
Abstract
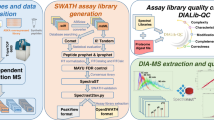
Targeted proteomics by selected/multiple reaction monitoring (S/MRM) or, on a larger scale, by SWATH (sequential window acquisition of all theoretical spectra) MS (mass spectrometry) typically relies on spectral reference libraries for peptide identification. Quality and coverage of these libraries are therefore of crucial importance for the performance of the methods. Here we present a detailed protocol that has been successfully used to build high-quality, extensive reference libraries supporting targeted proteomics by SWATH MS. We describe each step of the process, including data acquisition by discovery proteomics, assertion of peptide-spectrum matches (PSMs), generation of consensus spectra and compilation of MS coordinates that uniquely define each targeted peptide. Crucial steps such as false discovery rate (FDR) control, retention time normalization and handling of post-translationally modified peptides are detailed. Finally, we show how to use the library to extract SWATH data with the open-source software Skyline. The protocol takes 2–3 d to complete, depending on the extent of the library and the computational resources available.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout


Similar content being viewed by others


References
Domon, B. & Aebersold, R. Options and considerations when selecting a quantitative proteomics strategy. Nat. Biotechnol. 28, 710–721 (2010).
Picotti, P. & Aebersold, R. Selected reaction monitoring-based proteomics: workflows, potential, pitfalls and future directions. Nat. Methods 9, 555–566 (2012).
Gillet, L.C. et al. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol. Cell. Proteomics 11, O111.016717 (2012).
Venable, J.D., Dong, M.-Q., Wohlschlegel, J., Dillin, A. & Yates, J.R. III. Automated approach for quantitative analysis of complex peptide mixtures from tandem mass spectra. Nat. Methods 1, 39–45 (2004).
Chapman, J.D., Goodlett, D.R. & Masselon, C.D. Multiplexed and data-independent tandem mass spectrometry for global proteome profiling. Mass Spectrom. Rev. 33, 452–470 (2014).
Röst, H.L. et al. OpenSWATH enables automated, targeted analysis of data-independent acquisition MS data. Nat. Biotechnol. 32, 219–223 (2014).
Bernhardt, O.M. et al. Spectronaut: a fast and efficient algorithm for MRM-like processing of data independent acquisition (SWATH-MS) data. F1000Posters Presented at the 60th American Society for Mass Spectrometry Conference, 20–24 May 2012 5, 1092 (2014).
MacLean, B. et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968 (2010).
Reiter, L. et al. mProphet: automated data processing and statistical validation for large-scale SRM experiments. Nat. Methods 8, 430–435 (2011).
Zi, J. et al. Expansion of the ion library for mining SWATH-MS data through fractionation proteomics. Anal. Chem. 86, 7242–7246 (2014).
Lam, H. et al. Building consensus spectral libraries for peptide identification in proteomics. Nat. Methods 5, 873–875 (2008).
Hughes, M.A., Silva, J.C., Geromanos, S.J. & Townsend, C.A. Quantitative proteomic analysis of drug-induced changes in mycobacteria. J. Proteome Res. 5, 54–63 (2006).
Frewen, B.E., Merrihew, G.E., Wu, C.C., Noble, W.S. & MacCoss, M.J. Analysis of peptide MS/MS spectra from large-scale proteomics experiments using spectrum libraries. Anal. Chem. 78, 5678–5684 (2006).
Picotti, P. et al. A database of mass spectrometric assays for the yeast proteome. Nat. Methods 5, 913–914 (2008).
Prakash, A. et al. Expediting the development of targeted SRM assays: using data from shotgun proteomics to automate method development. J. Proteome Res. 8, 2733–2739 (2009).
Picotti, P. et al. High-throughput generation of selected reaction-monitoring assays for proteins and proteomes. Nat. Methods 7, 43–46 (2010).
Collins, B.C. et al. Quantifying protein interaction dynamics by SWATH mass spectrometry: application to the 14-3-3 system. Nat. Methods 10, 1246–1253 (2013).
Picotti, P. et al. A complete mass-spectrometric map of the yeast proteome applied to quantitative trait analysis. Nature 494, 266–270 (2013).
Schubert, O.T. et al. The Mtb Proteome library: a resource of assays to quantify the complete proteome of Mycobacterium tuberculosis. Cell Host Microbe 13, 602–612 (2013).
Karlsson, C., Malmström, L., Aebersold, R. & Malmström, J.A. Proteome-wide selected reaction monitoring assays for the human pathogen Streptococcus pyogenes. Nat. Commun. 3, 1301 (2012).
Hüttenhain, R. et al. N-Glycoprotein SRMAtlas: a resource of mass-spectrometric assays for N-glycosites enabling consistent and multiplexed protein quantification for clinical applications. Mol. Cell. Proteomics 12, 1005–1016 (2013).
Hüttenhain, R. et al. Reproducible quantification of cancer-associated proteins in body fluids using targeted proteomics. Sci. Transl. Med. 4, 142ra94 (2012).
Rosenberger, G. et al. A repository of assays to quantify 10,000 human proteins by SWATH-MS. Sci. Data 1, 140031 (2014).
Deutsch, E.W. et al. A guided tour of the trans-proteomic pipeline. Proteomics 10, 1150–1159 (2010).
Chambers, M.C. et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 30, 918–920 (2012).
Sturm, M. et al. OpenMS: an open-source software framework for mass spectrometry. BMC Bioinformatics 9, 163 (2008).
Cox, J. & Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 (2008).
Lam, H. et al. Development and validation of a spectral library searching method for peptide identification from MS/MS. Proteomics 7, 655–667 (2007).
Lam, H. & Aebersold, R. Building and searching tandem mass (MS/MS) spectral libraries for peptide identification in proteomics. Methods 54, 424–431 (2011).
Weisbrod, C.R., Eng, J.K., Hoopmann, M.R., Baker, T. & Bruce, J.E. Accurate peptide fragment mass analysis: multiplexed peptide identification and quantification. J. Proteome Res. 11, 1621–1632 (2012).
Selevsek, N. et al. Reproducible and consistent quantification of the Saccharomyces cerevisiae proteome by SWATH-MS. Mol. Cell. Proteomics, http://dx.doi.org/10.1074/mcp.M113.035550 (2015).
Heller, M. et al. Added value for tandem mass spectrometry shotgun proteomics data validation through isoelectric focusing of peptides. J. Proteome Res. 4, 2273–2282 (2005).
Stergachis, A.B., MacLean, B., Lee, K., Stamatoyannopoulos, J.A. & MacCoss, M.J. Rapid empirical discovery of optimal peptides for targeted proteomics. Nat. Methods 8, 1041–1043 (2011).
Qeli, E. et al. Improved prediction of peptide detectability for targeted proteomics using a rank-based algorithm and organism-specific data. Proteomics 108, 269–283 (2014).
Mallick, P. et al. Computational prediction of proteotypic peptides for quantitative proteomics. Nat. Biotechnol. 25, 125–131 (2006).
Eyers, C.E. et al. CONSeQuence: prediction of reference peptides for absolute quantitative proteomics using consensus machine learning approaches. Mol. Cell. Proteomics 10, M110.003384 (2011).
Fusaro, V.A., Mani, D.R., Mesirov, J.P. & Carr, S.A. Prediction of high-responding peptides for targeted protein assays by mass spectrometry. Nat. Biotechnol. 27, 190–198 (2009).
Tang, H. et al. A computational approach toward label-free protein quantification using predicted peptide detectability. Bioinformatics 22, e481–e488 (2006).
Webb-Robertson, B.-J.M. et al. A support vector machine model for the prediction of proteotypic peptides for accurate mass and time proteomics. Bioinformatics 24, 1503–1509 (2008).
Li, S., Arnold, R.J., Tang, H. & Radivojac, P. On the accuracy and limits of peptide fragmentation spectrum prediction. Anal. Chem. 83, 790–796 (2011).
Escher, C. et al. Using iRT, a normalized retention time for more targeted measurement of peptides. Proteomics 12, 1111–1121 (2012).
Toprak, U.H. et al. Conserved peptide fragmentation as a benchmarking tool for mass spectrometers and a discriminating feature for targeted proteomics. Mol. Cell. Proteomics 13, 2056–2071 (2014).
de Graaf, E.L., Altelaar, A.F.M., van Breukelen, B., Mohammed, S. & Heck, A.J.R. Improving SRM assay development: a global comparison between triple quadrupole, ion trap, and higher energy CID peptide fragmentation spectra. J. Proteome Res. 10, 4334–4341 (2011).
Deutsch, E. mzML: a single, unifying data format for mass spectrometer output. Proteomics 8, 2776–2777 (2008).
Keller, A., Eng, J., Zhang, N., Li, X.-J. & Aebersold, R. A uniform proteomics MS/MS analysis platform utilizing open XML file formats. Mol. Syst. Biol. 1, 2005.0017 (2005).
Elias, J.E. & Gygi, S.P. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–214 (2007).
Shteynberg, D. et al. iProphet: multi-level integrative analysis of shotgun proteomic data improves peptide and protein identification rates and error estimates. Mol. Cell. Proteomics 10, M111.007690 (2011).
Shteynberg, D., Nesvizhskii, A.I., Moritz, R.L. & Deutsch, E.W. Combining results of multiple search engines in proteomics. Mol. Cell. Proteomics 12, 2383–2393 (2013).
Picotti, P., Aebersold, R. & Domon, B. The implications of proteolytic background for shotgun proteomics. Mol. Cell. Proteomics 6, 1589–1598 (2007).
Walmsley, S.J. et al. Comprehensive analysis of protein digestion using six trypsins reveals the origin of trypsin as a significant source of variability in proteomics. J. Proteome Res. 12, 5666–5680 (2013).
Kim, J.-S., Monroe, M.E., Camp, D.G., Smith, R.D. & Qian, W.-J. In-source fragmentation and the sources of partially tryptic peptides in shotgun proteomics. J. Proteome Res. 12, 910–916 (2013).
Eng, J.K., Searle, B.C., Clauser, K.R. & Tabb, D.L. A face in the crowd: recognizing peptides through database search. Mol. Cell. Proteomics 10, R111.009522 (2011).
Keller, A., Nesvizhskii, A.I., Kolker, E. & Aebersold, R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 74, 5383–5392 (2002).
Reiter, L. et al. Protein identification false discovery rates for very large proteomics data sets generated by tandem mass spectrometry. Mol. Cell. Proteomics 8, 2405–2417 (2009).
Liu, J. et al. Methods for peptide identification by spectral comparison. Proteome Sci. 5, 3 (2007).
Röst, H.L., Malmström, L. & Aebersold, R. A computational tool to detect and avoid redundancy in selected reaction monitoring. Mol. Cell. Proteomics 11, 540–549 (2012).
Deutsch, E.W. et al. TraML--a standard format for exchange of selected reaction monitoring transition lists. Mol. Cell. Proteomics 11, R111.015040 (2012).
Vizcaíno, J.A. et al. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 32, 223–226 (2014).
Picotti, P., Bodenmiller, B., Mueller, L.N., Domon, B. & Aebersold, R. Full dynamic range proteome analysis of S. cerevisiae by targeted proteomics. Cell 138, 795–806 (2009).
Nesvizhskii, A.I., Keller, A., Kolker, E. & Aebersold, R. A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 75, 4646–4658 (2003).
Acknowledgements
We thank C. Ludwig and S. Bader for their discussions and feedback on the manuscript, L. Blum for implementation of iProphet support in MAYU, H. Röst for packaging msproteomicstools, J. Slagel for including the qtofpeakpicker and the new MAYU version in the TPP, and the PRIDE Team for maintaining the ProteomeXchange platform. This work has been financially supported by the Framework Programme 7 of the European Commission through SysteMTb (241587), UNICELLSYS (201142), PRIME-XS (262067) and ProteomeXchange (260558), a European Research Council advanced grant Proteomics v3.0 (233226), the Federal Ministry of Education and Research (e:Bio Express2Present, 0316179C) and the Forschungszentrum Immunologie of the University Medical Center Mainz.
Author information
Authors and Affiliations
Contributions
O.T.S., L.C.G. and B.C.C. developed the workflow and wrote the manuscript; P.N. developed the tools spectrast2tsv.py and spectrast_cluster.py; G.R. and H.L. developed and implemented the retention time normalization and iRT calibration in SpectraST; W.E.W. developed the qtofpeakpicker; D.A., P.M. and B.M. implemented automated SWATH library import into Skyline; and R.A. directed the project and contributed to writing the manuscript.
Corresponding author
Ethics declarations
Competing interests
R.A. holds shares of Biognosys AG, which operates in the field covered by the article (products are Spectronaut software and iRT-kit).
Integrated supplementary information
Supplementary Figure 1 Comparison of converters.
(a) Violin plots show the intrinsic fragment ion spectrum variability of three yeast sample injections converted with three DDA centroiding algorithms.
(b) Violin plots show how well the relative intensities of the six most intense fragment ions obtained by a certain centroiding algorithm compare to the relative intensities of the same six fragments in SWATH MS data.
The spectrum similarity was determined using the normalised spectral contrast angle as described by Toprak and co-workers41. N indicates the number of pairwise comparisons of fragment ion spectra for the same peptide precursors between each data file and S indicates the estimated level of dissimilarity obtained by comparing the experimental score distribution to the score distribution of a perturbation benchmark dataset (the lower S, the more similar the spectra).
(c) Depending on the centroiding algorithm implemented in the different peak pickers, the number of peptide and protein identifications from a database search vary slightly (after filtering for protein-level FDR=1%). The size of the circles is proportional to the number of identifications.
Supplementary Figure 2 MAYU-estimated FDRs with respect to iProphet probability thresholds.
The software MAYU estimates the FDRs on PSM (mFDR), peptide (pepFDR), and protein (protFDR) level with respect to the applied iProphet probability threshold. The data is based on the case study described in the protocol, which consists of three whole cell lysates of yeast. As comparison the protein FDR of a very large data set of 331 runs of different human cells and tissues is shown (Pan Human Library as described by Rosenberger and colleagues23).
Supplementary Figure 3 Comparison of consensus and best replicate spectral libraries.
a) Violin plots show the intrinsic fragment ion spectrum variability of three injections of a yeast sample converted with the qtofpeakpicker and summarised using either the best replicate fragment ion spectrum or consensus algorithm implemented in SpectraST11.
(b) Violin plots show how well the relative intensities of the six most intense fragment ions compare to the relative intensities of the same six fragments in SWATH MS data.
The spectrum similarity was determined using the normalised spectral contrast angle as described by Toprak and co-workers41. N indicates the number of pairwise comparisons of fragment ion spectra for the same peptide precursors between each data file and S indicates the estimated level of dissimilarity obtained by comparing the experimental score distribution to the score distribution of a perturbation benchmark dataset (the lower S, the more similar the spectra). Common neutral losses were included in the comparison.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–3, Supplementary Notes 1–5, Supplementary Tables 1–6 and Supplementary Tutorial (PDF 6958 kb)
Rights and permissions
About this article

Cite this article
Schubert, O., Gillet, L., Collins, B. et al. Building high-quality assay libraries for targeted analysis of SWATH MS data. Nat Protoc 10, 426–441 (2015). https://doi.org/10.1038/nprot.2015.015
Published:
Issue Date:
DOI: https://doi.org/10.1038/nprot.2015.015
This article is cited by
-
Prospective observational study on biomarkers of response in pancreatic ductal adenocarcinoma
Nature Medicine (2024)
-
A comprehensive spectral assay library to quantify the Halobacterium salinarum NRC-1 proteome by DIA/SWATH-MS
Scientific Data (2023)
-
NeuroLINCS Proteomics: Defining human-derived iPSC proteomes and protein signatures of pluripotency
Scientific Data (2023)
-
Calibr improves spectral library search for spectrum-centric analysis of data independent acquisition proteomics
Scientific Reports (2022)
-
Baseline proteomics characterisation of the emerging host biomanufacturing organism Halomonas bluephagenesis
Scientific Data (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
