93 Altogether, this is expected to create an environment where the most effective way of the cancer cell to deal with the p53 problem is by selection for direct mutational loss of wt p53 function.
At least in a model system based on mouse fibroblasts, the activation of p53 by deregulated β-catenin does not appear to involve apoptosis; instead, it is manifested in the form of a senescence-like growth arrest.94 This may be due to the antiapoptotic capabilities of β-catenin, which leave p53 only with the growth-inhibitory option at its disposal. In particular, β-catenin strongly induces the expression of the WISP-1 gene, whose product can effectively block p53-mediated apoptosis.97 It is of note that the antiapoptotic effects of WISP-1 rely on activation of the Akt kinase, a pivotal participant in another p53-regulatory network (see below).
Perhaps not surprisingly, the interplay between p53 and β-catenin turns out to be more complex. In particular, it was found that not only can β-catenin modulate p53 levels, but p53 can also modulate β-catenin levels (Figure 4). Thus activation of p53, as occurs in response to genotoxic stress, can lead to proteolytic degradation of β-catenin by enhancing its ubiquitination and subsequent demise in the proteasome.98,99,100 It follows that when p53 is induced in emerging β-catenin-driven tumors, for example, as a result of increased genomic stability, hypoxia or activation of additional oncogenes, it may feed back onto β-catenin and cause its downregulation. This will obviously impose a further strong selective pressure for the mutational inactivation of p53, particularly at advanced stages of the cancer process.
Recently, the crosstalk between p53 and β-catenin was found to be a family affair. The p53 family includes, besides p53 itself, also two additional genes, termed p63 and p73. Each of those genes gives rise to multiple protein products. Most notably, some of these products contain an intact N-terminal transactivation domain (TAD) and are thus, in principle, transactivation-competent, superficially resembling p53, whereas the others—denoted delta N or DN forms—lack this TAD and instead act as dominant-negative antagonists of p53 and of the transactivation proficient forms of p63 and p73, and are overexpressed in a variety of epithelial cancers.6,101 It now turns out that exaggerated expression of DN variants can contribute to cancer not only by inactivation of p53 (and presumably of TAD-containing p63 and p73 molecules), but also by upregulation of β-catenin (Figure 4). This is achieved by inhibition of the ubiquitination and proteasomal degradation of β-catenin, by a mechanism involving interference with the ability of the GSK-3β kinase to phosphorylate β-catenin and thereby signal its ubiquitination.102
In conclusion, the eventual outcome of β-catenin deregulation and its ability to drive cancer are greatly dictated through its interactions, both positive and negative, with p53 and p53 family members (Figure 4). When all components of the network are in place and intact, it is expected that p53 will win and cancer will be effectively prevented. However, the consecutive acquisition of defects and aberrations in this network, will shift the balance in favor of β-catenin and will eventually pave the road to cancer.
p53, Mdm2 and Akt: a matter of balance?
Recent work has established the existence of an additional crossregulatory network, involving p53 and the Akt/PKB kinase (Figure 5). Akt is a well-established antiapoptotic protein, implicated primarily in the normal response to a variety of survival signals, as well as in the aberrant survival of many types of cancer cells.103,104 Activation of Akt by survival signals is driven through a kinase cascade comprising PI3-kinase (PI3 K) and downstream kinases. As it turns out, Akt also has extensive dealings with the p53 pathway. The best worked out component of this crosstalk has to do not with p53 itself, but rather with Mdm2.105,106,107,108,109 Akt can engage in direct protein–protein interactions with Mdm2. Furthermore, it can phosphorylate Mdm2 on at least two residues, serines 166 and 186. This phosphorylation was shown to be required for the translocation of Mdm2 from the cytoplasm into the nucleus, where it can target p53 for inactivation and degradation. As a consequence, elevated Akt is expected to result in attenuation of p53 function, thereby disabling the tumor-suppressor activity of p53 and facilitating survival and cancer progression (Figure 5).

p53–Akt crossregulatory network. P and Ac stand for phosphorylation and acetylation events, respectively. See text for further details
There is also a second, opposite component to this network. In fact, activated p53 can cause a rapid decrease in the steady-state levels of Akt, through a mechanism involving caspase-mediated Akt degradation.106 Hence, pronounced accumulation of active p53, for instance upon extensive genotoxic stress, may result in downregulation of Akt and render the cells more prone not only to the direct apoptotic effects of p53, but also to all other concurrent apoptotic signals whose outcome can be normally blocked by Akt.
As is the case for β-catenin, there are many more players in the Akt-p53 game. Of particular note, p53 positively regulates the expression of the PTEN tumor-suppressor gene.110 PTEN, itself a frequent target for inactivation and loss in human cancer, encodes a phosphatidylinositide phosphatase, which counteracts the action of PI3 K. Thus, PTEN serves to prevent the activation of Akt, thereby facilitating apoptosis. The finding that PTEN is a transcriptional target of p53 establishes an additional link in the network: activation of p53 will augment PTEN expression, which will incapacitate Akt and in this manner will further facilitate apoptosis. To make things even more interesting, p53 also represses the expression of the catalytic subunit of PI3 K.111 Since PI3 K is a critical upstream activator of Akt, this inhibitory effect of p53 will also lead to Akt inactivation, which may cooperate with the induction of PTEN and the degradation of Akt to achieve effective p53-mediated attenuation of Akt function. The relative contribution of the different inhibitory mechanisms to Akt inactivation may vary among different cell types, as well as between normal and malignant cells.111 Once again, this brings in the picture of p53 as a master regulator, which pulls simultaneously many strings when apoptosis has to be orchestrated (Figure 5).
Beta catenin, Akt and p53: dancing together?
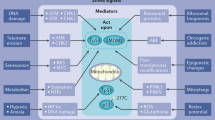
As complex as the β-catenin–p53 (Figure 4) and the Akt-p53 (Figure 5) interplays may be, this is still an oversimplification of the real picture. As a matter of fact, there exists an extensive crossfeeding between these two networks, whose impact on apoptosis and on cancer is likely to be high. In this intercalation of networks, the glycogen synthase kinase 3 beta (GSK-3β) kinase plays a pivotal role. This protein kinase is a central negative regulator of β-catenin, responsible for the phosphorylation events that signal β-catenin ubiquitination and drive its proteasomal degradation.87,89 In addition, GSK-3β is a favorite Akt target. Phosphorylation of GSK-3β by Akt inactivates the former, preventing it from further phosphorylating its downstream substrates.112 Of particular interest, GSK-3β was recently shown to interact directly with p53;113 this interaction boosts the activity of both proteins, presumably enabling them to act hand-in-hand in executing antiproliferative responses. Thus, GSK-3β serves as a strong link between Akt, p53 and β-catenin (Figure 6).

Crosstalk between β-catenin, Akt and p53. See text for further details
Indeed, phosphorylation and inactivation of GSK-3β by Akt lead to subsequent nuclear accumulation of transcriptionally active β-catenin, in response to a variety of physiological and pathological triggers,114,115,116,117 whereas PTEN has the opposite effect: it augments β-catenin degradation and downregulates its transcriptional activity.118,119
Interestingly, excess GSK-3β can trigger apoptosis;120 the downregulation of β-catenin by constitutively active GSK-3β, presumably leading to shut-off of antiapoptotic genes such as WISP-1, along with the activation of p53 through protein–protein interaction,113 offer two interesting mechanistic explanations for this apoptotic effect. In addition, GSK-3β can phosphorylate directly the p21waf1 protein, thereby accelerating its degradation.84 Given the antiapoptotic effect of p21 under many conditions (see above), this could provide yet a third contribution of GSK-3β to p53-mediated apoptosis. On the other hand, constitutive activation of β-catenin is expected to augment WISP-1 expression, leading to Akt activation 97 and subsequent inactivation of GSK-3β (Figure 6).
All in all, Figure 6 depicts an intricate balance between the key participants of the Akt network and the β-catenin network, with p53 featured in a very intimate cross-talk with all of them. The final balance may tilt differently in favor of the various players, depending on the cellular and tissue context. In particular, cancer cells that have acquired relevant genetic lesions many handle this delicate balance quite unlike their normal predecessors. For instance, the stabilizing effect of Akt on β-catenin is particularly pronounced in cells that have not yet undergone extensive tumor progression and thus still retain a normal network of signal transduction pathways.117 This suggests that, at more advanced stages of tumor progression, cells may accumulate additional lesions that endow them with constitutive β-catenin activity irrespective of Akt status. It is tempting to speculate that one type of such lesion may be mutational inactivation of p53.
Concluding Remarks
The almost unprecedented amount of research performed on p53 has equipped us with a stunning wealth of information. If there is a clear take-home message from all that information, it is that one should not expect simple answers, even if the question seems simple. This is perhaps best illustrated by the attempts to establish whether the apoptotic function of p53 is the main determinant of its tumor-suppressor capability. Very elegant experiments were performed to address this question in mouse cancer models. While these experiments have yielded unequivocal answers, these answers were strikingly different among different model systems. Thus, whereas apoptosis was proven to be the critical determinant of p53's protective effect in lymphoma and in choroid plexus tumors,121,122 it was deemed practically irrelevant in intestinal track cancer, where maintenance of genomic stability by p53 is probably implicated instead.123 As illustrated in this review, one may have to approach p53 not as a simple switch that determines cell fate single-handedly, but rather as a component, albeit an important one, in an intricate network of signals and molecular interactions.9 The actual output of this network, and the particular contribution of p53 to that output, will inevitably depend not only on p53, but largely also on its multiple interactions with the many other players in this complex game of life and death, normal growth control and cancer.
Abbreviations
- ARF:
-
alternative reading frame (tumor-suppressor protein)
- DN:
-
delta N-terminus
- GSK-3β:
-
Glycogen synthase kinase 3 beta
- HB-EGF:
-
heparin binding epidermal growth factor
- Mdm2:
-
mouse double minute 2
- NF-κB:
-
nuclear factor kappa B
- p53BS:
-
p53 binding site
- PI3K:
-
phosphoinositide 3-OH kinase
- PKB:
-
protein kinase B
- RB:
-
retinoblastoma susceptibility tumor-suppressor
- TAD:
-
transactivation domain
- wT1:
-
Wilm's tumor susceptibility gene 1
- Wt p53:
-
wild type p53.
References
Oren M and Rotter V (1999) Introduction: p53–the first twenty years. Cell Mol. Life Sci. 55: 9–11
Alarcon-Vargas D and Ronai Z (2002) p53-Mdm2 – the affair that never ends. Carcinogenesis 23: 541–547
Ashcroft M, Taya Y and Vousden KH (2000) Stress signals utilize multiple pathways to stabilize p53. Mol. Cell Biol. 20: 3224–3233
Bargonetti J and Manfredi JJ (2002) Multiple roles of the tumor-suppressor p53. Curr. Opin. Oncol. 14: 86–91
Hickman ES, Moroni MC and Helin K (2002) The role of p53 and pRB in apoptosis and cancer. Curr. Opin. Genet. Dev. 12: 60–66
Michael D and Oren M (2002) The p53 and Mdm2 families in cancer. Curr. Opin. Genet. Dev. 12: 53–59
Sears RC and Nevins JR (2002) Signaling networks that link cell proliferation and cell fate. J. Biol. Chem. 277: 11617–11620
Shen Y and White E (2001) p53-dependent apoptosis pathways. Adv. Cancer Res. 82: 55–84
Vogelstein B, Lane D and Levine AJ (2000) Surfing the p53 network. Nature 408: 307–310
Woods DB and Vousden KH (2001) Regulation of p53 function. Exp. Cell Res. 264: 56–66
Kaeser MD and Iggo RD (2002) Chromatin immunoprecipitation analysis fails to support the latency model for regulation of p53 DNA binding activity in vivo. Proc. Natl. Acad. Sci. USA 99: 95–100
Daujat S, Neel H and Piette J (2001) MDM2: life without p53. Trends Genet. 17: 459–464
Deb SP (2002) Function and dysfunction of the human oncoprotein MDM2. Front Bio. Sci. 7: d235–243
Momand J, Wu HH and Dasgupta G (2000) MDM2--master regulator of the p53 tumor-suppressor protein. Gene. 242: 15–29
Asher G, Lotem J, Kama R, Sachs L and Shaul Y (2002) NQO1 stabilizes p53 through a distinct pathway. Proc. Natl. Acad. Sci. USA 99: 3099–3104
Benetti R, Del Sal G, Monte M, Paroni G, Brancolini C and Schneider C (2001) The death substrate Gas2 binds m-calpain and increases susceptibility to p53-dependent apoptosis. EMBO J. 20: 2702–2714
Fuchs SY, Adler V, Buschmann T, Yin Z, Wu X, Jones SN and Ronai Z (1998) JNK targets p53 ubiquitination and degradation in nonstressed cells. Genes Dev. 12: 2658–2663
Kubbutat MH and Vousden KH (1997) Proteolytic cleavage of human p53 by calpain: a potential regulator of protein stability. Mol. Cell Biol. 17: 460–468
Weitzman JB, Fiette L, Matsuo K and Yaniv M (2000) JunD protects cells from p53-dependent senescence and apoptosis. Mol. Cell 6: 1109–1119
Tergaonkar V, Pando M, Vafa O, Wahl G and Verma I (2002) p53 stabilization is decreased upon NFkappaB activation. A role for NFkappaB in acquisition of resistance to chemotherapy. Cancer Cell 1: 493–503
Ryan KM, Ernst MK, Rice NR and Vousden KH (2000) Role of NF-kappaB in p53-mediated programmed cell death. Nature 404: 892–897
Chao C, Saito S, Kang J, Anderson CW, Appella E and Xu Y (2000) p53 transcriptional activity is essential for p53-dependent apoptosis following DNA damage. EMBO J. 19: 4967–4975
Jimenez GS, Nister M, Stommel JM, Beeche M, Barcarse EA, Zhang XQ, O'Gorman S and Wahl GM (2000) A transactivation-deficient mouse model provides insights into Trp53 regulation and function. Nat. Genet. 26: 37–43
Friedlander P, Haupt Y, Prives C and Oren M (1996) A mutant p53 that discriminates between p53 responsive genes cannot induce apoptosis. Mol. Cell Biol. 16: 4961–4971
Ludwig RL, Bates S and Vousden KH (1996) Differential activation of target cellular promoters by p53 mutants with impaired apoptotic function. Mol. Cell. Biol. 16: 4952–4960
Lohrum M and Scheidtmann KH (1996) Differential effects of phosphorylation of rat p53 on transactivation of promoters derived from different p53 responsive genes. Oncogene 13: 2527–2539
Oda K, Arakawa H, Tanaka T, Matsuda K, Tanikawa C, Mori T, Nishimori H, Tamai K, Tokino T, Nakamura Y and Taya Y (2000) p53AIP1, a potential mediator of p53-dependent apoptosis, and its regulation by Ser-46-phosphorylated p53. Cell 102: 849–862
Bulavin DV, Demidov ON, Saito S, Kauraniemi P, Phillips C, Amundson SA, Ambrosino C, Sauter G, Nebreda AR, Anderson CW, Kallioniemi A, Fornace Jr AJ and Appella E (2002) Amplification of PPM1D in human tumors abrogates p53 tumor-suppressor activity. Nat. Genet. 31: 210–215
Chehab NH, Malikzay A, Appel M and Halazonetis TD (2000) Chk2/hCds1 functions as a DNA damage checkpoint in G1 by stabilizing p53. Genes Dev. 14: 278–288
Jack MT, Woo RA, Hirao A, Cheung A, Mak TW and Lee PW (2002) Chk2 is dispensable for p53-mediated G1 arrest but is required for a latent p53-mediated apoptotic response. Proc. Natl. Acad. Sci. USA 99: 9825–9829
Costanzo A, Merlo P, Pediconi N, Fulco M, Sartorelli V, Cole PA, Fontemaggi G, Fanciulli M, Schiltz L, Blandino G, Balsano C and Levrero M (2002) DNA damage-dependent acetylation of p73 dictates the selective activation of apoptotic target genes. Mol. Cell 9: 175–186
Wang Y and Prives C (1995) Increased and altered DNA binding of human p53 by S and G2/M but not G1 cyclin-dependent kinases. Nature 376: 88–91
Samuels-Lev Y, O'Connor DJ, Bergamaschi D, Trigiante G, Hsieh JK, Zhong S, Campargue I, Naumovski L, Crook T and Lu X (2001) ASPP proteins specifically stimulate the apoptotic function of p53. Mol. Cell 8: 781–794
Shikama N, Lee CW, France S, Delavaine L, Lyon J, Krstic-Demonacos M and La Thangue NB (1999) A novel cofactor for p300 that regulates the p53 response. Mol. Cell 4: 365–376
Frade R, Balbo M and Barel M (2000) RB18A, whose gene is localized on chromosome 17q12–q21.1, regulates in vivo p53 transactivating activity. Cancer Res. 60: 6585–6589
Rozenfeld-Granot G, Krishnamurthy J, Kannan K, Toren A, Amariglio N, Givol D and Rechavi G (2002) A positive feedback mechanism in the transcriptional activation of Apaf-1 by p53 and the coactivator Zac-1. Oncogene 21: 1469–1476
Szak ST, Mays D and Pietenpol JA (2001) Kinetics of p53 binding to promoter sites in vivo. Mol. Cell Biol. 21: 3375–3386
Contente A, Dittmer A, Koch MC, Roth J and Dobbelstein M (2002) A polymorphic microsatellite that mediates induction of PIG3 by p53. Nat. Genet. 30: 315–320
Wu X and Levine AJ (1994) p53 and E2F-1 cooperate to mediate apoptosis. Proc. Natl. Acad. Sci. USA 91: 3602–3606
Hsieh JK, Yap D, O'Connor DJ, Fogal V, Fallis L, Chan F, Zhong S and Lu X (2002) Novel function of the cyclin A binding site of E2F in regulating p53-induced apoptosis in response to DNA damage. Mol. Cell Biol. 22: 78–93
Rogoff HA, Pickering MT, Debatis ME, Jones S and Kowalik TF (2002) E2F1 induces phosphorylation of p53 that is coincident with p53 accumulation and apoptosis. Mol. Cell Biol. 22: 5308–5318
Moroni MC, Hickman ES, Denchi EL, Caprara G, Colli E, Cecconi F, Muller H and Helin K (2001) Apaf-1 is a transcriptional target for E2F and p53. Nat. Cell Biol. 3: 552–558
Flores ER, Tsai KY, Crowley D, Sengupta S, Yang A, McKeon F and Jacks T (2002) p63 and p73 are required for p53-dependent apoptosis in response to DNA damage. Nature 416: 560–564
Maheswaran S, Englert C, Bennett P, Heinrich G and Haber DA (1995) The WT1 gene product stabilizes p53 and inhibits p53-mediated apoptosis. Genes Dev. 9: 2143–2156
MacLachlan TK, Takimoto R and El-Deiry WS (2002) BRCA1 directs a selective p53-dependent transcriptional response towards growth arrest and DNA repair targets. Mol. Cell Biol. 22: 4280–4292
Dumaz N and Meek DW (1999) Serine15 phosphorylation stimulates p53 transactivation but does not directly influence interaction with HDM2. EMBO J. 18: 7002–7010
Lambert PF, Kashanchi F, Radonovich MF, Shiekhattar R and Brady JN (1998) Phosphorylation of p53 serine 15 increases interaction with CBP. J. Biol. Chem. 273: 33048–33053
Wang T, Kobayashi T, Takimoto R, Denes AE, Snyder EL, el-Deiry WS and Brachmann RK (2001) hADA3 is required for p53 activity. EMBO J. 20: 6404–6413
Ginsberg D, Mechta F, Yaniv M and Oren M (1991) Wild-type p53 can down-modulate the activity of various promoters. Proc. Natl. Acad. Sci. USA 88: 9979–9983
Zilfou JT, Hoffman WH, Sank M, George DL and Murphy M (2001) The corepressor mSin3a interacts with the proline-rich domain of p53 and protects p53 from proteasome-mediated degradation. Mol. Cell Biol. 21: 3974–3985
Venot C, Maratrat M, Dureuil C, Conseiller E, Bracco L and Debussche L (1998) The requirement for the p53 proline-rich functional domain for mediation of apoptosis is correlated with specific PIG3 gene transactivation and with transcriptional repression. EMBO J. 17: 4668–4679
Sabbatini P, Chiou SK, Rao L and White E (1995) Modulation of p53-mediated transcriptional repression and apoptosis by the adenovirus E1B 19 K protein. Mol. Cell Biol. 15: 1060–1070
Shen YQ and Shenk T (1994) Relief of p53-mediated transcriptional repression by the adenovirus E1B 19-kDa protein or the cellular bcl-2 protein. Proc. Natl. Acad. Sci. USA 91: 8940–8944
Ryan KM and Vousden KH (1998) Characterization of structural p53 mutants which show selective defects in apoptosis but not cell cycle arrest. Mol. Cell Biol. 18: 3692–3698
Haldar S, Negrini M, Monne M, Sabbioni S and Croce CM (1994) Downregulation of bcl-2 by p53 in breast cancer cells. Cancer Res. 54: 2095–2097
Miyashita T, Harigai M, Hanada M and Reed JC (1994) Identification of a p53-dependent negative response element in the bcl-2 gene. Cancer Res. 54: 3131–3135
Sugars KL, Budhram-Mahadeo V, Packham G and Latchman DS (2001) A minimal Bcl-x promoter is activated by Brn-3a and repressed by p53. Nucleic Acids Res. 29: 4530–4540
Hoffman WH, Biade S, Zilfou JT, Chen J and Murphy M (2002) Transcriptional repression of the anti apoptotic survivin gene by wild type p53. J. Biol. Chem. 277: 3247–3257
Mirza A, McGuirk M, Hockenberry TN, Wu Q, Ashar H, Black S, Wen SF, Wang L, Kirschmeier P, Bishop WR, Nielsen LL, Pickett CB and Liu S (2002) Human survivin is negatively regulated by wild-type p53 and participates in p53-dependent apoptotic pathway. Oncogene 21: 2613–2622
Caelles C, Helmberg A and Karin M (1994) p53-dependent apoptosis in the absence of transcriptional activation of p53-target genes. Nature 370: 220–223
Haupt Y, Rowan S, Shaulian E, Vousden KH and Oren M (1995) Induction of apoptosis in HeLa cells by trans-activation-deficient p53. Gene Dev. 9: 2170–2183
Wagner AJ, Kokontis JM and Hay N (1994) Myc-mediated apoptosis requiRes wild-type p53 in a manner independent of cell cycle arrest and the ability of p53 to induce p21(waf1/cip1). Gene Develop. 8: 2817–2830
Baptiste N, Friedlander P, Chen X and Prives C (2002) The proline-rich domain of p53 is required for cooperation with anti neoplastic agents to promote apoptosis of tumor cells. Oncogene 21: 9–21
Matas D, Sigal A, Stambolsky P, Milyavsky M, Weisz L, Schwartz D, Goldfinger N and Rotter V (2001) Integrity of the N-terminal transcription domain of p53 is required for mutant p53 interference with drug-induced apoptosis. EMBO J. 20: 4163–4172
Bennett M, Macdonald K, Chan SW, Luzio JP, Simari R and Weissberg P (1998) Cell surface trafficking of Fas: a rapid mechanism of p53-mediated apoptosis [see comments]. Science 282: 290–293
Schuler M, Bossy-Wetzel E, Goldstein JC, Fitzgerald P and Green DR (2000) p53 induces apoptosis by caspase activation through mitochondrial cytochrome c release. J. Biol. Chem. 275: 7337–7342
Ding HF and Fisher DE (2001) p53, caspase 8, and regulation of apoptosis after ionizing radiation. J. Pediatr. Hematol. Oncol. 23: 185–188
Moll UM and Zaika A (2001) Nuclear and mitochondrial apoptotic pathways of p53. FEBS Lett. 493: 65–69
Regula KM and Kirshenbaum LA (2001) p53 activates the mitochondrial death pathway and apoptosis of ventricular myocytes independent of de novo gene transcription. J. Mol. Cell Cardiol. 33: 1435–1445
Vikhanskaya F, D'Incalci M and Broggini M (1995) Decreased cytotoxic effects of doxorubicin in a human ovarian cancer-cell line expressing wild-type p53 and WAF1/CIP1 genes. Int. J. Cancer 61: 397–401
Malcomson RDG, Oren M, Wyllie AH and Harrison DJ (1995) P53-independent death and p53-induced protection against apoptosis in fibroblasts treated with chemotherapeutic drugs. Br. J. Cancer 72: 952–957
Lassus P, Bertrand C, Zugasti O, Chambon JP, Soussi T, Mathieu-Mahul D and Hibner U (1999) Antiapoptotic activity of p53 maps to the COOH-terminal domain and is retained in a highly oncogenic natural mutant. Oncogene 18: 4699–4709
McKay BC, Becerril C and Ljungman M (2001) P53 plays a protective role against UV- and cisplatin-induced apoptosis in transcription-coupled repair proficient fibroblasts. Oncogene 20: 6805–6808
Gorospe M, Cirielli C, Wang XT, Seth P, Capogrossi MC and Holbrook NJ (1997) p21(Waf1/Cip1) protects against p53-mediated apoptosis of human melanoma cells. Oncogene 14: 929–935
Bissonnette N and Hunting DJ (1998) p21-induced cycle arrest in G1 protects cells from apoptosis induced by UV-irradiation or RNA polymerase II blockage. Oncogene 16: 3461–3469
Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW and Vogelstein B (1998) Requirement for p53 and p21 to Sustain G2 arrest after DNA damage. Science 282: 1497–1501
Bunz F, Hwang PM, Torrance C, Waldman T, Zhang Y, Dillehay L, Williams J, Lengauer C, Kinzler KW and Vogelstein B (1999) Disruption of p53 in human cancer cells alters the responses to therapeutic agents. J. Clin. Invest. 104: 263–269
Mariette X, Sibilia J, Roux S, Meignin V and Janin A (2002) A new defensive mechanism to prevent apoptosis in salivary ductal cells from patients with Sjogren's syndrome: over-expression of p53 and p21. Rheumatology (Oxford) 41: 96–99
Polyak K, Waldman T, He TC, Kinzler KW and Vogelstein B (1996) Genetic determinants of p53-induced apoptosis and growth arrest. Gene Develop. 10: 1945–1952
Wang Y, Blandino G and Givol D (1999) Induced p21waf expression in H1299 cell line promotes cell senescence and protects against cytotoxic effect of radiation and doxorubicin. Oncogene 18: 2643–2649
Roninson IB (2002) Oncogenic functions of tumour suppressor p21(Waf1/Cip1/Sdi1): association with cell senescence and tumour-promoting activities of stromal fibroblasts. Cancer Lett. 179: 1–14
Shaulian E, Schreiber M, Piu F, Beeche M, Wagner EF and Karin M (2000) The mammalian UV response: c-Jun induction is required for exit from p53-imposed growth arrest. Cell 103: 897–907
Li Y, Dowbenko D and Lasky LA (2002) AKT/PKB phosphorylation of p21Cip1/WAF1 enhances protein stability of p21Cip1/WAF1 and promotes cell survival. J. Biol. Chem. 277:11352–11361
Rossig L, Badorff C, Holzmann Y, Zeiher AM and Dimmeler S (2002) Glycogen synthase kinase-3 couples AKT-dependent signaling to the regulation of p21Cip1 degradation. J. Biol. Chem. 277: 9684–9689
Meng RD, McDonald III ER, Sheikh MS, Fornace Jr AJ and El-Deiry WS (2000) The TRAIL decoy receptor TRUNDD (DcR2, TRAIL-R4) is induced by adenovirus-p53 overexpression and can delay TRAIL-, p53-, and KILLER/DR5-dependent colon cancer apoptosis. Mol. Ther. 1: 130–144
Fang L, Li G, Liu G, Lee SW and Aaronson SA (2001) p53 induction of heparin-binding EGF-like growth factor counteracts p53 growth suppression through activation of MAPK and PI3K/Akt signaling cascades. EMBO J. 20: 1931–1939
Peifer M and Polakis P (2000) Wnt signaling in oncogenesis and embryogenesis -- a look outside the nucleus. Science 287: 1606–1609
Brantjes H, Barker N, van EJ and Clevers H (2002) TCF: Lady Justice casting the final verdict on the outcome of Wnt signalling. Biol. Chem. 383: 255–261
Polakis P (2000) Wnt signaling and cancer. Genes Dev. 14: 1837–1851
Damalas A, Ben-Ze'ev A, Simcha I, Shtutman M, Leal JF, Zhurinsky J, Geiger B and Oren M (1999) Excess beta-catenin promotes accumulation of transcriptionally active p53. EMBO J. 18: 3054–3063
Lowe SW (1999) Activation of p53 by oncogenes. Endocr. Relat. Cancer 6: 45–48
Sherr CJ and Weber JD (2000) The ARF/p53 pathway. Curr. Opin. Genet. Dev. 10: 94–99
Russell JL, Powers JT, Rounbehler RJ, Rogers PM, Conti CJ and Johnson DG (2002) ARF differentially modulates apoptosis induced by E2F1 and Myc. Mol. Cell Biol. 22: 1360–1368
Damalas A, Kahan S, Shtutman M, Ben-Ze'ev A and Oren M (2001) Deregulated beta-catenin induces a p53- and ARF-dependent growth arrest and cooperates with Ras in transformation. EMBO J. 20: 4912–4922
Esteller M, Tortola S, Toyota M, Capella G, Peinado MA, Baylin SB and Herman JG (2000) Hypermethylation-associated inactivation of p14(ARF) is independent of p16(INK4a) methylation and p53 mutational status. Cancer Res. 60: 129–133
Tolbert D, Lu X, Yin C, Tantama M and Van Dyke T (2002) p19(ARF) is dispensable for oncogenic stress-induced p53-mediated apoptosis and tumor suppression in vivo. Mol. Cell Biol. 22: 370–377
Su F, Overholtzer M, Besser D and Levine AJ (2002) WISP-1 attenuates p53-mediated apoptosis in response to DNA damage through activation of the Akt kinase. Genes Dev. 16: 46–57
Liu J, Stevens J, Rote CA, Yost HJ, Hu Y, Neufeld KL, White RL and Matsunami N (2001) Siah-1 mediates a novel beta-catenin degradation pathway linking p53 to the adenomatous polyposis coli protein. Mol. Cell 7: 927–936
Matsuzawa SI and Reed JC (2001) Siah-1, SIP, and Ebi collaborate in a novel pathway for beta-catenin degradation linked to p53 responses. Mol. Cell 7: 915–926
Sadot E, Geiger B, Oren M and Ben-Ze'ev A (2001) Downregulation of beta-catenin by activated p53. Mol. Cell Biol. 21: 6768–6781
Yang A, Kaghad M, Caput D and McKeon F (2002) On the shoulders of giants: p63, p73 and the rise of p53. Trends Genet. 18: 90–95
Patturajan M, Nomoto S, Sommer M, Fomenkov A, Hibi K, Zangen R, Poliak N, Califano J, Trink B, Ratovitski E and Sidransky D (2002) DeltaNp63 induces beta-catenin nuclear accumulation and signaling. Cancer Cell 1: 369–379
Nicholson KM and Anderson NG (2002) The protein kinase B/Akt signalling pathway in human malignancy. Cell Signal. 14: 381–395
Brazil DP and Hemmings BA (2001) Ten years of protein kinase B signalling: a hard Akt to follow. Trends Bio. Chem. Sci. 26: 657–664
Ashcroft M, Ludwig RL, Woods DB, Copeland TD, Weber HO, MacRae EJ and Vousden KH (2002) Phosphorylation of HDM2 by Akt. Oncogene 21: 1955–1962
Gottlieb TM, Leal JF, Seger R, Taya Y and Oren M . (2002) Cross-talk between Akt, p53 and Mdm2: possible implications for the regulation of apoptosis. Oncogene 21: 1299–1303
Mayo LD and Donner DB (2001) A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc. Natl. Acad. Sci. USA 98: 11598–11603
Ogawara Y, Kishishita S, Obata T, Isazawa Y, Suzuki T, Tanaka K, Masuyama N and Gotoh Y (2002) Akt enhances Mdm2-mediated ubiquitination and degradation of p53. J. Biol. Chem. 277: 21843–21850
Zhou BP, Liao Y, Xia W, Zou Y, Spohn B and Hung MC (2001) HER-2/neu induces p53 ubiquitination via Akt-mediated MDM2 phosphorylation. Nat. Cell Biol. 3: 973–982
Stambolic V, MacPherson D, Sas D, Lin Y, Snow B, Jang Y, Benchimol S and, Mak TW (2001) Regulation of PTEN Transcription by p53. Mol. Cell 8: 317–325
Singh B, Reddy PG, Goberdhan A, Walsh C, Dao S, Ngai I, Chou TC, Levine AJ, Rao PH and Stoffel A (2002) p53 regulates cell survival by inhibiting PIK3CA in squamous cell carcinomas. Genes Dev. 16: 984–893
Weston CR and Davis RJ (2001) Signal transduction: signaling specificity – a complex affair. Science 292: 2439–2440
Watcharasit P, Bijur GN, Zmijewski JW, Song L, Zmijewska A, Chen X, Johnson GV and Jope RS (2002) Direct, activating interaction between glycogen synthase kinase-3beta and p53 after DNA damage. Proc. Natl. Acad. Sci. USA 99: 7951–7955
Desbois-Mouthon C, Cadoret A, Blivet-Van Eggelpoel MJ, Bertrand F, Cherqui G, Perret C and Capeau J (2001) Insulin and IGF-1 stimulate the beta-catenin pathway through two signalling cascades involving GSK-3beta inhibition and Ras activation. Oncogene 20: 252–259
Fukumoto S, Hsieh CM, Maemura K, Layne MD, Yet SF, Lee KH, Matsui T, Rosenzweig A, Taylor WG, Rubin JS, Perrella MA and Lee ME (2001) Akt participation in the Wnt signaling pathway through Dishevelled. J. Biol. Chem. 276: 17479–17483
Monick MM, Carter AB, Robeff PK, Flaherty DM, Peterson MW and Hunninghake GW (2001) Lipopolysaccharide activates Akt in human alveolar macrophages resulting in nuclear accumulation and transcriptional activity of beta-catenin. J. Immunol. 166: 4713–4720
Satyamoorthy K, Li G, Vaidya B, Patel D and Herlyn M (2001) Insulin-like growth factor-1 induces survival and growth of biologically early melanoma cells through both the mitogen-activated protein kinase and beta-catenin pathways. Cancer Res. 61: 7318–7324
Persad S, Troussard AA, McPhee TR, Mulholland DJ and Dedhar S (2001) Tumor-suppressor PTEN inhibits nuclear accumulation of beta-catenin and T cell/lymphoid enhancer factor 1-mediated transcriptional activation. J. Cell Biol. 153: 1161–1174
Sharma M, Chuang WW and Sun Z (2002) Phosphatidylinosital 3-kinase/Akt stimulates androgen pathway through GSK3beta inhibition and nuclear beta -catenin accumulation. J. Biol. Chem. 277: 30935–30941
Pap M and Cooper GM (1998) Role of glycogen synthase kinase-3 in the phosphatidylinositol 3-Kinase/Akt cell survival pathway. J. Biol. Chem. 273: 19929–19932
Schmitt CA, Fridman JS, Yang M, Baranov E, Hoffman RM and Lowe SW (2002) Dissecting p53 tumor-suppressor functions in vivo. Cancer Cell 1: 289–298
Symonds H, Krall L, Remington L, Saenzrobles M, Lowe S, Jacks T and Vandyke T (1994) p53-dependent apoptosis suppresses tumor growth and progression in vivo. Cell 78: 703–711
Fazeli A, Steen RG, Dickinson SL, Bautista D, Dietrich WF, Bronson RT, Bresalier RS, Lander ES, Costa J and Weinberg RA (1997) Effects of p53 mutations on apoptosis in mouse intestinal and human colonic adenomas. Proc. Natl. Acad. Sci. USA 94: 10199–10204
Acknowledgements
The work of the Oren lab is supported in part by grants from the National Cancer Institute, USA (RO1 CA 40099), The European Community 5th Framework Program (grant QLG1-1999-00273). The USA–Israel Binational Science Foundation, the German–Israel Project Cooperation (DIP), the Cooperation Program in Cancer Research of the DKFZ and Israel's Ministry of Science (MOS), the Center for Excellence Program of the Israel Science Foundation, the Robert Bosch Foundation (Germany), the Kadoorie Charitable Foundations, and the Yad Abraham Center for Cancer Diagnosis and Therapy.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by G Melino
Rights and permissions
About this article
Cite this article
Oren, M. Decision making by p53: life, death and cancer. Cell Death Differ 10, 431–442 (2003). https://doi.org/10.1038/sj.cdd.4401183
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4401183
Keywords
This article is cited by
-
Loss of EPS8 sensitizes non-small-cell lung carcinoma to chemotherapy-induced DNA damage
Cancer Gene Therapy (2023)
-
Mutant p53 sustains serine-glycine synthesis and essential amino acids intake promoting breast cancer growth
Nature Communications (2023)
-
The MRN complex maintains the biliary-derived hepatocytes in liver regeneration through ATR-Chk1 pathway
npj Regenerative Medicine (2023)
-
Strong apoptotic response of testis tumor cells following cisplatin treatment
International Urology and Nephrology (2023)
-
A nomogram combining clinical factors and biomarkers for predicting the recurrence of high-risk cutaneous squamous cell carcinoma
BMC Cancer (2022)
