
Abstract
Cholangiocarcinoma (CCA) includes a cluster of highly heterogeneous biliary malignant tumours that can arise at any point of the biliary tree. Their incidence is increasing globally, currently accounting for ~15% of all primary liver cancers and ~3% of gastrointestinal malignancies. The silent presentation of these tumours combined with their highly aggressive nature and refractoriness to chemotherapy contribute to their alarming mortality, representing ~2% of all cancer-related deaths worldwide yearly. The current diagnosis of CCA by non-invasive approaches is not accurate enough, and histological confirmation is necessary. Furthermore, the high heterogeneity of CCAs at the genomic, epigenetic and molecular levels severely compromises the efficacy of the available therapies. In the past decade, increasing efforts have been made to understand the complexity of these tumours and to develop new diagnostic tools and therapies that might help to improve patient outcomes. In this expert Consensus Statement, which is endorsed by the European Network for the Study of Cholangiocarcinoma, we aim to summarize and critically discuss the latest advances in CCA, mostly focusing on classification, cells of origin, genetic and epigenetic abnormalities, molecular alterations, biomarker discovery and treatments. Furthermore, the horizon of CCA for the next decade from 2020 onwards is highlighted.
Similar content being viewed by others

Introduction
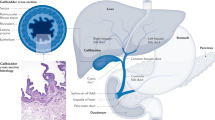
Cholangiocarcinoma (CCA) constitutes a diverse group of malignancies emerging in the biliary tree. CCAs are divided into three subtypes depending on their anatomical site of origin: intrahepatic (iCCA), perihilar (pCCA) and distal (dCCA) CCA1,2 (Fig. 1). Of note, considered as an independent entity, mixed HCC–CCA tumours are a rare type of liver malignancy sharing features of both iCCA and HCC and presenting an aggressive disease course and poor prognosis3,4. iCCAs arise above the second-order bile ducts, whereas the point of anatomical distinction between pCCA and dCCA is the insertion of the cystic duct. pCCA and dCCA can also be collectively referred to as ‘extrahepatic’ (eCCA)5. In the USA, pCCA is the single largest group, accounting for approximately 50–60% of all CCAs, followed by dCCA (20–30%) and iCCA (10–20%)1,6,7. CCA is the second most common primary hepatic malignancy after hepatocellular carcinoma (HCC), comprising approximately 15% of all primary liver tumours and 3% of gastrointestinal cancers1,6,7. CCAs are usually asymptomatic in early stages and, therefore, often diagnosed when the disease is already in advanced stages, which highly compromises therapeutic options, resulting in a dismal prognosis1,8. CCA is a rare cancer, but its incidence (0.3–6 per 100,000 inhabitants per year)1 and mortality (1–6 per 100,000 inhabitants per year, globally9, not taking into account specific regions with incidence >6 per 100,000 habitants such as South Korea, China and Thailand) have been increasing in the past few decades worldwide, representing a global health problem. Despite advances in CCA awareness, knowledge, diagnosis and therapies, patient prognosis has not improved substantially in the past decade, with 5-year survival (7–20%) and tumour recurrence rates after resection still disappointing10,11,12,13,14,15,16,17. Therefore, a detailed study of these types of cancers is urgently needed to improve patient welfare and outcomes. Considering the high heterogeneity of CCAs, individual characterization of these tumours at the genomic, epigenetic and molecular levels is an indispensable approach to ascertain their pathogenesis, paving the path for new therapeutic options and personalized medicine. In this expert Consensus Statement, which is endorsed by the European Network for the Study of Cholangiocarcinoma (ENS-CCA), we provide a comprehensive and critical overview of current knowledge and what is envisaged on the horizon for CCA, focusing on epidemiology, risk factors, clinical presentation, diagnosis, genetic and epigenetic landscape, molecular perturbations, chemoresistance and therapies.

On the basis of the anatomical site of origin, cholangiocarcinoma (CCA) is classified into intrahepatic CCA (iCCA), perihilar CCA (pCCA) and distal CCA (dCCA). iCCA is defined as a malignancy located in the periphery of the second-order bile ducts, pCCA arises in the right and/or left hepatic duct and/or at their junction, and dCCA involves the common bile duct (that is, the choledochus). Grossly, CCA can show three main patterns of growth: mass-forming, periductal-infiltrating, and intraductal-growing. Mass-forming CCA is a mass lesion in the hepatic parenchyma. Periductal-infiltrating iCCA grows inside the duct wall and spreads longitudinally along the wall. Intraductal-growing CCA is a polypoid or papillary tumour growing towards the duct lumen.
Methods
This international group of multidisciplinary experts in CCA (that is, oncologists, surgeons, hepatologists, geneticists, immunologists, basic scientists) has been intensively collaborating within the ENS-CCA since 2015 with the main aims of improving our understanding of CCA and the management of patients. In this regard, this expert consensus is endorsed by the ENS-CCA. The overall goal of this multidisciplinary statement is to provide a detailed critical overview of the current knowledge in this field, proposing some expert recommendations and highlighting what is envisaged for the next decade.
J.M.B. and G.J.G. identified the areas of interest, stratified the consensus statement into the sections presented in the document and assigned them to selected ENS-CCA members or non-European collaborators (L.R.R., S.G., S.R., J.H. and G.J.G.) who are expert in each field of knowledge and research. To write this document, a PubMed search was conducted by combining the term ‘cholangiocarcinoma’ with the following terms: ‘epidemiology’, ‘risk factors’, ‘classification’, ‘cells of origin’, ‘diagnosis’, ‘staging’, ‘genetics’, ‘epigenetics’, ‘signalling pathways’, ‘epithelial-to-mesenchymal transition’, ‘cancer stem cells’, ‘tumour microenvironment’, ‘immunobiology’, ‘in vitro and in vivo models’, ‘biomarkers’, ‘surgery’, ‘liver transplantation’, ‘therapies’, ‘clinical trials’ and ‘chemoresistance’. No specific search dates were used. All the sections were merged into a first draft by P.M.R and J.M.B. and then extensively revised to create the final document that was later circulated among all the authors for further correction, improvement, discussion and approval. The data presented in Fig. 2 were obtained by combining the values of mortality rates in men and women for both iCCA and eCCA reported in 2019 by Bertuccio et al.9. For the recommendations on CCA management and research priorities, ideas were proposed, discussed and approved after final revision by all the authors to reach a consensus.

Global age-standardized annual mortality rates for cholangiocarcinoma (CCA) (deaths per 100,000 inhabitants, including intrahepatic CCA, perihilar CCA and distal CCA) obtained from Bertuccio et al.9. Data refer to the periods 2000–2004 (2002), 2005–2009 (2007) and 2010–2014 (2012). Yellow indicates countries/regions with low mortality (<2 deaths per 100,000 people), orange indicates countries/regions with mortality between 2 and 4 deaths per 100,000 people, and red indicates countries/regions with high mortality (>4 deaths per 100,000 people). Mortality in eastern countries/regions in which CCA is highly prevalent (that is, Thailand, China, Taiwan and South Korea) have not yet been reported and, therefore, CCA incidence is shown for these countries1.
Epidemiology and risk factors
The global mortality for CCA increased worldwide during the periods 2000–2004, 2005–2009 and 2010–2014 (Fig. 2), according to the WHO and Pan American Health Organization databases for 32 selected locations in Europe, America, Asia and Oceania9. Furthermore, CCA mortality was higher in men than in women worldwide, and in countries/regions in Asia versus those in the West. Accordingly, Asian individuals were reported to have the highest mortality (2.81 per 100,000 men in Japan). However, in the USA, the more noticeable increases in mortality between 2004 and 2014 were found for African American individuals (45%), followed by Asian (22%) and white (20%) individuals18. The age-standardized incidence of CCA shows considerable geographical variation, with the highest incidence in Eastern countries/regions; incidence varies from 85 per 100,000 in northeastern Thailand (the highest reported value globally) to 0.4 per 100,000 in Canada12. Variations in incidence probably reflect differences in local risk factors and potential genetic predispositions1,2,19.
The three subtypes of CCA can have different risk factors, pathobiology, clinical presentations, management and prognosis, as well as distinct epidemiological trends1,2. Over the past few decades, the reported age-standardized incidence for iCCA has been steadily increasing in most locations worldwide, whereas the age-standardized incidence for eCCA has been decreasing2,19. However, these trends need cautious interpretation given that all versions of the main International Classification of Diseases (ICD) have so far failed to include a separate code for the largest group of CCA (pCCA) and previous versions of ICD–Oncology (ICD-O) have cross-referenced pCCA (technically extrahepatic) to iCCA. Importantly, for the first time, subsequent iterations of both ICD and ICD-O (ICD-11 and ICD-O-4, respectively) — which are due to come into effect by 2021 — will have separate codes for recording iCCA, pCCA and dCCA20. Having clearly defined codes for the three subtypes of CCA might facilitate more accurate and meaningful epidemiological data. In the meantime, reported epidemiological trends for CCA and/or biliary tract cancer need to be interpreted carefully.
Furthermore, in the USA, the incidence of iCCA is higher in older people (≥45 years old) than in younger people and in Hispanic individuals than in non-Hispanic individuals, and is associated with a worse 5-year survival in both these populations21. Worse overall survival (OS) rates have also been reported for African Americans, followed by American Indians and Alaska Native groups21,22. Of note, the hospital charges associated with iCCA management almost doubled from 2005 to 2014 in the USA22, and male patients with low annual incomes (<US$37,999) tended to show shorter OS, pinpointing socioeconomic treatment discrepancies that clearly affect outcome23. Other factors that affect the interpretation of CCA incidence trends include the unknown effects of improved diagnostics (imaging, endoscopy, histology), greater awareness and the acceptance of performing a biopsy even when imaging is highly suggestive of HCC (in accordance with current guidelines), and increasing the detection of combined HCC–CCA or iCCAs3,24,25,26,27,28. Furthermore, there is substantial global variation in rates of microscopically verified (that is, histologically or cytologically confirmed) cases of CCA reported to cancer registries worldwide. For example, Khon Kaen in Thailand, despite being believed to have the highest overall age-standardized incidence of CCA worldwide, has one of the lowest morphologically verified percentages (only 9% of all cases of liver disease)29. In this regard, CCA can be notoriously difficult to accurately diagnose due to its location often being inaccessible to histology or cytology, a lack of clear diagnostic imaging criteria, and inaccurate non-invasive tumour biomarkers1,2.
Several risk factors, both common and rare, have been linked to CCA (Table 1). Although some risk factors are shared by all forms of CCA, others seem to be more specific for one subtype and seem to be more important in different regions. A common characteristic amongst many of these risk factors is that they are associated with chronic inflammation of the biliary epithelium and bile stasis19. Several recognized risk factors have increased globally over recent decades (1990–2016) and could be contributing to increasing CCA rates. For instance, high alcohol consumption, tobacco smoking and viral infections (hepatitis B virus (HBV) and hepatitis C virus (HCV)) have been reported to increase the risk of CCA development30. Moreover, it is also important to highlight the global obesity pandemic, as well as the metabolic syndrome and/or presence of nonalcoholic fatty liver disease, as risk factors that deserve future central attention30. However, in most locations, the majority of CCA cases remain sporadic, without any identifiable risk factor present. A number of studies are examining the potential influence of commonly used drugs such as aspirin31,32,33 and lipid-lowering statins34,35 in the prevention of CCA. Notably, post-diagnosis aspirin usage has been found to be associated with a reduced risk of death (HR 0.71) among patients with CCA36. Polymorphisms of host genes encoding enzymes involved in xenobiotic detoxification, DNA repair, multidrug resistance, immune response and folate metabolism have been linked to CCA development19. There are currently no published genome-wide association studies (GWAS) in CCA, but an appropriately powered one is eagerly anticipated.
Classification and cells of origin
iCCA can emerge at any point of the intrahepatic biliary tree, ranging from bile ductules to the second-order bile ducts (segmental bile ducts). In contrast to HCC, iCCA usually develops in non-cirrhotic liver37. pCCA can arise in the right and/or left hepatic duct and/or at their junction (so-called perihilar bile ducts)38, and dCCA involves the common bile duct39. The current term eCCA is now discouraged as it combines subtypes with distinct clinicopathological features, prognosis and therapeutic options, and also due to the difficulties in discriminating between intrahepatic and extrahepatic origins of pCCA.
iCCA can show three main patterns of growth: mass-forming, periductal-infiltrating, and intraductal-growing1,38 (Fig. 1); pCCA and dCCA present as flat or poorly defined nodular sclerosing tumours or, less frequently, as intraductal papillary tumours40. CCA can be preceded by pre-invasive lesions39. Histologically, although the vast majority of pCCA and dCCA are conventional mucin-producing adenocarcinomas or papillary tumours40, iCCA shows several histological variants (that is, conventional, cholangiolocarcinoma and rare variants)41 (Fig. 3; Table 2). Conventional iCCA can be further classified into two main histological subtypes according to the level or size of the affected duct42,43,44,45,46 (Fig. 3; Table 2). Small bile duct iCCA presents as a small-sized tubular or acinar adenocarcinoma with nodular growth invading the liver parenchyma, and with no or minimal mucin production42,43,44,45,46. Large bile duct iCCA arises in large intrahepatic bile ducts and comprises mucin-producing columnar tumour cells arranged in a large duct or papillary architecture38,46,47,48,49. Remarkably, the histological subtyping parallels the high molecular heterogeneity of CCAs and can be ascribed to different cells of origin and pathogenesis41. Small bile duct iCCA can be characterized by isocitrate dehydrogenase (IDH1, IDH2) mutations or fibroblast growth factor receptor 2 (FGFR2) fusions40,50,51,52,53,54,55. By contrast, large bile duct iCCA, similar to pCCA and dCCA, shows a high frequency of mutations in KRAS and/or TP53 genes51,53,56,57. Interestingly, dCCA is also associated with ELF3 mutations58. Growing evidence demonstrates that distinct cells of origin within an organ can give rise to different subtypes of cancer, typically tissue-specific stem and progenitor cells59,60,61,62. Evidence regarding the cells of origin of CCA in humans was obtained by phenotyping the candidate tissues and/or cells of origin with respect to CCA subtypes through histological and gene expression analysis38,44,46,63,64,65,66,67,68, whereas indirect evidence might be derived from risk factors67,68.

Based on the duct size, the intrahepatic biliary tree can be further subdivided into small and large intrahepatic bile ducts (iBDs). Small iBDs are lined by small cuboidal cholangiocytes whereas columnar and mucous cholangiocytes line large iBDs. Typically, large iBDs contain peribiliary glands within their wall. The extrahepatic biliary tree shares anatomical features with large iBDs. Histological cholangiocarcinoma (CCA) variants reflect the phenotype of the involved duct and the putative cell of origin. Conventional intrahepatic CCA (iCCA) has two main variants: small duct-type iCCA arises in small iBDs with cuboidal cholangiocytes representing the putative cell of origin, and large duct-type iCCA involves large iBDs and is considered to be derived from columnar cholangiocytes and peribiliary glands (seromucous glands; mucous acini are shown in light pink, serous acini are shown in green). Cholangiolocarcinoma (CLC) is a frequent histological variant of iCCA and its phenotype suggests the origin from bile ductules or ductular reaction (DR) that occurs in chronic liver diseases. The vast majority of perihilar CCA (pCCA) and distal CCA (dCCA) are considered to originate from the lining epithelium and peribiliary glands. This histological subtyping underlies distinct clinicopathological and molecular features as summarized in Table 2. eBD, extrahepatic bile duct; HpSC, human pluripotent stem cell.
Small bile duct and cholangiolocarcinoma iCCA subtypes emerge at the level of smaller intrahepatic bile ducts, including bile ductules38,44,46,69. In these portions of the biliary tree, hepatic stem or progenitor cells (HpSCs) and cuboidal cholangiocytes represent surface epithelium and are the putative cells of origin of these malignancies38,44,46,69 (Fig. 3; Table 2). Interestingly, HpSCs have been implicated in CK19+ HCC70 and in combined HCC–CCA4,64,71. Notably, CCA-like HCC tumours display embryonic stem cell-like expression traits, further substantiating the involvement of bipotent hepatic progenitor cells, and in humans display a worse prognosis than HCC72. In line with these findings, nestin, a maker of bipotent progenitor oval cells, is greatly increased in HCC–CCA tumours and is associated with a worse prognosis, and has been proposed as a new possible diagnostic and prognostic biomarker3,26. Small bile duct and cholangiolocarcinoma iCCA usually develop on a background of chronic liver disease (such as chronic viral hepatitis and cirrhosis)38,46,70, characterized by HpSC activation71,73,74.
Large bile duct iCCA, pCCA and dCCA derive from columnar mucous cholangiocytes or peribiliary glands38,44,46,47,49,69 (Fig. 3), which are also implicated in the origin of precursor lesions (such as intraductal papillary neoplasm)46. These malignancies mainly develop in ducts affected by chronic inflammation as in primary sclerosing cholangitis (PSC) or liver fluke infection46,49,69. In PSC, peribiliary gland cell proliferation, mucinous metaplasia, and dysplasia to cancer progression take place within bile ducts and along the biliary tree, mimicking the cancerization field (‘field defect’)49,75.
Controversies exist regarding the cellular origins of iCCA based on lineage tracing studies in experimental carcinogenetic models76. Indeed, there is evidence in favour of HpSC, cholangiocyte or hepatocyte origin of iCCA from these experimental settings76,77,78,79,80,81. Thus, a definitive determination of the origin of iCCA in humans cannot be reached based on current evidence and requires further research. Moreover, it should be underlined that current experimental models of liver damage do not fully recapitulate the pathogenesis of human chronic liver disease, including proliferative senescence in hepatocytes66,82,83, and that lineage tracing studies must be conducted and interpreted cautiously76,84,85,86,87.
Clinical presentation
Diagnosis
CCAs are usually asymptomatic during early stages. The most frequent symptom of pCCA and dCCA is jaundice due to biliary tract obstruction88. In iCCA, jaundice is less frequent and mostly associated with advanced disease. Other symptoms of advanced disease include asthenia, abdominal pain, malaise, nausea, anorexia and weight loss. iCCA is an incidental finding in around 20–25% of cases88. In patients with cirrhosis, ultrasonography surveillance for HCC enables iCCA diagnosis at an asymptomatic, early stage89. Unfortunately, the majority of iCCA cases occur in the absence of known risk factors90, when the only chance for early diagnosis is by cross-sectional imaging performed for other reasons.
Imaging techniques, such as ultrasonography, contrast-enhanced ultrasonography (CEUS), CT and MRI, play a key part in the management of CCA in terms of diagnosis, staging, follow-up and assessment of treatment response. Their diagnostic accuracy is influenced by the anatomical location and growth patterns of CCA, and their use for staging varies according to tumour location91. CT is considered the standard imaging method for the preoperative assessment of both iCCA and pCCA; it provides a comprehensive evaluation of the primary tumour, the relationship with adjacent structures, and potential thoracic and abdominal spread91. MRI has similar accuracy to CT for diagnosis and staging, but it incorporates specific sequences such as diffusion-weighted imaging and the potential for performing magnetic resonance cholangiopancreatography (MRCP), which is critical for pCCA staging92. The most frequent imaging patterns displayed by iCCA on both CT and MRI are arterial peripheral rim enhancement with progressive homogeneous contrast agent uptake until the delayed or stable uptake during late dynamic phases93,94. A targetoid pattern defined as arterial rim enhancement, peripheral washout and delayed central enhancement can also be present in iCCA95. When gadoxetic acid is used, the washout should be read in the portal phase instead of in delayed phases to prevent misclassification between HCC and iCCA in a cirrhotic liver96. More controversial is the use of CEUS in iCCA, particularly in the setting of underlying chronic liver disease. iCCA exhibits homogeneous arterial hyperenhancement followed by venous washout in near 50% of patients, a pattern indistinguishable from that found in HCC94,97. However, in a relevant proportion of patients with iCCA, washout takes place earlier than 60 s after contrast agent injection; this feature is rarely observed in HCC, and the intensity of washout in the portal phase is more marked in iCCA than in HCC89. These refinements might decrease the risk of misdiagnosis in HCC98, and have been adopted by the Liver Imaging Reporting Data System (LI-RADS) for CEUS (LI-RADS-CEUS)99. No evidence supports the use of 18F-FDG PET for completion of staging, which could be of special value to exclude the presence of lymph node or distant metastases100.
As no specific CCA radiology pattern exists, histopathological or cytological analysis is mandatory to confirm the diagnosis1,28. This diagnosis is based on the WHO classification of biliary tract cancer showing an adenocarcinoma or mucinous carcinoma101, with tubular and/or papillary structures and a variable fibrous stroma102.
Staging
There is no widely used staging system for CCA, although it can be staged according to the American Joint Committee on Cancer (AJCC) TNM system103,104. Despite providing a clinically meaningful classification correlated with prognosis105, the current TNM classification has some limitations. First, it has limited discriminatory ability between T2 and T3 tumours in surgically resected iCCAs105,106. T2 tumours include multifocal disease or disease with intrahepatic vascular invasion that probably reflect disseminated disease and the OS in patients with these tumours does not differ from the OS in patients with extrahepatic metastatic disease105. Similarly, there is also evidence supporting the negative effect of the presence of multifocal iCCA (iCCA with liver metastases; T2) on prognosis (OS) when compared with other early stages, which might require consideration in future versions of the AJCC TNM classifications107. Second, although size has been included for the first time as a prognostic factor for iCCA in the eighth edition of the AJCC Cancer Staging Manual, the only cut-off size considered is 5 cm in T1 tumours. Several authors have shown that a 2 cm cut-off value might identify very early tumours with very low likelihood of dissemination and potential long-term survival with low recurrence rates24,108. Finally, the TNM classification misses relevant prognostic factors such as the presence of cancer-related symptoms (such as abdominal pain or malaise) or the degree of liver function impairment. As previously shown with HCC, future proposals from society guidelines should focus on stratifying non-surgical patients for clinical studies using clinical and imaging data. Notably, Chaiteerakij et al. proposed a new staging system for pCCA based on tumour size and number, vascular encasement, lymph node and peritoneal metastasis, Eastern Cooperative Oncology Group (ECOG) performance status (ECOG-PS), and CA19-9 level, which has shown a better performance in predicting survival than the TNM staging system109. Also, important for stratification in clinical trials, radiographic staging parameters need to be developed in the absence of histological staging, and a radiographic staging system has been proposed for pCCA109.
Genetics and epigenetics
Genomics
Initial efforts using integrative genomics approaches to stratify CCA based on prognosis have highlighted extensive deregulated transcriptomic landscapes showing augmented anti-apoptotic signalling, angiogenesis, signal transduction and transcriptional control8,110. The main oncogenic networks comprised WNT-CTNNB1, MYC, ERBB, TNF and VEGF signalling, emphasizing cell survival signalling pathways in patients with poor OS8. Regarding genomic alterations, CCA falls midway in the mutational spectrum of cancers111, with an approximately equal content of genomic alterations in iCCA (median 39 non-synonymous mutations per tumour) and eCCA (median 35 non-synonymous mutations per tumour)56. Massive sequencing studies56,112,113,114,115,116,117,118,119,120,121 have improved our understanding of the causal mechanisms in CCA, emphasizing the genomic complexity in prevalent oncogenic modules affecting: cell cycle regulation; DNA damage and genomic instability (TP53, CDKN2A, CCND1, ATM, ROBO2, BRCA1 and BRAC2); MYC amplification; epigenetic regulation including NADPH metabolism (IDH1 and IDH2), de-ubiquitination (BAP1), SWI–SNF complex (PBRM1, ARID1A, ARID1B, ARID2, SMARCA2, SMARCA4 and SMARCAD1) and histone (de-)methylation (MLL2, MML3, KMT2C, KDM4A, KDM5D, KDM6A and KDM6B); kinase signalling (KRAS, ERBB1–3, BRAF, PIK3CA, PTEN, STK11, SMAD4 and FGFR1–3); immune dysregulation (JAK–STAT3 signalling); FGFR2 and PRKCA–PRKCB fusions; the WNT–CTNNB1 pathway (APC); Hippo signalling (NF2, SAV1 deletion); METLL13 amplifications; and deregulated Notch signalling. Interestingly, the predominant genomic alterations in CCA are associated with epigenetic processes122. Indeed, the most clinically significant genomic breakthroughs in iCCA are the discovery of hotspot IDH mutations (IDH1R132 and IDH2R172) that cause an accumulation of the oncometabolite 2-hydroxyglutarate (2-HG)57, as well as the constitutive active gene fusion event between FGFR2 and many different partners, including the most prevalent (BICC1 (refs50,112,113,114), PPHLN1 (ref.115), TACC3 (ref.112) and MGEA5 (ref.112)). These alterations are important as they are driving current marker-based phase III clinical trials testing specific agents targeting these alterations in FGFR2 fusion-positive CCA (NCT03773302)123,124 and IDH-mutated CCA (NCT02989857).
To date, information on the inherited predisposing genetic risk factors causing CCA is very limited125. Data mostly stem from GWAS of patient cohorts diagnosed with PSC25,126, with increased risk of CCA. However, the only detailed genomic association with aetiological risk factors investigated by genome sequencing has been the association with liver fluke infection (Opisthorchis viverrini and Clonorchis sinensis), with fluke-positive tumours showing an overall higher mutational rate (median 4,700 versus 3,143 somatic mutations per tumour)116 with prevalent mutations in SMAD4 and TP53 as well as ERBB2 amplifications116,117,118. Furthermore, although not in a high proportion, KRAS mutations have been recurrently found in all CCA subtypes56,116,117. A statistically significant association has also been observed between TP53 mutation and HBV infection119,120. Few studies have investigated the molecular distinction between iCCA, pCCA and dCCA8,56,116,121. Nakamura et al. emphasized the difference in anatomical location of the tumour, highlighting IDH, EPHA2 and BAP1 mutations and FGFR2 fusions in iCCA, whereas extrahepatic tumours specifically show PRKACA and PRKACB fusions as well as mutations in ELF3 (similar to tumours in the ampulla of Vater)127 and ARID1B56. Based on these fundamental causal alterations, tumours in distinct anatomical sites should probably be treated differently. Besides linking IDH mutations with the response to ivosidenib128, few studies have related genomic alterations to high-throughput drug screening119,129,130. Among these, Nepal et al. used an approach of integrative genomics in a large cohort of iCCAs to elucidate unique mutational signatures, structural variants and epigenomic alterations, emphasizing specific oncogenetic mechanisms in four distinct subsets of patients with potential drug responses and categories: RNA synthesis inhibition, IDH mutant; microtubule modulator, KRAS mutant; topoisomerase inhibition, TP53 mutant; and mTOR inhibitors119.
Epigenetics
Epigenetics was shown to play an important part in the initiation and progression of CCA, affecting tumour phenotype in the absence of changes in DNA sequences131. Deregulated patterns of methylation, histone modifications and aberrant expression of non-coding RNAs promote unbalanced transcription and gene expression that impair cell homeostasis and sustain malignant transformation. Growing evidence supports deregulated methylation motifs in CCA cells compared with their normal counterparts, with a prevalent hypermethylation of multiple CpG sites occurring in CCA132,133. One of the largest studies of integrative genetic and epigenetic analyses in CCA, including 489 CCAs from ten countries/regions, has shown how the molecular make-up of CCA goes beyond the differentiation according to anatomical site116. Indeed, by combining DNA sequencing with transcriptomic and DNA methylation analyses, four clusters of CCA with different clinical outcomes were identified. Two sets of hypermethylated CCAs stood out, with an interesting association between CpG island hypermethylation and liver fluke-related tumours, increased mutation rate, downregulation of the DNA demethylation enzyme TET1, upregulation of the histone methyltransferase EZH2 and an increased level of deamination events. Conversely, the subgroup of iCCAs with enrichment in IDH1/2 and BAP1 mutations, as well as FGFR translocations, showed hypermethylation of the CpG shores (the regions immediately flanking CpG islands, up to 2 kb away). This different pattern suggests how early epigenetic deregulation caused by external carcinogenic events (for example, liver flukes) are at the basis of CCA development in the first cluster, whereas in the second cluster, epigenetic aberrations probably arise as a downstream consequence of somatic mutations (IDH) that produce oncometabolites responsible for the DNA hypermethylation. These differences have remarkable clinical implications, because on the one hand early epigenetic events might be used for early detection of tumours in the first cluster (by using quantitative DNA methylation markers in the bile of individuals at risk)80 and on the other hand, the tumour clonal mutations might be a marker of effective targeted therapies (such as IDH inhibitors).
Methylome data can also provide insights into the cells of origin of CCA. Tumours with high genetic and epigenetic occurrence seem to have an enrichment of events within embryonic stem cell-related bivalent regulation134,135,136. IDH-mutated tumours instead seem to resemble the profile of cholangiocellular CCAs that show gene expression traits of epithelial–mesenchymal transition (EMT)136. Histone modifications have been less studied in CCA. Histone deacetylase (HDAC) enzymes are responsible for regulation of histone acetylation that ultimately affects chromatin organization. HDAC were found to be upregulated in CCA in vitro137, and are being investigated as targets of treatment. Evidence also suggests that HDAC inhibitors, as well as dasatinib, might be particularly active in IDH-mutated tumour cells129,130. Non-coding RNAs account for around 98% human RNAs and include microRNAs (miRNAs) and long non-coding RNAs, among others. These non-coding RNAs regulate the expression of a plethora of target genes affecting all the hallmarks of the cancer phenotype from cell proliferation and migration to EMT and the regulation of the primary cilium in cholangiocytes138,139,140,141,142 (Fig. 4).

Non-coding RNAs (ncRNAs) that have been found to be dysregulated (up or down) in cholangiocarcinoma and that have key roles in the regulation of cellular processes, such as proliferation, cell cycle, ciliogenesis, epigenetics, inflammation, chemoresistance, survival, epithelial to mesenchymal transition (EMT), migration and invasion are shown.
Signalling and molecular networks
CCA often arises in the setting of prolonged biliary inflammation and/or cholestasis, which contribute to carcinogenesis. According to transcriptomic profiles, the ‘inflammation’ (38%) and ‘proliferation’ (62%) subtypes of iCCA were previously identified and reported to be differentially enriched with activation of the pro-inflammatory and oncogenic pathways, respectively110. The inflammation subclass of tumours was characterized by induction of immune-related signalling pathways. By contrast, the proliferation subclass was enriched in classic oncogenic pathways, including deregulated receptor tyrosine kinase (RTK) signalling, RAS–RAF–ERK, PI3K–AKT–mTOR, insulin growth factor receptor 1, MET, polo-like kinase 1, aurora kinase A, KRAS mutations and stem-like genomic traits as well as a focal deletion in the Hippo pathway (SAV1)8,110,143. Notably, patients with the proliferation subtype of iCCA displayed decreased OS (median 24.3 months versus 47.2 months for those with the inflammation subtype; P = 0.048).
Cholangiocarcinogenesis is orchestrated by a complex interplay of extracellular ligands (such as pro-inflammatory cytokines, growth factors and bile acids, among others), which are present in the tumour microenvironment (TME), and increased expression and/or aberrant activation of cell surface receptors and the deregulation of intracellular signalling pathways, finally leading to cell proliferation, survival and genetic and/or epigenetic alterations (Fig. 5).

The process of cholangiocarcinogenesis, and further tumour evolution and growth, involves complex and heterogeneous processes that include the interplay of extracellular ligands (such as pro-inflammatory cytokines, growth factors and bile acids, among others), which are present in the tumour microenvironment, and increased expression and/or aberrant activation of cell surface receptors and the deregulation of intracellular signalling pathways, finally leading to cell proliferation, survival and migration or invasion. The most common genes that might be mutated or amplified resulting in the overactivation of some of these pathways are KRAS, BRAF, ARID1, PBRM1, BAP1, IDH1 and IDH2. The activation of these signalling pathways might also occur as a result of the interaction between the tumour epithelia and the tumour reactive stroma. 2-HG, 2-hydroxyglutarate; ECM, extracellular matrix; RTK, receptor tyrosine kinase.
Chronic inflammation and fibrosis facilitate cholangiocyte transformation in a multistep manner, providing extracellular ligands that modulate several signalling pathways. In particular, sustained IL-6–STAT3 signalling was shown to contribute to mitogenesis by upregulating myeloid cell leukaemia 1 (MCL1) or altering EGFR promoter methylation144,145. Similarly, bile acids are not genotoxic but might also promote cholangiocarcinogenesis through a mechanism involving the activation of EGFR, induction of COX2, MCL1 and IL-6, and downregulation of farnesoid X receptor (FXR)146,147. Of note, FXR expression was reported to be decreased in human CCA tumours compared with surrounding normal liver tissue, correlating with tumour differentiation140. By contrast, the levels of TGR5, another bile acid receptor, were found to be increased in CCA tumours and to be correlated with a worse prognosis (perineural invasion)140. CCA tumours, and particularly iCCAs and pCCAs, are characterized by a reactive desmoplastic stroma containing cancer-associated fibroblasts (CAFs) that crosstalk with CCA cells secreting paracrine factors such as heparin-binding EGF-like growth factor, stromal-cell derived factor 1 (SDF1), platelet-derived growth factor (PDGF)-B and extracellular matrix (ECM) proteins148.
Although there are marked differences in the genomic features depending on the anatomical location and risk factors, activation of the RTK signalling pathway is a common event in CCA across subtypes. In this regard, aberrant EGFR, ERBB2 and MET RTK expression has been found in different CCA subclasses that are associated with worse prognosis8,110. RTK signalling mainly triggers the activation of the RAS–MAPK and PI3K–AKT–mTOR pathways. Furthermore, RAS–MAPK pathway activation due to KRAS-activating mutations is found in all CCAs without distinction, whereas BRAF mutations are more prevalent in iCCA149. Interestingly, chromosomal oncogenic gene fusion rearrangements involving FGFR2 RTK occur almost exclusively in iCCA50,52,56,112,113. Besides FGFR2 fusions, ROS1 kinase protein fusions have also been identified in iCCA150. Thus, RTK signalling pathways present actionable molecular alterations that are amenable for therapeutic targeting at multiple levels. IDH1 and IDH2 encode metabolic enzymes that interconvert isocitrate and α-ketoglutarate51,53,113,117,151. Mutations in IDH1 and IDH2 lead to the production of high levels of 2-hydroxyglutarate, an oncometabolite that interferes with histone and DNA demethylases and inhibits the mitochondrial electron transport chain. Indeed, IDH-mutant CCAs were shown to exhibit high levels of mitochondrial and low levels of chromatin modifier gene expression, such as low ARID1A expression due to DNA hypermethylation121. Besides epigenetic silencing, inactivating mutations in multiple chromatin-remodelling genes (including BAP1, ARID1A and PBRM1) are common in iCCA151.
Developmental pathways, including Notch, WNT and transforming growth factor-β (TGFβ) signalling pathways are prominently active in iCCA compared with HCC, as shown by integrated microarray analysis152. During liver repair and in inflammatory conditions (known risk factors for iCCA), signalling pathways involved in biliary development are activated in ductular reactive cells, including Notch, WNT, Hippo–YAP and Hedgehog. The Notch pathway is known to be involved in biliary repair, growth, tubulogenesis, fibrosis and maintenance of the stem cell niche; defective Notch function due to JAG1 or NOTCH2 mutations causes impaired regeneration and Alagille syndrome153, whereas increased Notch activity has been associated with primary liver tumours154. Overexpression or aberrant Notch receptor expression has been reported both in iCCAs and eCCA, including pCCA and dCCA155,156,157. Activation of Notch signalling was shown to mediate transdifferentiation of hepatocytes into cholangiocytes during carcinogenesis79,80,81,158. In this regard, experimental overexpression of the intracellular domain of NOTCH1 receptor (NICD1) in hepatocytes has been associated with the development of iCCA in mouse models79,80,158. Similarly, inhibition of NOTCH2, the expression of which has been shown to be related to well-differentiated iCCA155, markedly reduced tumour burden in various mouse models of liver cancer (including iCCA)81,159, whereas overexpression of NOTCH3 was associated with the development and progression of iCCA, promoting cell survival via PI3K–AKT signalling160. Several Notch inhibitors are being developed, and their availability increases interest in this pathway161.
The WNT–β-catenin signalling pathway is also known to be activated in most CCAs, in part as an effect of the release of Wnt ligands by inflammatory macrophages infiltrating the stroma162,163, but also as a consequence of DNA methylation alterations targeting this pathway133 and/or mutations encoding key components of the canonical WNT–β-catenin signalling pathway164. Notably, the promoter of the WNT–β-catenin pathway inhibitor SOX17 was hypermethylated in CCA tumour tissue compared with healthy tissue, correlating with a worse prognosis after tumour resection132. Noteworthy, SOX17 was shown to regulate cholangiocyte differentiation and to act as a tumour suppressor in CCA in vitro132. WNT inhibitors successfully inhibit tumour growth in experimental models163 and clinical trials with agents targeting this pathway are currently being explored164. The Hippo–YAP signalling pathway regulates organ size and cell proliferation, among other functions165. YAP is a transcriptional co-activator that is usually inhibited by Hippo (MST1 or MST2), but can be activated by Hippo-independent signals, such as inflammation and changes in ECM composition and stiffness166. Several groups have reported increased nuclear expression of YAP in CCA specimens and correlation with a worse prognosis167,168,169. In vitro studies on CCA cell lines have shown that YAP can be activated by IL-6, PDGF and fibroblast growth factor170,171. PDGF and fibroblast growth factor form a feed-forward loop activating YAP; YAP transcriptional targets are genes of these signalling pathways, such as FGFR1, FGFR2 and FGFR4 (refs170,171,172). Genetic alteration of the YAP pathway seems to be uncommon in CCA, according to an integrative genomic analysis of CCA specimens121. However, mutations in ARID1A have been reported in up to 14% of CCAs149. ARID1A encodes a subunit of the SWI–SNF chromatin-remodelling complex that among other functions reduces YAP transcriptional activity173.
EMT, stemness and plasticity
EMT is a cell plasticity-promoting phenomenon initially reported to occur during embryogenesis, but that also takes place in cancer, enabling epithelial cancer cells to acquire mesenchymal features with invasive properties that lead to metastatic colonization174. The prototype inducer of EMT is the TGFβ-dependent pathway, whose signature has been identified in iCCA stroma8,175. In CCA, TGFβ induces EMT directly or cooperates with other major EMT inducer pathways such as EGFR176,177. During this plastic EMT programme, tumour cells lose their epithelial traits and gain mesenchymal features178. Although initially considered as a binary process, it is now well established that epithelial cells undergoing EMT become mesenchymal in a gradual manner, known as partial EMT178,179. Thus, EMT is a dynamic process that gives rise to intermediate cellular states with both epithelial and mesenchymal traits, contributing to cell heterogeneity and a broad range of functions from cancer initiation to progression178,179. Notably, EMT is orchestrated by transcription factors (EMT-TFs), comprising SNAIL, ZEB and TWIST family, that regulate the expression of epithelial and mesenchymal genes180. CCAs express EMT-TFs, which are associated with poor prognosis regardless of anatomical localization181. Beyond the EMT programme, EMT-TFs display pleiotropic roles linking EMT to stemness, metabolic reprogramming, immune evasion and drug resistance178,182,183.
Increasing evidence suggests associations between EMT and acquisition of cancer stem cell (CSC) properties in different cancer types65,184, and this might also contribute to CCA heterogeneity as well as resistance to anticancer drugs. Importantly, CSCs represent a peculiar subcompartment of the tumour cell population crucially involved in recurrence, metastasis and drug resistance185,186,187. A growing body of evidence indicates that CSCs express EMT traits in human CCAs65,187,188,189. Interestingly, CCA emerging in patients with PSC are characterized by EMT features and high expression of stem and/or progenitor cell markers in peribiliary glands, suggesting a connection between EMT and stemness in tumour initiation49. Indeed, EMT-TFs, such as ZEB1, regulate expression of CSC markers by inhibiting miR-200 family members, well-known potent stemness repressors190. In stem-like iCCA, a signature linking miR-200c with EMT regulators such as ZEB1 and TGFβ has been identified191. Besides EMT, TGFβ is known to promote stemness in CCA cells in vitro (human CCA cell line TFK-1)192. A statistically significant correlation between TGFβ1 and aldehyde dehydrogenase 1 (ALDH1), a functional CSC marker, has been found in both iCCA and eCCA192. Furthermore, TGFβ-induced EMT resulted in acquisition of mesenchymal traits, ALDH expression and resistance to 5-fluorouracil (5-FU) in vitro192. Moreover, new evidence suggests that cell plasticity promoted by the EMT programme confers immunosuppressive effects on carcinoma cells by mechanisms not completely understood178; one mechanism identified so far is the regulation of the immune checkpoint PD1 ligand (PDL1) by ZEB1 in breast cancer cells193.
Tumour microenvironment
CCA tumours contain a diverse range of cellular types (Fig. 6). Although the tumour epithelium is considered as the coordinator of tumour growth, the importance of the TME cannot be understated. Histopathologically, CCA is typified by an extensive cellular and acellular stroma that can comprise the bulk of the tumour194. CCA shares many characteristics with scars that form around bile ducts in premalignant disease, as usually found in PSC and congenital hepatic fibrosis, suggesting that the origin of the tumour stroma can be found in the regenerative microenvironment during bile duct repair195. The CCA stroma consists of cancer-associated endothelial cells, CAFs and a complex group of inflammatory cells, including macrophages, neutrophils, natural killer (NK) and T cells196. In addition to this complex cellular microenvironment, the tumour stroma also contains an extensive network of ECM proteins such as collagens, laminin and fibronectin197,198. The TME directly interacts with the cancer epithelium to support epithelial proliferation and tumour growth, among which CAFs have been the most extensively investigated.

Cancer-associated fibroblasts (CAFs) are recruited and persistently activated by cholangiocarcinoma (CCA) cells, in response to the effects of PDGF-D, and of FGF and TGFβ1, also released by tumour-associated macrophages (TAMs). In turn, CAFs enhance cell proliferation and the invasive ability of CCA cells directly, or by influencing the activity of other cells in the tumour microenvironment. CAFs stimulate tumour-associated lymphangiogenesis (lymphatic endothelial cell (LEC)), support M2 polarization of TAMs and the activation of regulatory T (Treg) cells, while dampening the activity of CD8+ T cells, natural killer (NK) and dendritic cells. CAFs also induce heavy remodelling of the extracellular matrix (ECM), which becomes stiffer and affects mechanotransduction of CCA cells, leading to activation of intracellular pathways, including YAP–TAZ. Soluble factors mediating each cell–cell interplay are shown in boxes of different colours according to their origin (orange from CAFs, green from CCA cells, light blue from TAMs, red from ECM). Mediators in bold are those with proven effects, the rest are putative signalling molecules. CAF-derived short-range (Hedgehog (Hh)) and direct (NOTCH3) cell–cell developmental cues also underlie interactions with CCA cells (lower right corner).TH2 cell, T helper 2 cell.
Cancer-associated fibroblasts
CAFs are a heterogeneous population of spindle-shaped cells with mesenchymal origin that contribute to tumour progression in many human cancers199. In CCA, the abundance of CAFs positively correlates with tumour growth and poor survival200. CAFs most likely originate from several different cells types, namely tissue-resident portal fibroblasts, hepatic stellate cells, pericytes, bone marrow-derived mesenchymal stem cells and monocyte precursor-derived fibrocytes via transdifferentiation and activation181,201,202. This activation process also results in a metabolic reprogramming that enhances proliferation, cellular motility, as well as secretion of regulatory molecules and components of the ECM. Importantly, although CCA cells express mesenchymal markers they do not transdifferentiate into CAFs, but they do secrete PDGF-D to stimulate fibroblast migration203. In CCA, the persistent activation of fibroblasts is induced primarily by TGFβ, fibroblast growth factor and PDGF, which are released from tumour-associated macrophages and CCA cells204. TGFβ was reported to be pivotal in promoting an iCCA-desmoplastic phenotype in a 3D rat organotypic culture model205, and targeting the TGFβ pathway in thioacetamide-treated rats improved fibrosis and reduced CCA burden206. CAFs secrete a multitude of signalling molecules (such as IL-1β, PDGF-B, heparin-binding EGF-like growth factor and SDF1) that promote cancer progression by enhancing proliferation, survival, chemotaxis and angiogenesis148. Furthermore, CAFs have also been shown to promote CCA growth through short-range and direct cell–cell morphogenetic signals, such as NOTCH3 (ref.160) and Hedgehog207. By secreting immunomodulatory factors, CAFs can also promote an immunosuppressive TME208: they regulate innate immunity by supporting M2 macrophages, and decreasing NK cell activation. Regarding adaptive immunity, CAFs promote regulatory T cells and T helper 2 cells, and disable dendritic cells and cytotoxic T cells208. Data support the ability of CAFs to interact with lymphatic endothelial cells209. Following stimulation by PDGF-D originated from the tumoural cholangiocytes in vitro, CAFs secrete VEGF-A and VEGF-C, which recruit and assemble lymphatic endothelial cells in vascular structures susceptible to tumour cell intravasation209.
Cell interactions within the TME are favoured by the ECM, which is gradually ‘transformed’ into a compact and stiff scaffold, enabling mutual communications and exchange of paracrine factors between the different cell elements210. The ECM is continuously remodelled by deposition of newly synthesized matricellular proteins, including tenascin C, osteopontin and periostin, in concert with an intensive degradation by matrix metalloproteinases (MMPs; MMP1, MMP2, MMP3 and MMP9) that are copiously released by CAFs, tumour-associated macrophages and malignant cholangiocytes210. Thanks to these phenotypic changes, ECM boosts key pro-invasive functions of tumour cells. In cooperation with collagen I, tenascin C and integrins (α5β1, α5β3, α5β5 and α6β4), periostin stimulates cell proliferation of malignant cholangiocytes in a PI3K–AKT-dependent manner in vitro211. Increased ECM stiffening is also instrumental in the activation of intracellular mechanosensors, such as YAP–TAZ, involved in tumour initiation and progression. Whereas soft ECM inhibits YAP–TAZ activity by favouring its sequestration by the SWI–SNF chromatin-remodelling complex through ARID1A, stiff ECM induces YAP–TAZ to detach from SWI–SNF and to bind to TEAD, unfolding a transcriptional programme and promoting cell proliferation, CSC traits, plasticity and reprogramming173,212. Overall, the multifaceted interplay of CAFs with tumour cells, immune cells, lymphatic endothelial cells and ECM is continuously evolving (Fig. 6) and could offer potential therapeutic targets. Importantly, selective pro-apoptotic targeting of CAFs with subsequent reduction in tumour growth and lymph node metastases has been demonstrated in a CCA rat model213. Overall, the signalling networks that govern CCA tumours are the result of the intrinsic genomic and epigenetic alterations of tumour cholangiocytes, as well as their interplay with CAFs, immune cells and ECM. The secretion of proinflammatory, oncogenic and fibrogenic factors from CCA cells could contribute to the recruitment of other cells to the TME, which in turn will activate and sustain specific signalling pathways in cancer cells, thus perpetuating CCA growth and progression.
Immunobiology
Transcriptomic sequencing of CCA tumours has demonstrated that the subset of patients with the poorest prognosis have an elevated tumour mutational load and enhanced expression of immune checkpoint molecules56. Importantly, the presence of T cell-infiltrated TMEs, characterized by infiltration of CD8+ T cells, chemokines and molecules responsible for T cell priming and immune infiltration, is associated with higher response to immune checkpoint blockade, whereas non-T cell-infiltrated TMEs have poorer responses214,215.
Regarding innate immune responses, activated or ‘M2-like’ tumour-associated macrophages are anti-inflammatory and immunosuppressive. M2-like macrophages stimulate WNT signalling with consequent CCA progression163, and are associated with inferior patient outcomes216,217. High numbers of M2 tumour-associated macrophages are linked to poor disease-free survival in patients with iCCA217. Similarly, in a retrospective study of patients with pCCA who had undergone surgical resection, high density of tumour-associated macrophages in the tumour invasive front correlated with increased local and tumour recurrence216. Myeloid-derived suppressor cells are another immunosuppressive element in the TME. Fibroblast activation protein-positive (FAP+) CAFs promote myeloid-derived suppressor cell infiltration in desmoplastic tumours218. Moreover, increased stromal FAP expression in human resected CCA specimens has been linked to poor patient outcomes218. The presence of CD83+ dendritic cells in human resected CCA specimens was associated with better outcomes219. Although NK cells comprise 30–40% of all hepatic lymphocytes220, current knowledge on the role of these cells in CCA is limited. Culture of CCA cells (human CCA cell lines, Hucct1 cells and OZ cells) with the anti-EGFR monoclonal antibody cetuximab augmented CCA cell death via NK cell-induced antibody-dependent cellular cytotoxicity221. Similarly, infusion of ex vivo-expanded human NK cells in CCA mouse xenograft models resulted in tumour regression222.
CCA progression has been associated with a decrease in the components of the adaptive immune response223. Immunohistochemical analyses have demonstrated a preponderance of CD8+ T cells within the tumour and CD4+ T cells in the tumour–liver interface224, as well as an association with longer OS and the presence of tumour-infiltrating CD4+ or CD8+ T cells223,225,226,227. Similarly, the presence of B cells has been linked to a favourable prognosis in CCA223,224. Factors associated with a higher likelihood of response to immune checkpoint blockade include the presence of biomarkers such as PDL1, genetic aberrations such as DNA mismatch repair (MMR) deficiency and/or microsatellite instability (MSI), and the cumulative tumour mutational burden2. On the basis of small cohorts of patients with CCA (range 41–104 patients across the studies), PDL1 is expressed in 42–72% of tumours228,229,230, and seems to be present primarily on immune cells228,229. MMR deficiency has been reported in 5% of pCCA or dCCAs and 10% of iCCAs231. Of note, hypermutation was found in 6% of CCAs and MMR deficiency and/or MSI was present in 36% of these hypermutated tumours56. MSI-high tumours are generally ‘hot’ tumours with an increased number of neoepitopes, CD8+ T cell infiltration, and improved responses to immune checkpoint blockade in cancer generally215. In a cohort of 86 patients with MMR-deficient tumours, including four patients with CCA, immune checkpoint blockade with the anti-PDL1 monoclonal antibody pembrolizumab resulted in a complete response in one of the patients with CCA and stabilization of disease in the other three232. These data indicate that immune-directed therapies including immune checkpoint blockade are a promising approach for, at least, this subset of patients with CCA.
In vitro and in vivo experimental models
Over the past decade, a number of in vitro and in vivo models of cholangiocarcinogenesis have been generated to clarify the phenotypic, biochemical and biological events occurring during the transformation of normal cells into fully malignant cholangiocytes (Table 3). In vitro cell lines, mainly derived from human CCA specimens, have been used widely as a tool to study this disease76,233,234,235. Cell lines exhibit various advantages over animal models: they are free from non-tumourous and necrotic tissues, their growth can be synchronized, relatively high numbers of cells can be produced, cell proliferation and apoptosis can be accurately determined, and they can be molecularly modified (that is, by overexpression or silencing of genes, using antisense oligonucleotides, small interfering RNAs, CRISPR–Cas, and so on), therefore enabling the study of single genes or signal transduction pathways. Furthermore, cell lines can be subjected to drug administration. However, in vitro passaging renders cell lines increasingly different from the original tumours. Primary cultures of CCA cells from tumour tissue are used shortly after derivation and grown under serum-free growth factor-enhanced conditions; therefore, more closely resembling the in vivo situation236,237. Unfortunately, important shortcomings also apply to this system; in particular, primary cultures are time-consuming and elimination of non-tumour cells can be complicated. Furthermore, primary cultures can only be established from surgically resected specimens, limiting the applicability to a subset of patients with CCA who have undergone surgery. Also, primary culture cells lack realistic intercellular and cell–matrix interactions236,237. Importantly, preneoplastic (for example, PSC-derived cholangiocytes) and/or normal cholangiocyte primary cultures should be used as controls132,140,238,239.
To recapitulate more adequately the in vivo tumour tissue structure and to investigate the interaction between CCA and the TME, 3D model systems, known as tumour spheroids and organoids, were developed240,241. Tumour spheroids are self-assembled cultures of cancer cells in the presence or absence of stromal cells within a hydrogel, mimicking the basement membrane, where cell–cell interactions predominate over cell–substrate interactions241. By contrast, tumour organoids are self-organizing stem cell-like structures cultured and expanded in a hydrogel76,242,243. Organoids are successfully established from resected tissue biopsy and needle biopsy samples, faithfully recapitulating the patient tumour at the histopathological level, both in culture and as xenografts in immune-deficient mice242,244. However, to recapitulate CCA tumours in vivo, organoids should be co-cultured with stromal cells. Importantly, whole-exome sequencing revealed that the vast majority of the mutations are retained in liver cancer organoids derived from resected tissues, whereas mutation retention is heterogeneous in biopsy-derived liver cancer organoids76,242,243. Furthermore, CCA organoids have been shown to be a reliable system for drug testing and personalized medicine applications, and possess an almost negligible capacity to differentiate into hepatocytes76,245. As an alternative method, CCA organoids can be established by inducing genetic mutations in healthy organoids via viral transduction and CRISPR–Cas9 genome editing approaches, thus enabling the characterization and elucidation of the roles of oncogenes and/or tumour suppressor genes, either alone or in combination, in cholangiocarcinogenesis76,246.
Mouse models of CCA enable the investigation of the pathobiology of the disease and treatment response in a context that more closely recapitulates the human disease76,247,248,249. Multiple approaches have been used to induce CCA formation in mice and the principal mouse models can be classified into four major groups: chemically induced models, in which a chemotoxic drug is responsible for the oncogenic insult(s); genetically engineered mouse models (GEMM); implantation models; and transposon-based models. As human CCA can develop in the setting of a diseased liver, various methods have been developed to mimic liver alterations, such as those induced in humans by viral hepatitis, chronic inflammation and cholestasis, further increasing the similarity with the human situation. Another major advantage of in vivo models is that they enable the study of CCA starting from early pre-neoplastic to fully progressed lesions, meaning researchers can dissect the specific molecular events occurring at various stages of cholangiocarcinogenesis. In addition, in vivo models enable real-time monitoring of tumour development and response to therapies using imaging modalities such as CT or MRI, or other techniques involving bioluminescence.
Diagnostic and prognostic biomarkers
The current ‘omics’ era is enabling the discovery of new and promising biomarkers in biofluids (serum, urine, bile, saliva) and tumour tissue that could change the paradigm in disease diagnosis and management in the upcoming years (Supplementary Figure 1).
Circulating nucleic acids found in biofluids after active transport or resulting from dying cells are promising diagnostic and prognostic tools for human disorders250,251,252. Cell-free DNA (cfDNA) has been envisaged as mirroring changes in tumour aggressiveness and size, being found both in tumour tissue and plasma from patients with CCA253. Detection of cfDNA in plasma samples could also guide potential mutational-based therapeutic interventions as de novo multiple point mutations in FGFR2 kinase domain were detected in cfDNA, primary tumours and metastases from patients with CCA with acquired resistance to the pan-FGFR inhibitor BGJ398 (ref.254). On the other hand, miRNAs have received special attention due to their increased stability and abundance in biofluids. Two meta-analyses have evaluated their diagnostic value for CCA, and found a pooled area under the receiver operator curve (AUC) of ~0.9 (refs255,256). Notably, bile represented the biological fluid with the highest diagnostic capacity, followed by serum, tissue and urine (AUC 0.95, 0.913, 0.846 and 0.745, respectively)256. In this regard, some bile miRNAs have already been shown to display increased diagnostic capacity for CCA, in comparison with healthy individuals (miR-9, miR-145)257 and also when comparing patients with PSC-derived CCA and isolated PSC (miR-412, miR-640, miR-1537, miR-3189)258. Importantly, combining miR-1537 with CA19-9 resulted in higher diagnostic values than CA19-9 alone (AUC 0.91 versus 0.88; P > 0.05)258. In serum, the levels of miR-21 (refs259,260,261), a well-known onco-miR, were found to be increased in patients with CCA, compared with healthy individuals, positively correlating with clinical stage and poor survival, although the translation of this miRNA into clinics should be performed carefully since it is usually increased in serum and/or plasma of patients with HCC and other liver diseases and cancers262,263. Other miRNAs were also differentially found in the serum and/or plasma of patients with CCA compared with control individuals264,265,266,267,268,269,270, but with some inconsistency, which underscores the necessity for conducting further studies for validation in large, biopsy-proven and well-characterized cohorts of patients and adequate controls.
Proteins and cytokines are now regarded as potential diagnostic and/or prognostic biomarkers. A soluble fragment of cytokeratin-19 (CYFRA 21-1), MMP-7, osteopontin, periostin and IL-6, among others, were shown to be enriched in the serum of patients with CCA, when compared with healthy individuals as controls and/or patients with benign biliary diseases (such as PSC)271,272,273,274,275,276,277,278,279,280,281,282. Among these biomarkers, increased CYFRA 21-1 and osteopontin levels showed superior diagnostic capacity for identifying CCA compared with CA19-9 and CEA271,275, and also showed prognostic value. Of note, increased serum periostin levels were also associated with decreased OS, and the serum periostin level was an independent prognostic factor (HR 3.197)282. As cancer cells display marked metabolic alterations, measuring metabolites in distinct biological samples is now regarded as an encouraging alternative to find diagnostic and/or prognostic biomarkers. Up to now, only a limited number of studies have addressed this issue. Bile acids and phospholipids have been highlighted as promising metabolites in bile for the diagnosis of CCA, as their levels are increased in patients with CCA compared with healthy individuals and patients with HCC283,284,285,286,287. Serum metabolomics has also revealed promising diagnostic biomarkers288,289. An international collaborative study including patients with biopsy-proven iCCA, HCC or PSC and healthy individuals found that several metabolites had higher diagnostic capacity for iCCA than CA19-9, and the authors proposed an algorithm containing six metabolites that was able to differentially diagnose iCCA and HCC (AUC 0.9) in discovery (n = 20 per group) and validation phases (independent cohorts of 14–15 patients per group)289. Interestingly, proteomic analysis of serum extracellular vesicles from patients with CCA, HCC or PSC and healthy individuals as controls revealed candidate proteins with high accuracy for the differential diagnosis of these liver diseases, having higher AUC values than either CA19-9 or α-fetoprotein levels239. Furthermore, another study identified an extracellular vesicle-derived miRNA panel in bile (miRNAs miR-191, miR-486-3p, miR-1274b and miR-484) for the discrimination of CCA from non-malignant biliary diseases290. Although few studies have addressed the potential role of circulating tumour cells (CTCs) as diagnostic and/or prognostic biomarkers in CCA, in a study investigating the associations between numbers of CTCs, patient and tumour characteristics and survival in patients with biliary tract cancer, 17–25% of the patients showed elevated numbers of CTCs (two or more per 7.5 mL of blood)291,292,293, and <10% of the patients showed five or more CTCs per 7.5 mL of blood292. Elevated numbers of CTCs were correlated with greater tumour extent and with reduced overall and disease-free survival292,293. Nevertheless, novel isolation techniques are warranted since the available ones are mainly based on the presence of epithelial cell adhesion molecule, which is elevated in only ~10–20% of CCAs291.
Specific biomarkers in tumour tissue represent promising tools to predict prognosis and treatment response to potential adjuvant therapies in resected CCAs. In two large and independent cohorts of patients with iCCA who had undergone tumour resection (n = 137 in one study56; n = 292 in the other119), mutations in KRAS (12–16%) and TP53 (13–20%) were associated with shorter OS and an increased rate of tumour recurrence when compared with patients with IDH1 or IDH2 mutations or an ‘undetermined’ group (with none of the aforementioned mutations)56,119. According to the transcriptomic profile of iCCA tumours110, the proliferation type was linked with a worse prognosis. Furthermore, a specific 36-gene signature was strongly associated with poor survival in patients with resected iCCA8, and a meta-analysis of 73 studies (including 4,126 patients with CCA) revealed 77 prognostic protein biomarkers, of which fascin, EGFR, mucin 1 (MUC1), MUC4 and p27 were independently associated with OS: high levels of EGFR, MUC1, MUC4 and fascin expression were associated with reduced survival, whereas a high level of p27 expression was associated with increased survival294. Increased levels of miR-21 expression in iCCA were also positively correlated with clinical stage at diagnosis, tumour differentiation status and were linked with poor overall and progression-free survival259,295.
Management
Treatment of localized and advanced disease stages
Surgery is a potential curative option for CCA. However, most patients (∼70%) are diagnosed at late stages due to lack of specific symptoms104. When disease is unresectable, only palliative treatment is possible104,296. Figure 7 summarizes the experience and recommended management of patients diagnosed with CCA according to current guidelines, and lists upcoming potential treatments (see also Supplementary Table 1 for a summary of relevant clinical trials of drugs for CCA).

Flow chart of the presentation, management and outcome of patients with cholangiocarcinoma (CCA) according to current formal guidelines (Supplementary Table 1). BSC, best supportive care; CAR, chimeric antigen receptor; EBRT, external beam radiation therapy; ECOG-PS, Eastern Cooperative Oncology Group Performance Status; FOLFOX, folinic acid, 5-fluorouracil and oxaliplatin; MMR, DNA mismatch repair; OS, overall survival; PFS, progression-free survival; RFS, relapse-free survival; SBRT, stereotactic body radiation therapy.
Surgery
Most patients with CCA have metastatic or locally advanced (that is, unresectable) disease at presentation, and only ∼25% are eligible for resection297.
The majority of patients with iCCA present with large tumours (median size 6 cm)298. In ∼15% of patients the tumour grows towards the hepatic hilum causing biliary obstruction. A biopsy is not needed to confirm the diagnosis in patients with characteristic CCA imaging, elevated serum levels of CA19-9 and normal IgG4 levels, or after excluding other primary tumours (that is, colorectal, gastric and breast). The goal of surgery is a complete margin-negative resection (R0) with an adequate future liver remnant. Most patients require an (extended) hemi-hepatectomy with lymphadenectomy of at least six locoregional lymph nodes for adequate staging103. Staging laparoscopy is recommended by clinical guidelines, especially in patients with a high CA19-9 level or major vascular invasion299.
Regarding pCCA, pre-operative drainage of the future liver remnant is performed to improve liver function and avoid post-hepatectomy liver failure300,301. For this purpose, imaging (CT and/or MRCP) should be performed prior to biliary drainage for accurate staging and surgical planning. Surgery typically involves an (extended) hemi-hepatectomy, including the caudate lobe with en-bloc resection of the extrahepatic bile duct and regional lymph nodes. Staging laparoscopy should precede laparotomy to exclude occult metastatic disease that occurs in ∼15% of patients302. The 90-day postoperative mortality is up to 10% in experienced centres in Europe, with most (~48%) of those who die dying from post-hepatectomy liver failure303,304. In the largest centre in Asia, overall mortality was 4.7% for the period 1977–2010, with the rate markedly decreasing from 11.1% to 1.4% for the periods 1977–1990 and 2006–2010, respectively305. Patients with metastatic pCCA clearly do not benefit from resection305. However, patients with locally advanced disease undergo resection. The presence of Bismuth type IV pCCA (involving both the right and left intrahepatic ducts) is no longer an absolute contraindication for complete resection since it is associated with an OS similar to that in patients with less extensive biliary extension306. Moreover, resection and reconstruction of the portal vein and hepatic artery are increasingly performed307,308. However, tumour abutment on imaging of the main portal vein or common hepatic artery exceeding 180° is associated with a poor prognosis297. In most patients with extensive vascular involvement, the small potential benefit of resection might not justify the considerable surgical mortality rate. Future research should improve pre-operative assessment of the biliary extent of pCCA to reduce the number of R1 resections and of the function of the future liver remnant to reduce the likelihood of post-hepatectomy liver failure. Surgical strategies for dCCA usually require performing a pancreaticoduodenectomy, with removal of the head of the pancreas, the first part of the duodenum, the gallbladder and the bile duct296.
Patients with distant metastatic disease or involvement of aortocaval or truncal nodes are unlikely to benefit from resection309. In a 2018 SEER analysis, even patients with positive regional lymph nodes had similar OS after resection and adjuvant systemic chemotherapy310. Most guidelines recommend resection only for solitary tumours28,296,299. In a study investigating long-term outcomes after resection of iCCA, the median OS in patients with a solitary iCCA was 43.2 months, versus 21.2 months in those with two tumours and 15.3 months in those with three or more tumours311. Patients requiring major vascular resection for iCCA increasingly undergo resection with an acceptable median OS of 33 months312.
Resectable disease: role of adjuvant therapy
Frequent post-surgical relapse313,314 has led to multiple attempts to identify patients at increased risk of relapse315,316 and also to a number of studies of adjuvant therapy. Three phase III randomized clinical studies have been reported, and in all of them patients with resected biliary tract cancer (CCA and gallbladder cancer) were randomly assigned to observation alone or chemotherapy317,318,319. The chemotherapy arm was gemcitabine in the BCAT study (pCCA or dCCA only)317, gemcitabine and oxaliplatin (all biliary tract cancers) in the PRODIGE-12 study318, and capecitabine in the BILCAP study (all biliary tract cancers)319. A total of 226, 196 and 447 patients were randomly assigned in each study, respectively320. Although the BCAT and the PRODIGE-12 study failed to show a benefit from gemcitabine-based chemotherapy, the BILCAP study showed a benefit from adjuvant capecitabine in the pre-planned sensitivity analysis when compared with observation alone, in terms of OS (HR 0.71); however, no statistically significant benefit was observed in the intention-to-treat OS analysis. The BILCAP study did show a benefit in favour of capecitabine in terms of relapse-free survival (HR 0.75). Based on the partial benefits reported in the BILCAP trial, international guidelines published in 2019 recommend adjuvant capecitabine for a period of 6 months following curative resection of CCA as the current standard of care321. The role of chemoradiotherapy remains unclear and might be of benefit in patients with pCCA or dCCA with microscopic positive surgical margins (R1)321,322 or other high-risk factors, although this approach needs to be confirmed in prospective studies. Ongoing studies are evaluating the role of combination chemotherapy such as cisplatin and gemcitabine (ACTICCA-1 trial, NCT02170090; ClinicalTrials.gov) in the adjuvant setting.
Liver transplantation for intrahepatic and perihilar CCA
The inability to obtain a complete resection remains a limitation. Liver transplantation for pCCA was initially determined to be contraindicated due to a high rate of recurrence (~50%)323,324,325. However, following promising initial single-centre reports, a multicentre retrospective study in 216 patients with early-stage, unresectable pCCA treated with neoadjuvant chemoradiotherapy followed by liver transplantation in 12 centres in the USA demonstrated 5-year disease-free survival of 65%, with an intent-to-treat 5-year survival of 53%326,327,328. An area of uncertainty is that in a subgroup of patients, no malignancy was ever confirmed (either pre-operatively or in explanted specimens)329. Subsequent studies have replicated these findings, and identified risk factors for wait-list drop-out as well as for disease recurrence, thus identifying potential candidates for more effective future systemic therapies329,330,331,332. Still, in an Irish cohort, short-term mortality (10–58 months) was observed in patients with CCA undergoing liver transplantation and receiving neoadjuvant chemoradiotherapy330.
The efficacy of neoadjuvant chemoradiotherapy and liver transplantation in patients with unresectable disease has led to the question of whether similar therapy should be offered to patients with resectable pCCA. The extremely limited supply of liver allografts and the need for life-long immunosuppression are important obstacles to this strategy. However, a retrospective multicentre study found that patients with unresectable pCCA undergoing combined neoadjuvant therapy plus liver transplantation had longer 5-year survival (64% versus 18%; P < 0.001) than patients undergoing resection who otherwise met liver transplantation criteria, and this difference remained statistically significant in an intention-to-treat analysis, even after accounting for tumour size, nodal status and PSC333.
The data for liver transplantation in the setting of iCCA are more preliminary than for pCCA, but might be of great value for patients with cirrhosis and tumours smaller than 2 cm. In an initial Spanish study, 5-year survival following transplantation in patients with small, incidental iCCA (<2 cm) was 65%, and this was confirmed in a larger international retrospective analysis using similar selection criteria24,334. In a small series of patients (n = 6) with very large, unresectable iCCA treated with liver transplantation after a prolonged period of disease stability following treatment with neoadjuvant chemotherapy, the 5-year survival was 83% although, importantly, recurrence was noted in 50% of the patients335.
Palliative chemotherapy
At the time of assessment of patients with CCA for palliative treatment, the following three aspects need to be considered: patient fitness as assessed in terms of ECOG-PS (patients with an ECOG-PS of ≥3 are unlikely to benefit from treatment and should be managed with best supportive care); disease distribution (patients with oligometastatic disease or with liver-only disease might be suitable for specific treatment approaches); and accessibility of tumour profiling.
Robust data support the used of first-line cisplatin and gemcitabine chemotherapy in patients with advanced disease336,337. The ABC-02 trial randomly assigned 410 patients with ECOG-PS ≤2 to systemic chemotherapy with gemcitabine alone or cisplatin–gemcitabine336; the study showed an OS benefit in favour of cisplatin–gemcitabine (HR 0.64), a benefit confirmed in the Japanese randomized phase II BT22 study337. Although patients with bilirubin more than twice the upper limit of normal were excluded from the ABC-02 trial, safety and feasibility data support its use in patients with a good ECOG-PS (PS 0 or 1) with jaundice who have refractory biliary obstruction due to endoluminal disease338. More intensive triple-chemotherapy combinations are being explored in the first-line setting, such as cisplatin–gemcitabine combined with nab-paclitaxel339 or with S1 (tegafur, gimeracil and oteracil)340, and FOLFIRINOX (5-FU, oxaliplatin and irinotecan; AMEBICA study, NCT02591030). Acelarin (NUC-1031) is a first-in-class nucleotide analogue, which, unlike gemcitabine, is independent of hENT2 (also known as SLC29A2) cellular transport and is not metabolized by cytidine deaminase, resulting in greater intracellular concentrations. Acelarin with cisplatin341 will be compared with gemcitabine and cisplatin combination therapy in a phase III study (NCT04163900).
After progression on first-line chemotherapy, the benefit of second-line treatment remained unclear until the past few years342. The phase III ABC-06 clinical trial randomly assigned 162 patients diagnosed with advanced biliary tract cancer (72% CCA) who had already progressed on first-line cisplatin–gemcitabine to active symptom control (81 patients) or active symptom control with FOLFOX (folinic acid, 5-FU and oxaliplatin) second-line chemotherapy (81 patients), with OS as the primary end-point343. The ABC-06 trial showed a benefit from second-line chemotherapy (adjusted HR 0.69). Although differences in median OS were modest (5.3 versus 6.2 months) between study arms, differences in survival at 6 months (35.5% versus 50.6%) and 12 months (11.4% versus 25.9%) were clinically meaningful. Based on these findings, FOLFOX can be considered a new standard of care in the second-line setting.
Liver-directed therapies and management of oligometastatic disease
The benchmark for liver-directed therapies was set by a subgroup analysis of the ABC trials including only those 32 patients who received cisplatin and gemcitabine for unresectable iCCA without extrahepatic metastasis344. The median OS in cisplatin-treated and gemcitabine-treated patients with iCCA was 16.7 months and the 3-year OS was 0%. Patients diagnosed with iCCA in liver-predominant disease might be considered for liver-directed therapies345,346. Options for intra-arterial therapy include transarterial radioembolization (TARE) with yttrium-90 (ref.347) and liver chemosaturation232. TARE is the most developed approach but robust evidence supporting its activity is modest347,348,349,350, and the randomized SIRCCA clinical trial evaluating the benefit of adding TARE to gemcitabine and cisplatin in liver-only locally advanced iCCA was prematurely interrupted because of poor recruitment (NCT02807181). Options for local therapy, in the form of external beam radiation351, are also available. Data are awaited from prospective studies (such as the ABC-07 study; EudraCT 2014-003656-31) to evaluate the benefit derived from such approaches in combination with systemic chemotherapy. A phase II trial including 38 patients with unresectable iCCA who received hepatic intra-arterial pump chemotherapy with floxuridine found an impressive radiological response rate of 58% and a 3-year OS exceeding 40%352.
Targeted therapies
Inhibitors of IDH1 (AG120, IDH305), IDH2 (AG221) and pan-IDH1–IDH2 (AG881) are currently being tested in patients with iCCA. AG120 (ivosidenib) was tested in 73 patients with IDH1-mutant advanced CCA in a phase I study353. The only treatment-related grade 3 or worse adverse event present in more than one patient was fatigue (two patients, 3%), and 5% had a confirmed partial response. In a preliminary phase III trial in which 185 patients with IDH-1 mutant CCA were randomly assigned to ivosidenib or placebo128, ivosidenib showed a benefit in terms of progression free-survival (HR 0.37). Median OS was 10.8 months in patients receiving ivosidenib and 9.7 months in patients receiving placebo (HR 0.69); after adjustment of the OS estimation in the placebo arm for crossover (57% of patients in the placebo arm crossed over to ivosidenib at time of disease progression), the median OS in the placebo arm was 6 months. This landmark study provided level A evidence for the efficacy of targeted therapy in CCA and mandates the provision of molecular profiling in this cancer.
There have been promising preliminary data for FGFR inhibitors from phase II studies123,354,355,356,357. Hyperphosphataemia has been shown to be a class effect due to on-target blockade, and requires monitoring and active management356. Some FGFR inhibitors are currently being evaluated earlier in the disease course (first-line setting; for example, the FIGHT-302 study (NCT03656536), and the PROOF study (NCT03773302)). Furthermore, some pan-tumour studies including patients with neurotrophic RTK fusions (TRK inhibitors)358,359 or WNT pathway alterations such as RNF43 mutations (porcupine inhibitors; NCT03447470)118 are relevant to CCA, but only for a very small percentage of patients.
Role of immunotherapy
To date, the clinical data on immune-directed therapies in CCA are limited. Immunotherapy approaches such as vaccines have been tested in CCA without notable success360. Early data are also available for CAR T cell immunotherapy361. In some patients, immune checkpoint blockade with monoclonal antibodies has shown remarkable and durable response rates in a variety of human malignancies362. Checkpoint inhibitors were shown to be effective in patients with MMR-deficient tumours (including some patients with CCA) achieving objective responses in up to 40% of patients363. The KEYNOTE-028 basket trial of pembrolizumab included patients with advanced biliary tract cancer. The objective response rate in this subset was 17% (4 of 23) with a median progression-free survival of 1.8 months364. However, the KEYNOTE-158 trial failed to confirm such activity in biliary tract tumours, with only 6% of patients responding, with a median progression-free survival of 2 months (NCT02628067) according to a preliminary report230. The probable way forward for the development of immunotherapy in CCA (in tumours without MMR deficiency, which respond better) requires either combination immunotherapeutic approaches targeting both the innate and adaptive immune system and/or combined strategies involving chemotherapy or radiation, already planned as part of some of the ongoing clinical trials (ABC-09 trial; NCT03260712). Overall, recommendations for CCA management are summarized in Box 1.
Mechanisms of chemoresistance
A major limitation in the management of patients with CCA is the lack of response to pharmacological treatment. Mechanisms of chemoresistance (MOC) accounting for the marked multidrug resistance phenotype of CCA are still poorly understood365. Nevertheless, to identify the so-called resistome, including a set of proteins involved in the lack of response to chemotherapies, is required to predict treatment failure and to adapt the therapeutic strategy to the evolving defences of the tumour366. Genes involved in MOC are expressed by normal cholangiocytes, with different roles in their physiology, such as protection against potentially harmful compounds present in bile. Their expression during carcinogenesis accounts for intrinsic chemoresistance, whereas upregulation in response to treatment contributes to acquired chemoresistance365,367.
On the basis of their mechanism of action, MOC genes have been classified into seven groups365,366 (Fig. 8). For instance, impaired expression and/or function of plasma membrane solute carriers involved in drug uptake (MOC-1a) results in decreased sensitivity to drugs that cannot reach their intracellular targets. Downregulation of concentrative nucleoside transporters and equilibrative nucleoside transporters, involved in the uptake of nucleoside analogues, such as gemcitabine and 5-FU, or the copper transporter CTR1, involved in cisplatin uptake, lead to reduced sensitivity of CCA cells to these drugs368,369. OCT1 downregulation in CCA369,370,371 is involved in the lack of response to sorafenib372. ATP-binding cassette pumps are important factors accounting for the low intracellular concentrations of anticancer drugs (MOC-1b). The multidrug resistance protein 1, able to export etoposide, doxorubicin, paclitaxel and vinblastine, has been detected in gallbladder epithelium373, whereas the multidrug resistance-associated proteins MRP1 and MRP3 have been observed in CCA369. MRP1 expression has been associated with poor prognosis of patients with iCCA374. Changes in metabolic enzymes can reduce the proportion of active drugs inside tumour cells (MOC-2). Uridine monophosphate synthase, thymidine phosphorylase and uridine phosphorylase 1, which transforms 5-FU and gemcitabine into their active metabolites, are upregulated in many 5-FU-sensitive CCA tumours375. Glutathione S-transferase P, which inactivates drugs, such as cisplatin, by conjugation with glutathione, is highly expressed in CCA376.

Relevant genes and proteins involved in each type of mechanism of chemoresistance (MOC-1 to MOC-7) in cholangiocarcinoma (CCA) are shown, either because they are upregulated or downregulated or their function is enhanced or impaired. Drugs whose efficacy is affected by these changes in the resistome are shown. 5-FU, 5-fluorouracil; TKI, tyrosine-kinase inhibitor.
Importantly, the response to anticancer agents is dependent on the expression and/or function of their molecular targets (MOC-3). High expression of thymidylate synthase in human biliary tract carcinoma cells has been related to insensitivity to 5-FU377. Studies in CCA cells expressing oestrogen receptors have suggested that selective agonists could be a therapeutic option in patients with CCA378. Expression levels of EGFR have been associated with the sensitivity of CCA cells to targeted agents379. Moreover, increased ability of tumour cells to repair drug-induced DNA damage (MOC-4) can also contribute to chemoresistance. The endonuclease DNA excision repair protein 1 removes bulky DNA adducts, whose levels have been associated with the response to cisplatin in patients with CCA380. Changes in promoter methylation of protein complexes involved in DNA MMR, such as human MLH1, affect prognosis in CCA381. Upregulation of ribonucleotide reductase p53R2 has been proposed as a predictive marker of CCA resistance to gemcitabine382. Decreased expression and/or function of pro-apoptotic proteins results in reduced efficacy of chemotherapy (MOC-5a). Downregulation of NK4 in response to 5-FU treatment induces resistance in CCA cells to this drug in vitro55. Downregulation of BAX, BAK, caspase 3 and caspase 9 has been associated with drug resistance in cancer383. Interaction of Fas cell surface death receptor with calmodulin inhibits Fas-induced apoptosis and results in CCA chemoresistance384. By contrast, enhanced expression and/or function of anti-apoptotic proteins also reduces the efficacy of chemotherapy (MOC-5b). Overexpression of ERK and BCL-2 or the overactivation of the PI3K–AKT and RAF–MEK–ERK pathways have been associated with chemoresistance in CCA cells385. CCA usually shows lower vascularity than in HCC — probably as a result of its extensive stroma — which is associated with greater malignant potential386. Moreover, this reduced vascularity due to extensive stroma can limit the access of administered drugs to all tumour cells (MOC-7). This feature and other characteristics that affect the TME, including hypoxia and reduced pH, could reduce the effectiveness of anticancer drugs (MOC-6)387. It has been proposed that ECM proteins, such as laminin-332, induce resistance to doxorubicin and sorafenib in CCA197. Finally, activation of EMT is also involved in resistance to chemotherapy (MOC-7)366. High mobility group A1 protein promoted CCA tumorigenicity and conferred resistance to gemcitabine in vitro in CCA cell lines388.
Future directions and recommendations
The known risk factors for CCA are only involved in ∼20% of cases, indicating the urgent need to ascertain other causes of disease to improve awareness and screening policies for early diagnosis, which might substantially influence patient outcomes. However, considering some established risk factors, potential prevention strategies and lifestyle-modifying concerted actions should be developed to increase awareness. For instance, HBV vaccination, improvements in treatment of HBV infection, HCV infection and nonalcoholic fatty liver disease, and promotion of specific campaigns aiming to reduce alcohol and tobacco consumption, as well as obesity, might markedly influence both the incidence and mortality of CCA.
The accurate recording of epidemiological data (incidence and mortality of each subtype of CCA) and the elucidation of the environmental risk factors and their interplay with genetic and molecular determinants in cholangiocarcinogenesis are extremely important. In this regard, a new coding system was recently approved (ICD-11 and ICD-O-4)20 that better reflects the CCA subtypes — intrahepatic, perihilar and extrahepatic (distal) CCAs — and will start to be used in 2021. Importantly, diagnostic data need to be recorded uniformly and accurately by clinicians, administrators and cancer registries. Awareness of the historical miscoding of CCA should be raised amongst all members of the multidisciplinary team, and at all levels. We need to ensure the appropriate education of coding personnel, who should have senior clinician input to check the accuracy of coding data. Furthermore, accuracy of coding data should be regularly audited. In the future, given advances in our understanding of the genetic drivers for subtypes of CCA, perhaps coding of CCA might also involve molecular profiling.
CCAs are highly heterogeneous at both the intertumoural and intratumoural levels, and have a very poor prognosis. The high heterogeneity and chemoresistance of CCAs represent a limitation for common therapeutic strategies, but it is a unique opportunity for personalized, targeted therapies. Up to 50% of CCAs have current druggable mutations, amplifications or fusions (for example, IDH1, IDH2, BRAF, FGFR, HER2, PIK3CA, MET, among others), opening a new opportunity for therapeutic intervention that deserves intense basic and clinical research. In fact, targeting these mutations is amenable and is already a reality in other types of cancer389,390. In this scenario, the treatment of patients with breast cancer or colorectal cancer with trastuzumab (anti-HER2)391 and cetuximab–panitumumab (anti-EGFR)392 is an example of the successful use of targeted therapy. Therefore, exploration of targeted therapies on a background of standard of care chemotherapy should be continued for CCA. In addition, combined efforts should be made to develop curative therapies. Cost is a major drawback and funding opportunities should be revisited and improved, in parallel with increased awareness amongst the research community, general society, funding agencies and the pharmaceutical industry. Concerted action aimed at increasing the cooperation of these entities should be realized to achieve new effective therapies.
Interactions between cancer cells, CSCs and the TME, as well as the evident clonal evolution and cellular aberrations (genomic, genetic, epigenetic and molecular) contribute to CCA heterogeneity. New technical approaches such as single-cell RNA or DNA sequencing could provide novel critical information about cellular heterogeneity, in both the tumour compartment and the stroma, by capturing genomic and/or genetic alteration with a resolution at the level of the single cell.
New classifications of CCAs based on the combination of clinical, radiological, histological, genomic and molecular features, and later evaluation of their associations with prognosis and treatment response, are mandatory. In addition, it is fundamental to include the resistome in this equation, since the baseline and acquired MOCs will undoubtedly contribute to the success of the therapies tested. The potential determination of the resistome in liquid biopsies (that is, in cfDNA) would open a new avenue for personalized treatment. Future clinical trials should consider the stratification of patients considering clinicopathological subtyping and risk factors, as well as the genomic landscape. Moreover, patient selection for surgery, local therapies, chemotherapy and targeted therapies must be improved. Similar to the tumour compartment, a better stratification of these alterations within the microenvironment could help in the design of innovative treatment options including immunotherapies (such as immune checkpoints and CAR T cell therapy) and ECM-oriented treatments. The majority of clinical trials performed so far for advanced CCA did not take all these considerations into account, which might explain, at least in part, the disappointing results obtained. Thus, action needs to be taken to bring together experts across different fields. In addition, robust circulating biomarkers are needed for the accurate diagnosis of CCA, as well as to predict prognosis and treatment response. Non-coding RNA, specifically miRNAs, long non-coding RNAs and circular RNAs393,394 and circulating proteins and/or metabolites could represent such promising biomarkers, due to their easy detection and stability in biological fluids, either free or encapsulated into extracellular vesicles. In this regard, international collaborative projects, such as the ESCALON project (competitively funded by the European Union as part of the Horizon 2020 programme), are warranted to improve the understanding, prediction (risk factors) and diagnosis (biomarkers) of CCA not only in Europe and North America, but also in Latin America and other continents.
CCA management nowadays requires dedicated centres with multidisciplinary expertise that enable the proper translation of basic investigations to clinical practice. International collaborative networks of multidisciplinary scientists such as the ENS-CCA are especially important as they are accelerating acquisition of scientific knowledge on this cancer, which then influences clinical practice. In particular, it is important to highlight the ENS-CCA Action EURO-CHOLANGIO-NET, a European Horizon 2020 competitive programme (2019–2023) that has the objective to create and boost multidisciplinary and cross-sectional studies to decipher the biological jigsaw of CCA. This open, structured initiative has received the support of the European Commission and is endorsed by the European Association for the Study of the Liver, the International Liver Foundation, several research and development companies, and the CCA patient associations (including the Cholangiocarcinoma Foundation in the USA and the Alan Morement Memorial Fund (AMMF) in the UK). During the coming years, EURO-CHOLANGIO-NET will concentrate in the following objectives: shorten the current gaps in CCA knowledge and applications by overcoming the limitation of the small number of cases through the development of international clinical, histological and radiological registries, which are necessary to dissect the multilevel heterogeneity of CCAs; improve translation by generating consensus on appropriate experimental models of CCA, diagnostic and/or prognostic biomarkers and imaging techniques, and clinical management; dissect intertumoural and intratumoural heterogeneity to define specific features for early diagnosis of each CCA subtype; rationalize cost-efficient, personalized, targeted therapies for CCA by defining the driver mutations, epigenetic alterations, and transcriptome of each CCA histomorphological subtype; and develop novel drugs and therapeutic strategies. The ENS-CCA, together with dedicated foundations such as the AMMF and the Cholangiocarcinoma Foundation, as well as other international networks and collaborators have contributed to the creation of the Global Cholangiocarcinoma Alliance, which has the aim of joining forces to increase awareness of this cancer and to establish a global voice in CCA through community collaborations. In Asia, an important consortium was created — the Thailand Initiative in Genomics and Expression Research for Liver Cancer (TIGER-LC) — to identify genomic and endemic factors that could modify CCA and HCC susceptibility and progression395. Combining the efforts from experts worldwide will definitely contribute to improved understanding of CCA and will reinforce the necessary link between basic and clinical science, and therefore hopefully improve patient welfare.
Conclusions
CCAs are highly aggressive and heterogeneous, at both the intertumoural and intratumoural levels, resulting in poor prognosis. Different risk factors, interactions between cancer cells, CSCs and the TME, as well as the evident clonal evolution and genetic and/or epigenetic aberrations contribute to CCA heterogeneity. Different CCA molecular subtypes, with distinct prognoses and responses to therapy, have already been described. Tumour resection is still the only potentially curative option for these patients, although only a small percentage of patients are eligible and the percentage recurrence is high. We still lack of accurate noninvasive biomarkers for the diagnosis and to estimate the prognosis in patients with CCA. Furthermore, knowledge of the MOCs of these cancers needs to be expanded, but current information should be included in the future treatment decisions. Although important information has already been unveiled, CCA is still an open field of research, with important gaps that need to be filled (Table 4). Therefore, all the efforts must be gathered to try to go beyond and decipher the complexity of CCA.
References
Banales, J. M. et al. Expert consensus document: cholangiocarcinoma: current knowledge and future perspectives consensus statement from the European Network for the Study of Cholangiocarcinoma (ENS-CCA). Nat. Rev. Gastroenterol. Hepatol. 13, 261–280 (2016).
Rizvi, S., Khan, S. A., Hallemeier, C. L., Kelley, R. K. & Gores, G. J. Cholangiocarcinoma - evolving concepts and therapeutic strategies. Nat. Rev. Clin. Oncol. 15, 95–111 (2018).
Munoz-Garrido, P. & Rodrigues, P. M. The jigsaw of dual hepatocellular-intrahepatic cholangiocarcinoma tumours. Nat. Rev. Gastroenterol. Hepatol. 16, 653–655 (2019).
Brunt, E. et al. cHCC-CCA: consensus terminology for primary liver carcinomas with both hepatocytic and cholangiocytic differentation. Hepatology 68, 113–126 (2018).
Khan, A. S. & Dageforde, L. A. Cholangiocarcinoma. Surg. Clin. North. Am. 99, 315–335 (2019).
DeOliveira, M. L. et al. Cholangiocarcinoma: thirty-one-year experience with 564 patients at a single institution. Ann. Surg. 245, 755–762 (2007).
Nakeeb, A. et al. Cholangiocarcinoma. A spectrum of intrahepatic, perihilar, and distal tumors. Ann. Surg. 224, 463–473 (1996).
Andersen, J. B. et al. Genomic and genetic characterization of cholangiocarcinoma identifies therapeutic targets for tyrosine kinase inhibitors. Gastroenterology 142, 1021–1031.e15 (2012).
Bertuccio, P. et al. Global trends in mortality from intrahepatic and extrahepatic cholangiocarcinoma. J. Hepatol. 71, 104–114 (2019).
Lindner, P., Rizell, M. & Hafstrom, L. The impact of changed strategies for patients with cholangiocarcinoma in this millenium. HPB Surg. 2015, 736049 (2015).
Kamsa-Ard, S. et al. Cholangiocarcinoma trends, incidence, and relative survival in Khon Kaen, Thailand from 1989 through 2013: a population-based cancer registry study. J. Epidemiol. 29, 197–204 (2019).
Strijker, M. et al. Treatment and survival of resected and unresected distal cholangiocarcinoma: a nationwide study. Acta Oncol. 58, 1048–1055 (2019).
Alabraba, E. et al. Increased multimodality treatment options has improved survival for hepatocellular carcinoma but poor survival for biliary tract cancers remains unchanged. Eur. J. Surg. Oncol. 45, 1660–1667 (2019).
Groot Koerkamp, B. et al. Recurrence rate and pattern of perihilar cholangiocarcinoma after curative intent resection. J. Am. Coll. Surg. 221, 1041–1049 (2015).
Komaya, K. et al. Recurrence after curative-intent resection of perihilar cholangiocarcinoma: analysis of a large cohort with a close postoperative follow-up approach. Surgery 163, 732–738 (2018).
Cambridge, W. A. et al. Meta-analysis and meta-regression of survival after liver transplantation for unresectable perihilar cholangiocarcinoma. Ann. Surg. https://doi.org/10.1097/SLA.0000000000003801 (2020).
Spolverato, G. et al. Management and outcomes of patients with recurrent intrahepatic cholangiocarcinoma following previous curative-intent surgical resection. Ann. Surg. Oncol. 23, 235–243 (2016).
Yao, K. J., Jabbour, S., Parekh, N., Lin, Y. & Moss, R. A. Increasing mortality in the United States from cholangiocarcinoma: an analysis of the National Center for Health Statistics Database. BMC Gastroenterol. 16, 117 (2016).
Khan, S. A., Tavolari, S. & Brandi, G. Cholangiocarcinoma: epidemiology and risk factors. Liver Int. 39 (Suppl. 1), 19–31 (2019).
Bosman, F. T., Carneiro, F., Hruban, R. & Theise N. WHO Classification of Tumours: Digestive System Tumours 5th edn Vol. 1 (IARC, 2019).
Antwi, S. O., Mousa, O. Y. & Patel, T. Racial, ethnic, and age disparities in incidence and survival of intrahepatic cholangiocarcinoma in the United States; 1995–2014. Ann. Hepatol. 17, 604–614 (2018).
Ransome, E. et al. Trends in surgery and disparities in receipt of surgery for intrahepatic cholangiocarcinoma in the US: 2005–2014. J. Gastrointest. Oncol. 10, 339–347 (2019).
Uhlig, J. et al. Intrahepatic cholangiocarcinoma: socioeconomic discrepancies, contemporary treatment approaches and survival trends from the National Cancer Database. Ann. Surg. Oncol. 26, 1993–2000 (2019).
Sapisochin, G. et al. Liver transplantation for “very early” intrahepatic cholangiocarcinoma: international retrospective study supporting a prospective assessment. Hepatology 64, 1178–1188 (2016).
Alberts, R. et al. Genetic association analysis identifies variants associated with disease progression in primary sclerosing cholangitis. Gut 67, 1517–1524 (2018).
Xue, R. et al. Genomic and transcriptomic profiling of combined hepatocellular and intrahepatic cholangiocarcinoma reveals distinct molecular subtypes. Cancer Cell 35, 932–947.e8 (2019).
Saha, S. K., Zhu, A. X., Fuchs, C. S. & Brooks, G. A. Forty-year trends in cholangiocarcinoma incidence in the U.S.: intrahepatic disease on the rise. Oncologist 21, 594–599 (2016).
Bridgewater, J. et al. Guidelines for the diagnosis and management of intrahepatic cholangiocarcinoma. J. Hepatol. 60, 1268–1289 (2014).
Bray, F. et al. (eds) Cancer incidence in five continents (CI5) volume XI. IARC https://ci5.iarc.fr (2017).
Clements, O., Eliahoo, J., Kim, J. U., Taylor-Robinson, S. D. & Khan, S. A. Risk factors for intrahepatic and extrahepatic cholangiocarcinoma: a systematic review and meta-analysis. J. Hepatol. 72, 95–103 (2020).
Xiong, J. et al. Aspirin use is associated with a reduced risk of cholangiocarcinoma: a systematic review and meta-analysis. Cancer Manag. Res. 10, 4095–4104 (2018).
Burr, N. E. et al. Aspirin may prevent cholangiocarcinoma: a case-control study from the United Kingdom. Dig. Dis. Sci. 59, 1567–1572 (2014).
Choi, J. et al. Aspirin use and the risk of cholangiocarcinoma. Hepatology 64, 785–796 (2016).
Liu, Z. et al. Statin use and reduced risk of biliary tract cancers in the UK clinical practice research datalink. Gut 68, 1458–1464 (2019).
Peng, Y. C. et al. Statins are associated with a reduced risk of cholangiocarcinoma: a population-based case-control study. Br. J. Clin. Pharmacol. 80, 755–761 (2015).
Jackson, S. S. et al. Association between aspirin use and biliary tract cancer survival. JAMA Oncol. 5, 1802–1804 (2019).
Lee, D. H. & Lee, J. M. Primary malignant tumours in the non-cirrhotic liver. Eur. J. Radiol. 95, 349–361 (2017).
Nakanuma, Y. et al. Pathological classification of intrahepatic cholangiocarcinoma based on a new concept. World J. Hepatol. 2, 419–427 (2010).
Deoliveira, M. L. et al. New staging system and a registry for perihilar cholangiocarcinoma. Hepatology 53, 1363–1371 (2011).
Nakanuma, Y. & Kakuda, Y. Pathologic classification of cholangiocarcinoma: new concepts. Best Pract. Res. Clin. Gastroenterol. 29, 277–293 (2015).
Kendall, T. et al. Anatomical, histomorphological and molecular classification of cholangiocarcinoma. Liver Int. 39 (Suppl. 1), 7–18 (2019).
Aishima, S. et al. Proposal of progression model for intrahepatic cholangiocarcinoma: clinicopathologic differences between hilar type and peripheral type. Am. J. Surg. Pathol. 31, 1059–1067 (2007).
Akita, M. et al. Dichotomy in intrahepatic cholangiocarcinomas based on histologic similarities to hilar cholangiocarcinomas. Mod. Pathol. 30, 986–997 (2017).
Liau, J. Y. et al. Morphological subclassification of intrahepatic cholangiocarcinoma: etiological, clinicopathological, and molecular features. Mod. Pathol. 27, 1163–1173 (2014).
Hayashi, A. et al. Distinct clinicopathologic and genetic features of 2 histologic subtypes of intrahepatic cholangiocarcinoma. Am. J. Surg. Pathol. 40, 1021–1030 (2016).
Komuta, M. et al. Histological diversity in cholangiocellular carcinoma reflects the different cholangiocyte phenotypes. Hepatology 55, 1876–1888 (2012).
Cardinale, V. et al. Mucin-producing cholangiocarcinoma might derive from biliary tree stem/progenitor cells located in peribiliary glands. Hepatology 55, 2041–2042 (2012).
Igarashi, S. et al. Participation of peribiliary glands in biliary tract pathophysiologies. World J. Hepatol. 5, 425–432 (2013).
Carpino, G. et al. Neoplastic transformation of the peribiliary stem cell niche in cholangiocarcinoma arisen in primary sclerosing cholangitis. Hepatology 69, 622–638 (2019).
Arai, Y. et al. Fibroblast growth factor receptor 2 tyrosine kinase fusions define a unique molecular subtype of cholangiocarcinoma. Hepatology 59, 1427–1434 (2014).
Borger, D. R. et al. Frequent mutation of isocitrate dehydrogenase (IDH)1 and IDH2 in cholangiocarcinoma identified through broad-based tumor genotyping. Oncologist 17, 72–79 (2012).
Graham, R. P. et al. Fibroblast growth factor receptor 2 translocations in intrahepatic cholangiocarcinoma. Hum. Pathol. 45, 1630–1638 (2014).
Kipp, B. R. et al. Isocitrate dehydrogenase 1 and 2 mutations in cholangiocarcinoma. Hum. Pathol. 43, 1552–1558 (2012).
Lowery, M. A. et al. Comprehensive molecular profiling of intrahepatic and extrahepatic cholangiocarcinomas: potential targets for intervention. Clin. Cancer Res. 24, 4154–4161 (2018).
Ge, X. et al. NK4 regulates 5-fluorouracil sensitivity in cholangiocarcinoma cells by modulating the intrinsic apoptosis pathway. Oncol. Rep. 30, 448–454 (2013).
Nakamura, H. et al. Genomic spectra of biliary tract cancer. Nat. Genet. 47, 1003–1010 (2015).
Wang, P. et al. Mutations in isocitrate dehydrogenase 1 and 2 occur frequently in intrahepatic cholangiocarcinomas and share hypermethylation targets with glioblastomas. Oncogene 32, 3091–3100 (2013).
Yachida, S. et al. Genomic sequencing identifies ELF3 as a driver of ampullary carcinoma. Cancer Cell 29, 229–240 (2016).
Visvader, J. E. Cells of origin in cancer. Nature 469, 314–322 (2011).
Lytle, N. K., Barber, A. G. & Reya, T. Stem cell fate in cancer growth, progression and therapy resistance. Nat. Rev. Cancer 18, 669–680 (2018).
Sutherland, K. D. & Visvader, J. E. Cellular mechanisms underlying intertumoral heterogeneity. Trends Cancer 1, 15–23 (2015).
Hoadley, K. A. et al. Cell-of-origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell 173, 291–304.e6 (2018).
Coulouarn, C. et al. Combined hepatocellular-cholangiocarcinomas exhibit progenitor features and activation of Wnt and TGFβ signaling pathways. Carcinogenesis 33, 1791–1796 (2012).
Moeini, A. et al. Mixed hepatocellular cholangiocarcinoma tumors: cholangiolocellular carcinoma is a distinct molecular entity. J. Hepatol. 66, 952–961 (2017).
Cardinale, V. et al. Profiles of cancer stem cell subpopulations in cholangiocarcinomas. Am. J. Pathol. 185, 1724–1739 (2015).
Lanzoni, G., Cardinale, V. & Carpino, G. The hepatic, biliary, and pancreatic network of stem/progenitor cell niches in humans: a new reference frame for disease and regeneration. Hepatology 64, 277–286 (2016).
Bragazzi, M. C. et al. New insights into cholangiocarcinoma: multiple stems and related cell lineages of origin. Ann. Gastroenterol. 31, 42–55 (2018).
Cardinale, V., Carpino, G., Reid, L., Gaudio, E. & Alvaro, D. Multiple cells of origin in cholangiocarcinoma underlie biological, epidemiological and clinical heterogeneity. World J. Gastrointest. Oncol. 4, 94–102 (2012).
Komuta, M. et al. Clinicopathological study on cholangiolocellular carcinoma suggesting hepatic progenitor cell origin. Hepatology 47, 1544–1556 (2008).
Lee, J. S. et al. A novel prognostic subtype of human hepatocellular carcinoma derived from hepatic progenitor cells. Nat. Med. 12, 410–416 (2006).
Sia, D., Villanueva, A., Friedman, S. L. & Llovet, J. M. Liver cancer cell of origin, molecular class, and effects on patient prognosis. Gastroenterology 152, 745–761 (2017).
Woo, H. G. et al. Identification of a cholangiocarcinoma-like gene expression trait in hepatocellular carcinoma. Cancer Res. 70, 3034–3041 (2010).
Tomasetti, C. & Vogelstein, B. Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 347, 78–81 (2015).
Zhu, L. et al. Multi-organ mapping of cancer risk. Cell 166, 1132–1146.e7 (2016).
Carpino, G. et al. Activation of biliary tree stem cells within peribiliary glands in primary sclerosing cholangitis. J. Hepatol. 63, 1220–1228 (2015).
Vicent, S. et al. Experimental models to unravel the molecular pathogenesis, cell of origin and stem cell properties of cholangiocarcinoma. Liver Int. 39 (Suppl. 1), 79–97 (2019).
Dill, M. T. et al. Constitutive Notch2 signaling induces hepatic tumors in mice. Hepatology 57, 1607–1619 (2013).
Guest, R. V. et al. Cell lineage tracing reveals a biliary origin of intrahepatic cholangiocarcinoma. Cancer Res. 74, 1005–1010 (2014).
Sekiya, S. & Suzuki, A. Intrahepatic cholangiocarcinoma can arise from Notch-mediated conversion of hepatocytes. J. Clin. Invest. 122, 3914–3918 (2012).
Fan, B. et al. Cholangiocarcinomas can originate from hepatocytes in mice. J. Clin. Invest. 122, 2911–2915 (2012).
Wang, J. et al. Notch2 controls hepatocyte-derived cholangiocarcinoma formation in mice. Oncogene 37, 3229–3242 (2018).
Lu, W. Y. et al. Hepatic progenitor cells of biliary origin with liver repopulation capacity. Nat. Cell Biol. 17, 971–983 (2015).
Raven, A. et al. Cholangiocytes act as facultative liver stem cells during impaired hepatocyte regeneration. Nature 547, 350–354 (2017).
Rios, A. C., Fu, N. Y., Cursons, J., Lindeman, G. J. & Visvader, J. E. The complexities and caveats of lineage tracing in the mammary gland. Breast Cancer Res. 18, 116 (2016).
Tarlow, B. D., Finegold, M. J. & Grompe, M. Clonal tracing of Sox9+ liver progenitors in mouse oval cell injury. Hepatology 60, 278–289 (2014).
Lemaigre, F. P. Determining the fate of hepatic cells by lineage tracing: facts and pitfalls. Hepatology 61, 2100–2103 (2015).
De Minicis, S. et al. Liver carcinogenesis: rodent models of hepatocarcinoma and cholangiocarcinoma. Dig. Liver Dis. 45, 450–459 (2013).
Alvaro, D. et al. Cholangiocarcinoma in Italy: a national survey on clinical characteristics, diagnostic modalities and treatment. Results from the “Cholangiocarcinoma” committee of the Italian Association for the Study of Liver Disease. Dig. Liver Dis. 43, 60–65 (2011).
Wildner, D. et al. CEUS in hepatocellular carcinoma and intrahepatic cholangiocellular carcinoma in 320 patients – early or late washout matters: a subanalysis of the DEGUM multicenter trial. Ultraschall Med. 36, 132–139 (2015).
Sirica, A. E. et al. Intrahepatic cholangiocarcinoma: continuing challenges and translational advances. Hepatology 69, 1803–1815 (2019).
Joo, I., Lee, J. M. & Yoon, J. H. Imaging diagnosis of intrahepatic and perihilar cholangiocarcinoma: recent advances and challenges. Radiology 288, 7–13 (2018).
Jhaveri, K. S. & Hosseini-Nik, H. MRI of cholangiocarcinoma. J. Magn. Reson. Imaging 42, 1165–1179 (2015).
Rimola, J. et al. Cholangiocarcinoma in cirrhosis: absence of contrast washout in delayed phases by magnetic resonance imaging avoids misdiagnosis of hepatocellular carcinoma. Hepatology 50, 791–798 (2009).
Galassi, M. et al. Patterns of appearance and risk of misdiagnosis of intrahepatic cholangiocarcinoma in cirrhosis at contrast enhanced ultrasound. Liver Int. 33, 771–779 (2013).
Chernyak, V. et al. Liver Imaging Reporting and Data System (LI-RADS) version 2018: imaging of hepatocellular carcinoma in at-risk patients. Radiology 289, 816–830 (2018).
Choi, S. H. et al. Intrahepatic cholangiocarcinoma in patients with cirrhosis: differentiation from hepatocellular carcinoma by using gadoxetic acid-enhanced MR imaging and dynamic CT. Radiology 282, 771–781 (2017).
Vilana, R. et al. Intrahepatic peripheral cholangiocarcinoma in cirrhosis patients may display a vascular pattern similar to hepatocellular carcinoma on contrast-enhanced ultrasound. Hepatology 51, 2020–2029 (2010).
Terzi, E. et al. Contrast ultrasound LI-RADS LR-5 identifies hepatocellular carcinoma in cirrhosis in a multicenter restropective study of 1,006 nodules. J. Hepatol. 68, 485–492 (2018).
American College of Radiology. CEUS LI-RADS v2017. ACR https://www.acr.org/Clinical-Resources/Reporting-and-Data-Systems/LI-RADS/CEUS-LI-RADS-v2017 (2017).
Lamarca, A. et al. (18)F-fluorodeoxyglucose positron emission tomography ((18)FDG-PET) for patients with biliary tract cancer: systematic review and meta-analysis. J. Hepatol. 71, 115–129 (2019).
Hamilton, S. R. A. et al. WHO Classification of Tumours: Pathology and Genetics of Tumours of the Digestive System 3rd edn Vol. 2 (IARC, 2000).
Ishak, K. G., Goodman, Z. D. & Stocker, J. T. Atlas of Tumor Pathology: Tumors of the Liver and Intrahepatic Bile Ducts (Armed Forces Institute of Pathology, 2001).
Amin, M. B. et al. AJCC Cancer Staging Manual (Springer, 2017).
Forner, A. et al. Clinical presentation, diagnosis and staging of cholangiocarcinoma. Liver Int. 39 (Suppl. 1), 98–107 (2019).
Spolverato, G. et al. Comparative performances of the 7th and the 8th editions of the American Joint Committee on Cancer staging systems for intrahepatic cholangiocarcinoma. J. Surg. Oncol. 115, 696–703 (2017).
Kang, S. H. et al. Prognostic comparison of the 7th and 8th editions of the American Joint Committee on Cancer staging system for intrahepatic cholangiocarcinoma. J. Hepatobiliary Pancreat. Sci. 25, 240–248 (2018).
Lamarca, A. et al. Liver metastases (LM) from intrahepatic cholangiocarcinoma (iCCA): outcomes from the European Network for the Study of Cholangiocarcinoma (ENS-CCA) registry and implications on current American Joint Committee on Cancer (AJCC) staging [abstract 731P]. Ann. Oncol. 30 (Suppl. 5), v280–v281 (2019).
Sapisochin, G. et al. “Very early” intrahepatic cholangiocarcinoma in cirrhotic patients: should liver transplantation be reconsidered in these patients? Am. J. Transpl. 14, 660–667 (2014).
Chaiteerakij, R. et al. A new clinically based staging system for perihilar cholangiocarcinoma. Am. J. Gastroenterol. 109, 1881–1890 (2014).
Sia, D. et al. Integrative molecular analysis of intrahepatic cholangiocarcinoma reveals 2 classes that have different outcomes. Gastroenterology 144, 829–840 (2013).
Lawrence, M. S. et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499, 214–218 (2013).
Borad, M. J. et al. Integrated genomic characterization reveals novel, therapeutically relevant drug targets in FGFR and EGFR pathways in sporadic intrahepatic cholangiocarcinoma. PLoS Genet. 10, e1004135 (2014).
Ross, J. S. et al. New routes to targeted therapy of intrahepatic cholangiocarcinomas revealed by next-generation sequencing. Oncologist 19, 235–242 (2014).
Wu, Y. M. et al. Identification of targetable FGFR gene fusions in diverse cancers. Cancer Discov. 3, 636–647 (2013).
Sia, D. et al. Massive parallel sequencing uncovers actionable FGFR2-PPHLN1 fusion and ARAF mutations in intrahepatic cholangiocarcinoma. Nat. Commun. 6, 6087 (2015).
Jusakul, A. et al. Whole-genome and epigenomic landscapes of etiologically distinct subtypes of cholangiocarcinoma. Cancer Discov. 7, 1116–1135 (2017).
Chan-On, W. et al. Exome sequencing identifies distinct mutational patterns in liver fluke-related and non-infection-related bile duct cancers. Nat. Genet. 45, 1474–1478 (2013).
Ong, C. K. et al. Exome sequencing of liver fluke-associated cholangiocarcinoma. Nat. Genet. 44, 690–693 (2012).
Nepal, C. et al. Genomic perturbations reveal distinct regulatory networks in intrahepatic cholangiocarcinoma. Hepatology 68, 949–963 (2018).
Zou, S. et al. Mutational landscape of intrahepatic cholangiocarcinoma. Nat. Commun. 5, 5696 (2014).
Farshidfar, F. et al. Integrative genomic analysis of cholangiocarcinoma identifies distinct IDH-mutant molecular profiles. Cell Rep. 18, 2780–2794 (2017).
O’Rourke, C. J., Munoz-Garrido, P., Aguayo, E. L. & Andersen, J. B. Epigenome dysregulation in cholangiocarcinoma. Biochim. Biophys. Acta Mol. Basis Dis. 1864, 1423–1434 (2018).
Javle, M. et al. Phase II study of BGJ398 in patients with FGFR-altered advanced cholangiocarcinoma. J. Clin. Oncol. 36, 276–282 (2018).
Mazzaferro, V. et al. Derazantinib (ARQ 087) in advanced or inoperable FGFR2 gene fusion-positive intrahepatic cholangiocarcinoma. Br. J. Cancer 120, 165–171 (2019).
Braconi, C. et al. Molecular perturbations in cholangiocarcinoma: is it time for precision medicine? Liver Int. 39 (Suppl. 1), 32–42 (2019).
Melum, E. et al. Genome-wide association analysis in primary sclerosing cholangitis identifies two non-HLA susceptibility loci. Nat. Genet. 43, 17–19 (2011).
Gingras, M. C. et al. Ampullary cancers harbor ELF3 tumor suppressor gene mutations and exhibit frequent WNT dysregulation. Cell Rep. 14, 907–919 (2016).
Abou-Alfa, G. K. et al. ClarIDHy: a global, phase 3, randomized, double-blind study of ivosidenib (IVO) vs placebo in patients with advanced cholangiocarcinoma (CC) with an isocitrate dehydrogenase 1 (IDH1) mutation. Ann. Oncol. 30 (Suppl. 5), v851–v934 (2019).
Saha, S. K. et al. Isocitrate dehydrogenase mutations confer dasatinib hypersensitivity and SRC dependence in intrahepatic cholangiocarcinoma. Cancer Discov. 6, 727–739 (2016).
Lampis, A. et al. MIR21 drives resistance to heat shock protein 90 inhibition in cholangiocarcinoma. Gastroenterology 154, 1066–1079.e5 (2018).
O’Rourke, C. J., Lafuente-Barquero, J. & Andersen, J. B. Epigenome remodeling in cholangiocarcinoma. Trends Cancer 5, 335–350 (2019).
Merino-Azpitarte, M. et al. SOX17 regulates cholangiocyte differentiation and acts as a tumor suppressor in cholangiocarcinoma. J. Hepatol. 67, 72–83 (2017).
Goeppert, B. et al. Global alterations of DNA methylation in cholangiocarcinoma target the Wnt signaling pathway. Hepatology 59, 544–554 (2014).
Easwaran, H. et al. A DNA hypermethylation module for the stem/progenitor cell signature of cancer. Genome Res. 22, 837–849 (2012).
Bernhart, S. H. et al. Changes of bivalent chromatin coincide with increased expression of developmental genes in cancer. Sci. Rep. 6, 37393 (2016).
Goeppert, B. et al. Integrative analysis defines distinct prognostic subgroups of intrahepatic cholangiocarcinoma. Hepatology 69, 2091–2106 (2019).
Morine, Y. et al. Role of histone deacetylase expression in intrahepatic cholangiocarcinoma. Surgery 151, 412–419 (2012).
Salati, M. & Braconi, C. Noncoding RNA in cholangiocarcinoma. Semin. Liver Dis. 39, 13–25 (2019).
Gradilone, S. A., O’Hara, S. P., Masyuk, T. V., Pisarello, M. J. & LaRusso, N. F. MicroRNAs and benign biliary tract diseases. Semin. Liver Dis. 35, 26–35 (2015).
Erice, O. et al. Differential effects of FXR or TGR5 activation in cholangiocarcinoma progression. Biochim. Biophys. Acta Mol. Basis Dis. 1864, 1335–1344 (2018).
Mansini, A. P. et al. MicroRNA (miR)-433 and miR-22 dysregulations induce histone-deacetylase-6 overexpression and ciliary loss in cholangiocarcinoma. Hepatology 68, 561–573 (2018).
Carotenuto, P. et al. Wnt signalling modulates transcribed-ultraconserved regions in hepatobiliary cancers. Gut 66, 1268–1277 (2017).
Andersen, J. B. & Thorgeirsson, S. S. Genomic decoding of intrahepatic cholangiocarcinoma reveals therapeutic opportunities. Gastroenterology 144, 687–690 (2013).
Isomoto, H. et al. Interleukin 6 upregulates myeloid cell leukemia-1 expression through a STAT3 pathway in cholangiocarcinoma cells. Hepatology 42, 1329–1338 (2005).
Wehbe, H., Henson, R., Meng, F., Mize-Berge, J. & Patel, T. Interleukin-6 contributes to growth in cholangiocarcinoma cells by aberrant promoter methylation and gene expression. Cancer Res. 66, 10517–10524 (2006).
Sirica, A. E. Role of ErbB family receptor tyrosine kinases in intrahepatic cholangiocarcinoma. World J. Gastroenterol. 14, 7033–7058 (2008).
Lozano, E. et al. Cocarcinogenic effects of intrahepatic bile acid accumulation in cholangiocarcinoma development. Mol. Cancer Res. 12, 91–100 (2014).
Brivio, S., Cadamuro, M., Strazzabosco, M. & Fabris, L. Tumor reactive stroma in cholangiocarcinoma: the fuel behind cancer aggressiveness. World J. Hepatol. 9, 455–468 (2017).
Moeini, A., Sia, D., Bardeesy, N., Mazzaferro, V. & Llovet, J. M. Molecular pathogenesis and targeted therapies for intrahepatic cholangiocarcinoma. Clin. Cancer Res. 22, 291–300 (2016).
Gu, T. L. et al. Survey of tyrosine kinase signaling reveals ROS kinase fusions in human cholangiocarcinoma. PLoS One 6, e15640 (2011).
Jiao, Y. et al. Exome sequencing identifies frequent inactivating mutations in BAP1, ARID1A and PBRM1 in intrahepatic cholangiocarcinomas. Nat. Genet. 45, 1470–1473 (2013).
Xue, T. C., Zhang, B. H., Ye, S. L. & Ren, Z. G. Differentially expressed gene profiles of intrahepatic cholangiocarcinoma, hepatocellular carcinoma, and combined hepatocellular-cholangiocarcinoma by integrated microarray analysis. Tumour Biol. 36, 5891–5899 (2015).
Turnpenny, P. D. & Ellard, S. Alagille syndrome: pathogenesis, diagnosis and management. Eur. J. Hum. Genet. 20, 251–257 (2012).
Geisler, F. & Strazzabosco, M. Emerging roles of Notch signaling in liver disease. Hepatology 61, 382–392 (2015).
Wu, W. R. et al. Clinicopathological significance of aberrant Notch receptors in intrahepatic cholangiocarcinoma. Int. J. Clin. Exp. Pathol. 7, 3272–3279 (2014).
Wu, W. R. et al. Notch1 is overexpressed in human intrahepatic cholangiocarcinoma and is associated with its proliferation, invasiveness and sensitivity to 5-fluorouracil in vitro. Oncol. Rep. 31, 2515–2524 (2014).
Aoki, S. et al. Aberrant activation of Notch signaling in extrahepatic cholangiocarcinoma: clinicopathological features and therapeutic potential for cancer stem cell-like properties. BMC Cancer 16, 854 (2016).
Zender, S. et al. A critical role for notch signaling in the formation of cholangiocellular carcinomas. Cancer Cell 23, 784–795 (2013).
Huntzicker, E. G. et al. Differential effects of targeting Notch receptors in a mouse model of liver cancer. Hepatology 61, 942–952 (2015).
Guest, R. V. et al. Notch3 drives development and progression of cholangiocarcinoma. Proc. Natl Acad. Sci. USA 113, 12250–12255 (2016).
Cigliano, A., Wang, J., Chen, X. & Calvisi, D. F. Role of the Notch signaling in cholangiocarcinoma. Expert Opin. Ther. Targets 21, 471–483 (2017).
Loilome, W. et al. Activated macrophages promote Wnt/β-catenin signaling in cholangiocarcinoma cells. Tumour Biol. 35, 5357–5367 (2014).
Boulter, L. et al. WNT signaling drives cholangiocarcinoma growth and can be pharmacologically inhibited. J. Clin. Invest. 125, 1269–1285 (2015).
Perugorria, M. J. et al. Wnt-β-catenin signalling in liver development, health and disease. Nat. Rev. Gastroenterol. Hepatol. 16, 121–136 (2019).
Pan, D. Hippo signaling in organ size control. Genes Dev. 21, 886–897 (2007).
Dupont, S. et al. Role of YAP/TAZ in mechanotransduction. Nature 474, 179–183 (2011).
Li, H. et al. Deregulation of Hippo kinase signalling in human hepatic malignancies. Liver Int. 32, 38–47 (2012).
Tao, J. et al. Activation of β-catenin and Yap1 in human hepatoblastoma and induction of hepatocarcinogenesis in mice. Gastroenterology 147, 690–701 (2014).
Pei, T. et al. YAP is a critical oncogene in human cholangiocarcinoma. Oncotarget 6, 17206–17220 (2015).
Smoot, R. L. et al. Platelet-derived growth factor regulates YAP transcriptional activity via Src family kinase dependent tyrosine phosphorylation. J. Cell Biochem. 119, 824–836 (2018).
Rizvi, S. et al. A hippo and fibroblast growth factor receptor autocrine pathway in cholangiocarcinoma. J. Biol. Chem. 291, 8031–8047 (2016).
Sugihara, T., Isomoto, H., Gores, G. & Smoot, R. YAP and the Hippo pathway in cholangiocarcinoma. J. Gastroenterol. 54, 485–491 (2019).
Chang, L. et al. The SWI/SNF complex is a mechanoregulated inhibitor of YAP and TAZ. Nature 563, 265–269 (2018).
Thiery, J. P., Acloque, H., Huang, R. Y. & Nieto, M. A. Epithelial-mesenchymal transitions in development and disease. Cell 139, 871–890 (2009).
Sulpice, L. et al. Molecular profiling of stroma identifies osteopontin as an independent predictor of poor prognosis in intrahepatic cholangiocarcinoma. Hepatology 58, 1992–2000 (2013).
Claperon, A. et al. Hepatic myofibroblasts promote the progression of human cholangiocarcinoma through activation of epidermal growth factor receptor. Hepatology 58, 2001–2011 (2013).
Claperon, A. et al. EGF/EGFR axis contributes to the progression of cholangiocarcinoma through the induction of an epithelial-mesenchymal transition. J. Hepatol. 61, 325–332 (2014).
Dongre, A. & Weinberg, R. A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 20, 69–84 (2019).
Pastushenko, I. & Blanpain, C. EMT transition states during tumor progression and metastasis. Trends Cell Biol. 29, 212–226 (2019).
Puisieux, A., Brabletz, T. & Caramel, J. Oncogenic roles of EMT-inducing transcription factors. Nat. Cell Biol. 16, 488–494 (2014).
Vaquero, J. et al. Epithelial-mesenchymal transition in cholangiocarcinoma: from clinical evidence to regulatory networks. J. Hepatol. 66, 424–441 (2017).
Goossens, S., Vandamme, N., Van Vlierberghe, P. & Berx, G. EMT transcription factors in cancer development re-evaluated: beyond EMT and MET. Biochim. Biophys. Acta Rev. Cancer 1868, 584–591 (2017).
Baulida, J. Epithelial-to-mesenchymal transition transcription factors in cancer-associated fibroblasts. Mol. Oncol. 11, 847–859 (2017).
Findlay, V. J., Wang, C., Watson, D. K. & Camp, E. R. Epithelial-to-mesenchymal transition and the cancer stem cell phenotype: insights from cancer biology with therapeutic implications for colorectal cancer. Cancer Gene Ther. 21, 181–187 (2014).
Cadamuro, M. et al. Autocrine and paracrine mechanisms promoting chemoresistance in cholangiocarcinoma. Int. J. Mol. Sci. 18, 149 (2017).
Correnti, M. & Raggi, C. Stem-like plasticity and heterogeneity of circulating tumor cells: current status and prospect challenges in liver cancer. Oncotarget 8, 7094–7115 (2017).
Raggi, C. et al. Dysregulation of iron metabolism in cholangiocarcinoma stem-like cells. Sci. Rep. 7, 17667 (2017).
Raggi, C. et al. Cholangiocarcinoma stem-like subset shapes tumor-initiating niche by educating associated macrophages. J. Hepatol. 66, 102–115 (2017).
Raggi, C., Invernizzi, P. & Andersen, J. B. Impact of microenvironment and stem-like plasticity in cholangiocarcinoma: molecular networks and biological concepts. J. Hepatol. 62, 198–207 (2015).
Wellner, U. et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat. Cell Biol. 11, 1487–1495 (2009).
Oishi, N. et al. Transcriptomic profiling reveals hepatic stem-like gene signatures and interplay of miR-200c and epithelial-mesenchymal transition in intrahepatic cholangiocarcinoma. Hepatology 56, 1792–1803 (2012).
Shuang, Z. Y. et al. Transforming growth factor-β1-induced epithelial-mesenchymal transition generates ALDH-positive cells with stem cell properties in cholangiocarcinoma. Cancer Lett. 354, 320–328 (2014).
Noman, M. Z. et al. The immune checkpoint ligand PD-L1 is upregulated in EMT-activated human breast cancer cells by a mechanism involving ZEB-1 and miR-200. Oncoimmunology 6, e1263412 (2017).
Sirica, A. E. & Gores, G. J. Desmoplastic stroma and cholangiocarcinoma: clinical implications and therapeutic targeting. Hepatology 59, 2397–2402 (2014).
Lorenzini, S. et al. Characterisation of a stereotypical cellular and extracellular adult liver progenitor cell niche in rodents and diseased human liver. Gut 59, 645–654 (2010).
Tamma, R. et al. Inflammatory cells infiltrate and angiogenesis in locally advanced and metastatic cholangiocarcinoma. Eur. J. Clin. Invest. 49, e13087 (2019).
Govaere, O. et al. Laminin-332 sustains chemoresistance and quiescence as part of the human hepatic cancer stem cell niche. J. Hepatol. 64, 609–617 (2016).
Szendroi, M. & Lapis, K. Distribution of fibronectin and laminin in human liver tumors. J. Cancer Res. Clin. Oncol. 109, 60–64 (1985).
Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 16, 582–598 (2016).
Chuaysri, C. et al. Alpha-smooth muscle actin-positive fibroblasts promote biliary cell proliferation and correlate with poor survival in cholangiocarcinoma. Oncol. Rep. 21, 957–969 (2009).
LeBleu, V. S. & Kalluri, R. A peek into cancer-associated fibroblasts: origins, functions and translational impact. Dis. Model. Mech. 11, dmm029447 (2018).
Itou, R. A. et al. Immunohistochemical characterization of cancer-associated fibroblasts at the primary sites and in the metastatic lymph nodes of human intrahepatic cholangiocarcinoma. Hum. Pathol. 83, 77–89 (2019).
Cadamuro, M. et al. Platelet-derived growth factor-D and Rho GTPases regulate recruitment of cancer-associated fibroblasts in cholangiocarcinoma. Hepatology 58, 1042–1053 (2013).
Tsuchida, T. & Friedman, S. L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 14, 397–411 (2017).
Manzanares, M. A. et al. Transforming growth factors α and β are essential for modeling cholangiocarcinoma desmoplasia and progression in a three-dimensional organotypic culture model. Am. J. Pathol. 187, 1068–1092 (2017).
Ling, H. et al. Transforming growth factor beta neutralization ameliorates pre-existing hepatic fibrosis and reduces cholangiocarcinoma in thioacetamide-treated rats. PLoS One 8, e54499 (2013).
El Khatib, M. et al. Inhibition of hedgehog signaling attenuates carcinogenesis in vitro and increases necrosis of cholangiocellular carcinoma. Hepatology 57, 1035–1045 (2013).
Ziani, L., Chouaib, S. & Thiery, J. Alteration of the antitumor immune response by cancer-associated fibroblasts. Front. Immunol. 9, 414 (2018).
Cadamuro, M. et al. Platelet-derived growth factor-D enables liver myofibroblasts to promote tumor lymphangiogenesis in cholangiocarcinoma. J. Hepatol. 70, 700–709 (2019).
Fabris, L. et al. The tumour microenvironment and immune milieu of cholangiocarcinoma. Liver Int. 39 (Suppl. 1), 63–78 (2019).
Utispan, K. et al. Periostin activates integrin α5β1 through a PI3K/AKTdependent pathway in invasion of cholangiocarcinoma. Int. J. Oncol. 41, 1110–1118 (2012).
Zanconato, F., Cordenonsi, M. & Piccolo, S. YAP/TAZ at the roots of cancer. Cancer Cell 29, 783–803 (2016).
Mertens, J. C. et al. Therapeutic effects of deleting cancer-associated fibroblasts in cholangiocarcinoma. Cancer Res. 73, 897–907 (2013).
Woo, S. R., Corrales, L. & Gajewski, T. F. Innate immune recognition of cancer. Annu. Rev. Immunol. 33, 445–474 (2015).
Binnewies, M. et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 24, 541–550 (2018).
Atanasov, G. et al. Prognostic significance of macrophage invasion in hilar cholangiocarcinoma. BMC Cancer 15, 790 (2015).
Hasita, H. et al. Significance of alternatively activated macrophages in patients with intrahepatic cholangiocarcinoma. Cancer Sci. 101, 1913–1919 (2010).
Yang, X. et al. FAP promotes immunosuppression by cancer-associated fibroblasts in the tumor microenvironment via STAT3-CCL2 signaling. Cancer Res. 76, 4124–4135 (2016).
Takagi, S. et al. Dendritic cells, T-cell infiltration, and Grp94 expression in cholangiocellular carcinoma. Hum. Pathol. 35, 881–886 (2004).
Bjorkstrom, N. K., Ljunggren, H. G. & Michaelsson, J. Emerging insights into natural killer cells in human peripheral tissues. Nat. Rev. Immunol. 16, 310–320 (2016).
Morisaki, T. et al. Combining cetuximab with killer lymphocytes synergistically inhibits human cholangiocarcinoma cells in vitro. Anticancer Res. 32, 2249–2256 (2012).
Jung, I. H. et al. In vivo study of natural killer (NK) cell cytotoxicity against cholangiocarcinoma in a nude mouse model. Vivo 32, 771–781 (2018).
Goeppert, B. et al. Prognostic impact of tumour-infiltrating immune cells on biliary tract cancer. Br. J. Cancer 109, 2665–2674 (2013).
Kasper, H. U., Drebber, U., Stippel, D. L., Dienes, H. P. & Gillessen, A. Liver tumor infiltrating lymphocytes: comparison of hepatocellular and cholangiolar carcinoma. World J. Gastroenterol. 15, 5053–5057 (2009).
Lim, Y. J. et al. High ratio of programmed cell death protein 1 (PD-1)(+)/CD8(+) tumor-infiltrating lymphocytes identifies a poor prognostic subset of extrahepatic bile duct cancer undergoing surgery plus adjuvant chemoradiotherapy. Radiother. Oncol. 117, 165–170 (2015).
Miura, T. et al. Prognostic impact of CD163+ macrophages in tumor stroma and CD8+ T-cells in cancer cell nests in invasive extrahepatic bile duct cancer. Anticancer Res. 37, 183–190 (2017).
Oshikiri, T. et al. Prognostic value of intratumoral CD8+ T lymphocyte in extrahepatic bile duct carcinoma as essential immune response. J. Surg. Oncol. 84, 224–228 (2003).
Fontugne, J. et al. PD-L1 expression in perihilar and intrahepatic cholangiocarcinoma. Oncotarget 8, 24644–24651 (2017).
Gani, F. et al. Program death 1 immune checkpoint and tumor microenvironment: implications for patients with intrahepatic cholangiocarcinoma. Ann. Surg. Oncol. 23, 2610–2617 (2016).
Ueno, M. et al. Pembrolizumab for advanced biliary adenocarcinoma: results from the multicohort, phase 2 KEYNOTE-158 study. Ann. Oncol. 29 (Suppl. 8), viii205–viii270 (2018).
Silva, V. W. et al. Biliary carcinomas: pathology and the role of DNA mismatch repair deficiency. Chin. Clin. Oncol. 5, 62 (2016).
Le, D. T. et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 357, 409–413 (2017).
Zabron, A., Edwards, R. J. & Khan, S. A. The challenge of cholangiocarcinoma: dissecting the molecular mechanisms of an insidious cancer. Dis. Model. Mech. 6, 281–292 (2013).
Tepsiri, N. et al. Drug sensitivity and drug resistance profiles of human intrahepatic cholangiocarcinoma cell lines. World J. Gastroenterol. 11, 2748–2753 (2005).
Domcke, S., Sinha, R., Levine, D. A., Sander, C. & Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 4, 2126 (2013).
Massani, M. et al. Isolation and characterization of biliary epithelial and stromal cells from resected human cholangiocarcinoma: a novel in vitro model to study tumor-stroma interactions. Oncol. Rep. 30, 1143–1148 (2013).
Miserocchi, G. et al. Management and potentialities of primary cancer cultures in preclinical and translational studies. J. Transl. Med. 15, 229 (2017).
Tabibian, J. H. et al. Characterization of cultured cholangiocytes isolated from livers of patients with primary sclerosing cholangitis. Lab. Invest. 94, 1126–1133 (2014).
Arbelaiz, A. et al. Serum extracellular vesicles contain protein biomarkers for primary sclerosing cholangitis and cholangiocarcinoma. Hepatology 66, 1125–1143 (2017).
Friedrich, J., Seidel, C., Ebner, R. & Kunz-Schughart, L. A. Spheroid-based drug screen: considerations and practical approach. Nat. Protoc. 4, 309–324 (2009).
Nath, S. & Devi, G. R. Three-dimensional culture systems in cancer research: focus on tumor spheroid model. Pharmacol. Ther. 163, 94–108 (2016).
Broutier, L. et al. Human primary liver cancer-derived organoid cultures for disease modeling and drug screening. Nat. Med. 23, 1424–1435 (2017).
Nuciforo, S. et al. Organoid models of human liver cancers derived from tumor needle biopsies. Cell Rep. 24, 1363–1376 (2018).
Lau, H. C. H. et al. Organoid models of gastrointestinal cancers in basic and translational research. Nat. Rev. Gastroenterol. Hepatol. 17, 203–222 (2020).
Saito, Y. et al. Induction of differentiation of intrahepatic cholangiocarcinoma cells to functional hepatocytes using an organoid culture system. Sci. Rep. 8, 2821 (2018).
Saborowski, A. et al. Murine liver organoids as a genetically flexible system to study liver cancer in vivo and in vitro. Hepatol. Commun. 3, 423–436 (2019).
Chen, X. & Calvisi, D. F. Hydrodynamic transfection for generation of novel mouse models for liver cancer research. Am. J. Pathol. 184, 912–923 (2014).
Loeuillard, E., Fischbach, S. R., Gores, G. J. & Rizvi, S. Animal models of cholangiocarcinoma. Biochim. Biophys. Acta Mol. Basis Dis. 1865, 982–992 (2019).
Mariotti, V. et al. Animal models of cholestasis: an update on inflammatory cholangiopathies. Biochim. Biophys. Acta Mol. Basis Dis. 1865, 954–964 (2019).
Macias, R. I. R. et al. The search for novel diagnostic and prognostic biomarkers in cholangiocarcinoma. Biochim. Biophys. Acta Mol. Basis Dis. 1864, 1468–1477 (2018).
Olaizola, P. et al. MicroRNAs and extracellular vesicles in cholangiopathies. Biochim. Biophys. Acta Mol. Basis Dis. 1864, 1293–1307 (2018).
Esparza-Baquer, A., Labiano, I., Bujanda, L., Perugorria, M. J. & Banales, J. M. MicroRNAs in cholangiopathies: potential diagnostic and therapeutic tools. Clin. Res. Hepatol. Gastroenterol. 40, 15–27 (2016).
Andersen, R. F. & Jakobsen, A. Screening for circulating RAS/RAF mutations by multiplex digital PCR. Clin. Chim. Acta 458, 138–143 (2016).
Goyal, L. et al. Polyclonal secondary FGFR2 mutations drive acquired resistance to FGFR inhibition in patients with FGFR2 fusion-positive cholangiocarcinoma. Cancer Discov. 7, 252–263 (2017).
Liang, Z., Liu, X., Zhang, Q., Wang, C. & Zhao, Y. Diagnostic value of microRNAs as biomarkers for cholangiocarcinoma. Dig. Liver Dis. 48, 1227–1232 (2016).
Zhou, J., Liu, Z., Yang, S. & Li, X. Identification of microRNAs as biomarkers for cholangiocarcinoma detection: a diagnostic meta-analysis. Clin. Res. Hepatol. Gastroenterol. 41, 156–162 (2017).
Shigehara, K. et al. Real-time PCR-based analysis of the human bile microRNAome identifies miR-9 as a potential diagnostic biomarker for biliary tract cancer. PLoS One 6, e23584 (2011).
Voigtlander, T. et al. MicroRNAs in serum and bile of patients with primary sclerosing cholangitis and/or cholangiocarcinoma. PLoS One 10, e0139305 (2015).
Wang, L. J. et al. MiR-21 promotes intrahepatic cholangiocarcinoma proliferation and growth in vitro and in vivo by targeting PTPN14 and PTEN. Oncotarget 6, 5932–5946 (2015).
Correa-Gallego, C. et al. Circulating plasma levels of microRNA-21 and microRNA-221 are potential diagnostic markers for primary intrahepatic cholangiocarcinoma. PLoS One 11, e0163699 (2016).
Liu, C. H. et al. Circulating microRNA-21 as a prognostic, biological marker in cholangiocarcinoma. J. Cancer Res. Ther. 14, 220–225 (2018).
Afonso, M. B., Rodrigues, P. M., Simao, A. L. & Castro, R. E. Circulating microRNAs as potential biomarkers in non-alcoholic fatty liver disease and hepatocellular carcinoma. J. Clin. Med. 5, 30 (2016).
Wang, Y. et al. Diagnostic and prognostic value of circulating miR-21 for cancer: a systematic review and meta-analysis. Gene 533, 389–397 (2014).
Wu, X. et al. Profiling of downregulated blood-circulating miR-150-5p as a novel tumor marker for cholangiocarcinoma. Tumour Biol. 37, 15019–15029 (2016).
Silakit, R. et al. Circulating miR-192 in liver fluke-associated cholangiocarcinoma patients: a prospective prognostic indicator. J. Hepatobiliary Pancreat. Sci. 21, 864–872 (2014).
Plieskatt, J. et al. A microRNA profile associated with Opisthorchis viverrini-induced cholangiocarcinoma in tissue and plasma. BMC Cancer 15, 309 (2015).
Wang, L. J. et al. Serum miR-26a as a diagnostic and prognostic biomarker in cholangiocarcinoma. Oncotarget 6, 18631–18640 (2015).
Wang, S. et al. Upregulated circulating miR-150 is associated with the risk of intrahepatic cholangiocarcinoma. Oncol. Rep. 33, 819–825 (2015).
Bernuzzi, F. et al. Serum microRNAs as novel biomarkers for primary sclerosing cholangitis and cholangiocarcinoma. Clin. Exp. Immunol. 185, 61–71 (2016).
Cheng, Q. et al. Circulating miR-106a is a novel prognostic and lymph node metastasis indicator for cholangiocarcinoma. Sci. Rep. 5, 16103 (2015).
Huang, L. et al. Serum CYFRA 21-1 in biliary tract cancers: a reliable biomarker for gallbladder carcinoma and intrahepatic cholangiocarcinoma. Dig. Dis. Sci. 60, 1273–1283 (2015).
Uenishi, T. et al. Serum cytokeratin 19 fragment (CYFRA21-1) as a prognostic factor in intrahepatic cholangiocarcinoma. Ann. Surg. Oncol. 15, 583–589 (2008).
Leelawat, K., Sakchinabut, S., Narong, S. & Wannaprasert, J. Detection of serum MMP-7 and MMP-9 in cholangiocarcinoma patients: evaluation of diagnostic accuracy. BMC Gastroenterol. 9, 30 (2009).
Leelawat, K., Narong, S., Wannaprasert, J. & Ratanashu-ek, T. Prospective study of MMP7 serum levels in the diagnosis of cholangiocarcinoma. World J. Gastroenterol. 16, 4697–4703 (2010).
Loosen, S. H. et al. Elevated levels of circulating osteopontin are associated with a poor survival after resection of cholangiocarcinoma. J. Hepatol. 67, 749–757 (2017).
Kobayashi, S., Werneburg, N. W., Bronk, S. F., Kaufmann, S. H. & Gores, G. J. Interleukin-6 contributes to Mcl-1 up-regulation and TRAIL resistance via an Akt-signaling pathway in cholangiocarcinoma cells. Gastroenterology 128, 2054–2065 (2005).
Cheon, Y. K. et al. Diagnostic utility of interleukin-6 (IL-6) for primary bile duct cancer and changes in serum IL-6 levels following photodynamic therapy. Am. J. Gastroenterol. 102, 2164–2170 (2007).
Onsurathum, S. et al. Proteomics detection of S100A6 in tumor tissue interstitial fluid and evaluation of its potential as a biomarker of cholangiocarcinoma. Tumour Biol. 40, 1010428318767195 (2018).
Shi, R. Y. et al. High expression of Dickkopf-related protein 1 is related to lymphatic metastasis and indicates poor prognosis in intrahepatic cholangiocarcinoma patients after surgery. Cancer 119, 993–1003 (2013).
Xu, H. et al. Elevation of serum KL-6 mucin levels in patients with cholangiocarcinoma. Hepatogastroenterology 55, 2000–2004 (2008).
Shen, J. et al. Comparative proteomic profiling of human bile reveals SSP411 as a novel biomarker of cholangiocarcinoma. PLoS One 7, e47476 (2012).
Thuwajit, C. et al. Clustering of patients with intrahepatic cholangiocarcinoma based on serum periostin may be predictive of prognosis. Oncol. Lett. 14, 623–634 (2017).
Park, J. Y. et al. Bile acid analysis in biliary tract cancer. Yonsei Med. J. 47, 817–825 (2006).
Albiin, N. et al. Detection of cholangiocarcinoma with magnetic resonance spectroscopy of bile in patients with and without primary sclerosing cholangitis. Acta Radiol. 49, 855–862 (2008).
Nagana Gowda, G. A., Shanaiah, N., Cooper, A., Maluccio, M. & Raftery, D. Visualization of bile homeostasis using (1)H-NMR spectroscopy as a route for assessing liver cancer. Lipids 44, 27–35 (2009).
Sharif, A. W. et al. Metabolic profiling of bile in cholangiocarcinoma using in vitro magnetic resonance spectroscopy. HPB 12, 396–402 (2010).
Satriano, L., Lewinska, M., Rodrigues, P. M., Banales, J. M. & Andersen, J. B. Metabolic rearrangements in primary liver cancers: cause and consequences. Nat. Rev. Gastroenterol. Hepatol. 16, 748–766 (2019).
Liang, Q. et al. Serum metabolomics uncovering specific metabolite signatures of intra- and extrahepatic cholangiocarcinoma. Mol. Biosyst. 12, 334–340 (2016).
Banales, J. M. et al. Serum metabolites as diagnostic biomarkers for cholangiocarcinoma, hepatocellular carcinoma, and primary sclerosing cholangitis. Hepatology 70, 547–562 (2019).
Severino, V. et al. Extracellular vesicles in bile as markers of malignant biliary stenoses. Gastroenterology 153, e498 (2017).
Al Ustwani, O., Iancu, D., Yacoub, R. & Iyer, R. Detection of circulating tumor cells in cancers of biliary origin. J. Gastrointest. Oncol. 3, 97–104 (2012).
Yang, J. D. et al. Circulating tumor cells are associated with poor overall survival in patients with cholangiocarcinoma. Hepatology 63, 148–158 (2016).
Valle, J. W. et al. Cediranib or placebo in combination with cisplatin and gemcitabine chemotherapy for patients with advanced biliary tract cancer (ABC-03): a randomised phase 2 trial. Lancet Oncol. 16, 967–978 (2015).
Ruys, A. T. et al. Prognostic biomarkers in patients with resected cholangiocarcinoma: a systematic review and meta-analysis. Ann. Surg. Oncol. 21, 487–500 (2014).
Chusorn, P. et al. Overexpression of microRNA-21 regulating PDCD4 during tumorigenesis of liver fluke-associated cholangiocarcinoma contributes to tumor growth and metastasis. Tumour Biol. 34, 1579–1588 (2013).
Valle, J. W. et al. Biliary cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 27, v28–v37 (2016).
van Vugt, J. L. A. et al. The prognostic value of portal vein and hepatic artery involvement in patients with perihilar cholangiocarcinoma. HPB 20, 83–92 (2018).
Doussot, A. et al. Outcomes after resection of intrahepatic cholangiocarcinoma: external validation and comparison of prognostic models. J. Am. Coll. Surg. 221, 452–461 (2015).
Weber, S. M. et al. Intrahepatic cholangiocarcinoma: expert consensus statement. HPB 17, 669–680 (2015).
Coelen, R. J. S. et al. Endoscopic versus percutaneous biliary drainage in patients with resectable perihilar cholangiocarcinoma: a multicentre, randomised controlled trial. Lancet Gastroenterol. Hepatol. 3, 681–690 (2018).
Kawashima, H. et al. Preoperative endoscopic nasobiliary drainage in 164 consecutive patients with suspected perihilar cholangiocarcinoma: a retrospective study of efficacy and risk factors related to complications. Ann. Surg. 257, 121–127 (2013).
Bird, N. et al. Role of staging laparoscopy in the stratification of patients with perihilar cholangiocarcinoma. Br. J. Surg. 104, 418–425 (2017).
Farges, O. et al. Multicentre European study of preoperative biliary drainage for hilar cholangiocarcinoma. Br. J. Surg. 100, 274–283 (2013).
Nuzzo, G. et al. Improvement in perioperative and long-term outcome after surgical treatment of hilar cholangiocarcinoma: results of an Italian multicenter analysis of 440 patients. Arch. Surg. 147, 26–34 (2012).
Nagino, M. et al. Evolution of surgical treatment for perihilar cholangiocarcinoma: a single-center 34-year review of 574 consecutive resections. Ann. Surg. 258, 129–140 (2013).
Ebata, T. et al. Surgical resection for Bismuth type IV perihilar cholangiocarcinoma. Br. J. Surg. 105, 829–838 (2018).
Abbas, S. & Sandroussi, C. Systematic review and meta-analysis of the role of vascular resection in the treatment of hilar cholangiocarcinoma. HPB 15, 492–503 (2013).
de Jong, M. C. et al. The impact of portal vein resection on outcomes for hilar cholangiocarcinoma: a multi-institutional analysis of 305 cases. Cancer 118, 4737–4747 (2012).
Groot Koerkamp, B. et al. American joint committee on cancer staging for resected perihilar cholangiocarcinoma: a comparison of the 6th and 7th editions. HPB 16, 1074–1082 (2014).
Kizy, S. et al. Surgical resection of lymph node positive intrahepatic cholangiocarcinoma may not improve survival. HPB 21, 235–241 (2019).
Buettner, S. et al. Survival after resection of multiple tumor foci of intrahepatic cholangiocarcinoma. J. Gastrointest. Surg. 23, 2239–2246 (2019).
Reames, B. N. et al. Impact of major vascular resection on outcomes and survival in patients with intrahepatic cholangiocarcinoma: a multi-institutional analysis. J. Surg. Oncol. 116, 133–139 (2017).
Wang, Y. et al. Prognostic nomogram for intrahepatic cholangiocarcinoma after partial hepatectomy. J. Clin. Oncol. 31, 1188–1195 (2013).
Wang, S. J. et al. Nomogram for predicting the benefit of adjuvant chemoradiotherapy for resected gallbladder cancer. J. Clin. Oncol. 29, 4627–4632 (2011).
Zhang, X. F. et al. Early versus late recurrence of intrahepatic cholangiocarcinoma after resection with curative intent. Br. J. Surg. 105, 848–856 (2018).
Zhang, X. F. et al. Defining early recurrence of hilar cholangiocarcinoma after curative-intent surgery: a multi-institutional study from the US extrahepatic biliary malignancy consortium. World J. Surg. 42, 2919–2929 (2018).
Ebata, T. et al. Randomized clinical trial of adjuvant gemcitabine chemotherapy versus observation in resected bile duct cancer. Br. J. Surg. 105, 192–202 (2018).
Edeline, J. et al. Gemcitabine and oxaliplatin chemotherapy or surveillance in resected biliary tract cancer (PRODIGE 12-ACCORD 18-UNICANCER GI): a randomized phase III study. J. Clin. Oncol. 37, 658–667 (2019).
Primrose, J. N. et al. Capecitabine compared with observation in resected biliary tract cancer (BILCAP): a randomised, controlled, multicentre, phase 3 study. Lancet Oncol. 20, 663–673 (2019).
Lamarca, A. et al. Current standards and future perspectives in adjuvant treatment for biliary tract cancers. Cancer Treat. Rev. 84, 101936 (2020).
Shroff, R. T. et al. Adjuvant therapy for resected biliary tract cancer: ASCO clinical practice guideline. J. Clin. Oncol. 37, 1015–1027 (2019).
Ben-Josef, E. et al. SWOG S0809: a phase II intergroup trial of adjuvant capecitabine and gemcitabine followed by radiotherapy and concurrent capecitabine in extrahepatic cholangiocarcinoma and gallbladder carcinoma. J. Clin. Oncol. 33, 2617–2622 (2015).
Meyer, C. G., Penn, I. & James, L. Liver transplantation for cholangiocarcinoma: results in 207 patients. Transplantation 69, 1633–1637 (2000).
Robles, R. et al. Spanish experience in liver transplantation for hilar and peripheral cholangiocarcinoma. Ann. Surg. 239, 265–271 (2004).
Seehofer, D. et al. Extended bile duct resection and [corrected] liver and transplantation in patients with hilar cholangiocarcinoma: long-term results. Liver Transpl. 15, 1499–1507 (2009).
Darwish Murad, S. et al. Efficacy of neoadjuvant chemoradiation, followed by liver transplantation, for perihilar cholangiocarcinoma at 12 US centers. Gastroenterology 143, 88–98 e83 (2012).
Rea, D. J. et al. Liver transplantation with neoadjuvant chemoradiation is more effective than resection for hilar cholangiocarcinoma. Ann. Surg. 242, 451–458 (2005).
Sudan, D. et al. Radiochemotherapy and transplantation allow long-term survival for nonresectable hilar cholangiocarcinoma. Am. J. Transpl. 2, 774–779 (2002).
Darwish Murad, S. et al. Predictors of pretransplant dropout and posttransplant recurrence in patients with perihilar cholangiocarcinoma. Hepatology 56, 972–981 (2012).
Duignan, S. et al. Neoadjuvant chemoradiotherapy followed by liver transplantation for unresectable cholangiocarcinoma: a single-centre national experience. HPB 16, 91–98 (2014).
Lehrke, H. D. et al. Prognostic significance of the histologic response of perihilar cholangiocarcinoma to preoperative neoadjuvant chemoradiation in liver explants. Am. J. Surg. Pathol. 40, 510–518 (2016).
Sio, T. T. et al. Outcome of transplant-fallout patients with unresectable cholangiocarcinoma. Am. J. Clin. Oncol. 39, 271–275 (2016).
Ethun, C. G. et al. Transplantation versus resection for hilar cholangiocarcinoma: an argument for shifting treatment paradigms for resectable disease. Ann. Surg. 267, 797–805 (2018).
Sapisochin, G. et al. Intrahepatic cholangiocarcinoma or mixed hepatocellular-cholangiocarcinoma in patients undergoing liver transplantation: a Spanish matched cohort multicenter study. Ann. Surg. 259, 944–952 (2014).
Lunsford, K. E. et al. Liver transplantation for locally advanced intrahepatic cholangiocarcinoma treated with neoadjuvant therapy: a prospective case-series. Lancet Gastroenterol. Hepatol. 3, 337–348 (2018).
Valle, J. et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N. Engl. J. Med. 362, 1273–1281 (2010).
Okusaka, T. et al. Gemcitabine alone or in combination with cisplatin in patients with biliary tract cancer: a comparative multicentre study in Japan. Br. J. Cancer 103, 469–474 (2010).
Lamarca, A., Benafif, S., Ross, P., Bridgewater, J. & Valle, J. W. Cisplatin and gemcitabine in patients with advanced biliary tract cancer (ABC) and persistent jaundice despite optimal stenting: effective intervention in patients with luminal disease. Eur. J. Cancer 51, 1694–1703 (2015).
Shroff, R. T. et al. Gemcitabine, cisplatin, and nab-paclitaxel for the treatment of advanced biliary tract cancers: a phase 2 clinical trial. JAMA Oncol. 5, 824–830 (2019).
Sakai, D. et al. Randomized phase III study of gemcitabine, cisplatin plus S-1 (GCS) versus gemcitabine, cisplatin (GC) for advanced biliary tract cancer [abstract 615O]. Ann. Oncol. 29 (Suppl. 8), viii205 (2018).
McNamara, M. G. et al. A new ProTide, NUC-1031, combined with cisplatin for the first-line treatment of advanced biliary tract cancer (ABC-08) [abstract 758P]. Ann. Oncol. 29 (Suppl. 8), viii259 (2018).
Lamarca, A., Hubner, R. A., David Ryder, W. & Valle, J. W. Second-line chemotherapy in advanced biliary cancer: a systematic review. Ann. Oncol. 25, 2328–2338 (2014).
Lamarca, A. et al. A randomised phase III, multi-centre, open-label study of Active Symptom Control (ASC) alone or ASC with oxaliplatin/5-FU chemotherapy (ASC+mFOLFOX) for patients (pts) with locally advanced/metastatic biliary tract cancers (ABC) previously-treated with cisplatin/gemcitabine (CisGem) chemotherapy. [abstract ABC-06]. J. Clin. Oncol. 37 (Suppl. 15), 4003 (2019).
Lamarca, A. et al. Advanced intrahepatic cholangiocarcinoma: post hoc analysis of the ABC-01, -02, and -03 clinical trials. J. Natl Cancer Inst. 112, 200–210 (2020).
Hyder, O. et al. Intra-arterial therapy for advanced intrahepatic cholangiocarcinoma: a multi-institutional analysis. Ann. Surg. Oncol. 20, 3779–3786 (2013).
National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology (NCCN, 2015).
Al-Adra, D. P. et al. Treatment of unresectable intrahepatic cholangiocarcinoma with yttrium-90 radioembolization: a systematic review and pooled analysis. Eur. J. Surg. Oncol. 41, 120–127 (2015).
Burger, I. et al. Transcatheter arterial chemoembolization in unresectable cholangiocarcinoma: initial experience in a single institution. J. Vasc. Interv. Radiol. 16, 353–361 (2005).
Ibrahim, S. M. et al. Treatment of unresectable cholangiocarcinoma using yttrium-90 microspheres: results from a pilot study. Cancer 113, 2119–2128 (2008).
Edeline, J. T. et al. Selective internal radiation therapy (SIRT) with yttrium-90-glass-microspheres plus chemotherapy in first-line treatment of advanced cholangiocarcinoma (MISPHEC study). Ann. Oncol. 28 (Suppl. 5), v209–v268 (2017).
Hong, T. S. et al. Multi-institutional phase II study of high-dose hypofractionated proton beam therapy in patients with localized, unresectable hepatocellular carcinoma and intrahepatic cholangiocarcinoma. J. Clin. Oncol. 34, 460–468 (2016).
Cercek, A. et al. Assessment of hepatic arterial infusion of floxuridine in combination with systemic gemcitabine and oxaliplatin in patients with unresectable intrahepatic cholangiocarcinoma: a phase 2 clinical trial. JAMA Oncol 6, 60–67 (2019).
Lowery, M. A. et al. Phase I study of AG-120, an IDH1 mutant enzyme inhibitor: Results from the cholangiocarcinoma dose escalation and expansion cohorts [abstract]. J. Clin. Oncol. 35 (Suppl. 15), 4015 (2017).
Javle, M. M. et al. A phase 2 study of BGJ398 in patients (pts) with advanced or metastatic FGFR-altered cholangiocarcinoma (CCA) who failed or are intolerant to platinum-based chemotherapy [abstract]. J. Clin. Oncol. 34 (Suppl. 4), 335 (2016).
Mazzaferro, V. et al. ARQ 087, an oral pan-fibroblast growth factor receptor (FGFR) inhibitor, in patients with advanced and/or metastatic intrahepatic cholangiocarcinoma (iCCA) [abstract PD-019]. Ann. Oncol. 27 (Suppl. 2), ii109 (2016).
Liu, S. & Quarles, L. D. How fibroblast growth factor 23 works. J. Am. Soc. Nephrol. 18, 1637–1647 (2007).
Meric-Bernstam, F. et al. Efficacy of TAS-120, an irreversible fibroblast growth factor receptor (FGFR) inhibitor, in cholangiocarcinoma patients with FGFR pathway alterations who were previously treated with chemotherapy and other FGFR inhibitors [abstract O-001]. Ann. Oncol. 29 (Suppl. 5), v100 (2018).
Drilon, A. et al. Safety and antitumor activity of the multitargeted Pan-TRK, ROS1, and ALK inhibitor entrectinib: combined results from two phase I trials (ALKA-372-001 and STARTRK-1). Cancer Discov. 7, 400–409 (2017).
Drilon, A. et al. Efficacy of larotrectinib in TRK fusion-positive cancers in adults and children. N. Engl. J. Med. 378, 731–739 (2018).
Yamamoto, K. et al. MUC1 peptide vaccination in patients with advanced pancreas or biliary tract cancer. Anticancer. Res. 25, 3575–3579 (2005).
Guo, Y. et al. Phase I study of chimeric antigen receptor-modified T cells in patients with EGFR-positive advanced biliary tract cancers. Clin. Cancer Res. 24, 1277–1286 (2018).
Wei, S. C., Duffy, C. R. & Allison, J. P. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 8, 1069–1086 (2018).
Le, D. T. et al. PD-1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med. 372, 2509–2520 (2015).
Ott, P. A. et al. T-cell-inflamed gene-expression profile, programmed death ligand 1 expression, and tumor mutational burden predict efficacy in patients treated with pembrolizumab across 20 cancers: KEYNOTE-028. J. Clin. Oncol. 37, 318–327 (2019).
Marin, J. J. G. et al. Molecular bases of chemoresistance in cholangiocarcinoma. Curr. Drug Targets 18, 889–900 (2017).
Marin, J. J. G. et al. Chemoresistance and chemosensitization in cholangiocarcinoma. Biochim. Biophys. Acta Mol. Basis Dis. 1864, 1444–1453 (2018).
Fouassier, L. et al. Signalling networks in cholangiocarcinoma: molecular pathogenesis, targeted therapies and drug resistance. Liver Int. 39 (Suppl. 1), 43–62 (2019).
Borbath, I. et al. Human equilibrative nucleoside transporter 1 (hENT1) expression is a potential predictive tool for response to gemcitabine in patients with advanced cholangiocarcinoma. Eur. J. Cancer 48, 990–996 (2012).
Martinez-Becerra, P. et al. No correlation between the expression of FXR and genes involved in multidrug resistance phenotype of primary liver tumors. Mol. Pharm. 9, 1693–1704 (2012).
Herraez, E. et al. Expression of SLC22A1 variants may affect the response of hepatocellular carcinoma and cholangiocarcinoma to sorafenib. Hepatology 58, 1065–1073 (2013).
Lautem, A. et al. Downregulation of organic cation transporter 1 (SLC22A1) is associated with tumor progression and reduced patient survival in human cholangiocellular carcinoma. Int. J. Oncol. 42, 1297–1304 (2013).
Al-Abdulla, R. et al. Genetic and epigenetic bases of the relationship between reduced OCT1 expression and poor response to sorafenib in hepatocellular carcinoma and cholangiocarcinoma. J. Hepatology 66, S462–S463 (2017).
Cao, L. et al. Expression of MDR1 mRNA and encoding P-glycoprotein in archival formalin-fixed paraffin-embedded gall bladder cancer tissues. Eur. J. Cancer 34, 1612–1617 (1998).
Srimunta, U. et al. High expression of ABCC1 indicates poor prognosis in intrahepatic cholangiocarcinoma. Asian Pac. J. Cancer Prev. 13 (Suppl.), 125–130 (2012).
Hahnvajanawong, C. et al. Orotate phosphoribosyl transferase mRNA expression and the response of cholangiocarcinoma to 5-fluorouracil. World J. Gastroenterol. 18, 3955–3961 (2012).
Nakajima, T. et al. Reversal of multiple drug resistance in cholangiocarcinoma by the glutathione S-transferase-pi-specific inhibitor O1-hexadecyl-gamma-glutamyl-S-benzylcysteinyl-D-phenylglycine ethylester. J. Pharmacol. Exp. Ther. 306, 861–869 (2003).
Habara, K., Ajiki, T., Kamigaki, T., Nakamura, T. & Kuroda, Y. High expression of thymidylate synthase leads to resistance to 5-fluorouracil in biliary tract carcinoma in vitro. Jpn. J. Cancer Res. 92, 1127–1132 (2001).
Marzioni, M. et al. An oestrogen receptor β-selective agonist exerts anti-neoplastic effects in experimental intrahepatic cholangiocarcinoma. Dig. Liver Dis. 44, 134–142 (2012).
Jimeno, A. et al. Epidermal growth factor receptor dynamics influences response to epidermal growth factor receptor targeted agents. Cancer Res. 65, 3003–3010 (2005).
Hwang, I. G. et al. Different relation between ERCC1 overexpression and treatment outcomes of two platinum agents in advanced biliary tract adenocarcinoma patients. Cancer Chemother. Pharmacol. 68, 935–944 (2011).
Limpaiboon, T. et al. Promoter hypermethylation is a major event of hMLH1 gene inactivation in liver fluke related cholangiocarcinoma. Cancer Lett. 217, 213–219 (2005).
Sato, J. et al. Gene expression analysis for predicting gemcitabine resistance in human cholangiocarcinoma. J. Hepatobiliary Pancreat. Sci. 18, 700–711 (2011).
Wilson, T. R., Johnston, P. G. & Longley, D. B. Anti-apoptotic mechanisms of drug resistance in cancer. Curr. Cancer Drug Targets 9, 307–319 (2009).
Fernandez, T. F., Samal, A. B., Bedwell, G. J., Chen, Y. & Saad, J. S. Structural and biophysical characterization of the interactions between the death domain of Fas receptor and calmodulin. J. Biol. Chem. 288, 21898–21908 (2013).
Yokoi, K. et al. Survival pathway of cholangiocarcinoma via AKT/mTOR signaling to escape RAF/MEK/ERK pathway inhibition by sorafenib. Oncol. Rep. 39, 843–850 (2018).
Fujita, N. et al. Mass-forming intrahepatic cholangiocarcinoma: enhancement patterns in the arterial phase of dynamic hepatic CT – correlation with clinicopathological findings. Eur. Radiol. 27, 498–506 (2017).
Leyva-Illades, D., McMillin, M., Quinn, M. & Demorrow, S. Cholangiocarcinoma pathogenesis: role of the tumor microenvironment. Transl. Gastrointest. Cancer 1, 71–80 (2012).
Quintavalle, C. et al. High mobility group A1 enhances tumorigenicity of human cholangiocarcinoma and confers resistance to therapy. Mol. Carcinog. 56, 2146–2157 (2017).
Park, J. J. H., Hsu, G., Siden, E. G., Thorlund, K. & Mills, E. J. An overview of precision oncology basket and umbrella trials for clinicians. CA Cancer J. Clin. 70, 125–137 (2020).
Padma, V. V. An overview of targeted cancer therapy. Biomedicine 5, 19 (2015).
Maximiano, S., Magalhaes, P., Guerreiro, M. P. & Morgado, M. Trastuzumab in the treatment of breast cancer. BioDrugs 30, 75–86 (2016).
Geng, F., Wang, Z., Yin, H., Yu, J. & Cao, B. Molecular targeted drugs and treatment of colorectal cancer: recent progress and future perspectives. Cancer Biother. Radiopharm. 32, 149–160 (2017).
Merdrignac, A. et al. A novel transforming growth factor beta-induced long noncoding RNA promotes an inflammatory microenvironment in human intrahepatic cholangiocarcinoma. Hepatol. Commun. 2, 254–269 (2018).
Moirangthem, A., Wang, X., Yan, I. K. & Patel, T. Network analyses-based identification of circular ribonucleic acid-related pathways in intrahepatic cholangiocarcinoma. Tumour Biol. 40, 1010428318795761 (2018).
Chaisaingmongkol, J. et al. Common molecular subtypes among Asian hepatocellular carcinoma and cholangiocarcinoma. Cancer Cell 32, 57–70.e3 (2017).
Petrick, J. L. et al. Risk factors for intrahepatic and extrahepatic cholangiocarcinoma in the United States: a population-based study in SEER-Medicare. PLoS One 12, e0186643 (2017).
Shin, H. R. et al. Epidemiology of cholangiocarcinoma: an update focusing on risk factors. Cancer Sci. 101, 579–585 (2010).
Jing, W. et al. Diabetes mellitus and increased risk of cholangiocarcinoma: a meta-analysis. Eur. J. Cancer Prev. 21, 24–31 (2012).
Wongjarupong, N. et al. Non-alcoholic fatty liver disease as a risk factor for cholangiocarcinoma: a systematic review and meta-analysis. BMC Gastroenterol. 17, 149 (2017).
Ishikawa, Y., Wada, I. & Fukumoto, M. Alpha-particle carcinogenesis in Thorotrast patients: epidemiology, dosimetry, pathology, and molecular analysis. J. Env. Pathol. Toxicol. Oncol. 20, 311–315 (2001).
Kato, I. & Kido, C. Increased risk of death in thorotrast-exposed patients during the late follow-up period. Jpn. J. Cancer Res. 78, 1187–1192 (1987).
Kumagai, S. et al. Relationship between cumulative exposure to 1,2-dichloropropane and incidence risk of cholangiocarcinoma among offset printing workers. Occup. Env. Med. 73, 545–552 (2016).
Brandi, G. et al. Asbestos: a hidden player behind the cholangiocarcinoma increase? Findings from a case-control analysis. Cancer Causes Control 24, 911–918 (2013).
Farioli, A. et al. Occupational exposure to asbestos and risk of cholangiocarcinoma: a population-based case-control study in four Nordic countries. Occup. Env. Med. 75, 191–198 (2018).
Ertel, A., Verghese, A., Byers, S. W., Ochs, M. & Tozeren, A. Pathway-specific differences between tumor cell lines and normal and tumor tissue cells. Mol. Cancer 5, 55 (2006).
Rizvi, S. et al. YAP-associated chromosomal instability and cholangiocarcinoma in mice. Oncotarget 9, 5892–5905 (2018).
Campbell, D. J., Dumur, C. I., Lamour, N. F., Dewitt, J. L. & Sirica, A. E. Novel organotypic culture model of cholangiocarcinoma progression. Hepatol. Res. 42, 1119–1130 (2012).
Cadamuro, M. et al. Animal models of cholangiocarcinoma: what they teach us about the human disease. Clin. Res. Hepatol. Gastroenterol. 42, 403–415 (2018).
Sirica, A. E. et al. A novel “patient-like” model of cholangiocarcinoma progression based on bile duct inoculation of tumorigenic rat cholangiocyte cell lines. Hepatology 47, 1178–1190 (2008).
Acknowledgements
J.M.B. received EASL Registry Awards 2016 and 2019 (European CCA Registry, ENS-CCA). J.M.B. and M.J.P. were supported by: the Spanish Ministry of Economy and Competitiveness (J.M.B.: FIS PI12/00380, FIS PI15/01132, FIS PI18/01075 and Miguel Servet Programme CON14/00129; M.J.P.: FIS PI14/00399, FIS PI17/00022 and Ramon y Cajal Programme RYC-2015-17755, co-financed by “Fondo Europeo de Desarrollo Regional” (FEDER)); ISCIII CIBERehd; “Diputación Foral de Gipuzkoa” (J.M.B: DFG15/010, DFG16/004), and BIOEF (Basque Foundation for Innovation and Health Research: EiTB Maratoia BIO15/CA/016/BD); the Department of Health of the Basque Country (M.J.P.: 2015111100; J.M.B.: 2017111010), and “Fundación Científica de la Asociación Española Contra el Cancer” (AECC Scientific Foundation) (J.M.B.). J.M.B. and J.W.V. were supported by the European Commission Horizon 2020 programme (ESCALON project 825510). The laboratory of J.B.A. is supported by competitive grants from the Danish Medical Research Council, the Danish Cancer Society, and the Novo Nordisk and A.P. Møller Foundations. J.J.G.M. and R.I.R.M. were supported by the Carlos III Institute of Health, Spain (PI16/00598 and PI18/00428) and were co-financed by the European Regional Development Fund. J.M.B. and J.J.G.M. were supported by the Ministry of Science and Innovation, Spain (SAF2016-75197-R), and the “Asociación Española Contra el Cancer”, Spain (AECC-2017). R.I.R.M. was supported by the “Centro Internacional sobre el Envejecimiento”, Spain (OLD-HEPAMARKER, 0348-CIE-6-E). A.L. received funding from the Christie Charity. M.M. was supported by the Università Politecnica delle Marche, Ancona, Italy (040020_R.SCIENT.A_2018_MARZIONI_M_STRATEGICO_2017). M.S. was supported by the Yale Liver Center Clinical and Translational Core and the Cellular and Molecular Core (DK034989 Silvio O. Conte Digestive Diseases Research Center). C.C. is supported by grants from INSERM, Université de Rennes, INCa, and ITMO Cancer AVIESAN dans le cadre du Plan Cancer (Non-coding RNA in Cancerology: Fundamental to Translational), Ligue Contre le Cancer and Région Bretagne. J.Bruix was supported by grants from Instituto de Salud Carlos III (PI18/00763), AECC (PI044031) and WCR (AICR) 16-0026. A.F. was supported by grants from ISCIII (PI13/01229 and PI18/00542). CIBERehd is funded by the Instituto de Salud Carlos III. V.C., D.M., J. Bridgewater and P.I. are members of the European Reference Network - Hepatological Diseases (ERN RARE-LIVER). J.M.B. is a collaborator of the ERN RARE-LIVER.
Author information
Authors and Affiliations
Contributions
The authors contributed equally to all aspects of the article.
Corresponding author
Ethics declarations
Competing interests
A.L. received travel and educational support from Ipsen, Pfizer, Bayer, AAA, Sirtex, Novartis, Mylan and Delcath; speaker honoraria from Merck, Pfizer, Ipsen and Incyte; and advisory honoraria from EISAI, Nutricia and QED; she is also a member of the Knowledge Network and NETConnect Initiatives funded by Ipsen. J.W.V. declares consulting or advisory roles for Agios, AstraZeneca, Delcath Systems, Keocyt, Genoscience Pharma, Incyte, Ipsen, Merck, Mundipharma EDO, Novartis, PCI Biotech, Pfizer, Pieris Pharmaceuticals, QED and Wren Laboratories; Speakers’ Bureau for Imaging Equipment Limited, Ipsen, Novartis and Nucana; and travel grants from Celgene and Nucana. J. Bridgewater declares consulting or advisory roles for Merck Serono, SERVIER, Roche, Bayer, AstraZeneca, Incyte and Basilea; travel support from MSD Oncology, Merck Serono, Servier and BMS. J.M.B. is scientific advisor to OWL Metabolomics. M.M. is speaker for Intercept Pharma and advisor to IQVIA srl and Simon & Cutcher Ltd. M.S. is a member of the Advisory Board for Bayer, Esiai/Merk and Engitix. A.F. received lecture fees from Bayer, Gilead and MSD; and consultancy fees from Bayer, AstraZeneca and Guerbert. J. Bruix received consultancy lecture fees from Bayer, Gilead and MSD; consultancy fees from Bayer, AstraZeneca and Guerbert; research grants from Bayer, BTG; educational grants from Bayer, BTG; conferences fees from Bayer, BTG and Ipsen; and fees for talks from Bayer-Shering Pharma, BTG- Biocompatibles, Eisai, Terumo, Sirtex and Ipsen. P.I. receives funding from AMAF Monza ONLUS and AIRCS. The remaining authors declare no competing interests.
Additional information
Peer review information
Nature Reviews Gastroenterology & Hepatology thank the anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
AMMF – The Cholangiocarcinoma Charity: https://ammf.org.uk
ClinicalTrials.gov: https://www.clinicaltrials.gov/
ESCALON project: https://escalon.eu/
European Network for the Study of Cholangiocarcinoma (ENS-CCA): http://www.enscca.org
Global Cholangiocarcinoma Alliance: https://globalccaalliance.com/en
The Cholangiocarcinoma Foundation: https://cholangiocarcinoma.org
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Banales, J.M., Marin, J.J.G., Lamarca, A. et al. Cholangiocarcinoma 2020: the next horizon in mechanisms and management. Nat Rev Gastroenterol Hepatol 17, 557–588 (2020). https://doi.org/10.1038/s41575-020-0310-z
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41575-020-0310-z
This article is cited by
-
Cholangiocyte organoids to study drug-induced injury
Stem Cell Research & Therapy (2024)
-
Survival analysis of patients with extrahepatic cholangiocarcinoma: a nomogram for clinical and MRI features
BMC Medical Imaging (2024)
-
RPL35A promotes the progression of cholangiocarcinoma by mediating HSPA8 ubiquitination
Biology Direct (2024)
-
Overcoming tight perihilar malignant biliary obstructions during percutaneous biliary intervention in Nigerian patients: case reports
Egyptian Journal of Radiology and Nuclear Medicine (2024)
-
The role of kinesin family members in hepatobiliary carcinomas: from bench to bedside
Biomarker Research (2024)
