
Abstract
Autophagy is a catabolic process aimed at recycling cellular components and damaged organelles in response to diverse conditions of stress, such as nutrient deprivation, viral infection and genotoxic stress. A growing amount of evidence in recent years argues for oxidative stress acting as the converging point of these stimuli, with reactive oxygen species (ROS) and reactive nitrogen species (RNS) being among the main intracellular signal transducers sustaining autophagy. This review aims at providing novel insight into the regulatory pathways of autophagy in response to glucose and amino acid deprivation, as well as their tight interconnection with metabolic networks and redox homeostasis. The role of oxidative and nitrosative stress in autophagy is also discussed in the light of its being harmful for both cellular biomolecules and signal mediator through reversible posttranslational modifications of thiol-containing proteins. The redox-independent relationship between autophagy and antioxidant response, occurring through the p62/Keap1/Nrf2 pathway, is also addressed in order to provide a wide perspective upon the interconnection between autophagy and oxidative stress. Herein, we also attempt to afford an overview of the complex crosstalk between autophagy and DNA damage response (DDR), focusing on the main pathways activated upon ROS and RNS overproduction. Along these lines, the direct and indirect role of autophagy in DDR is dissected in depth.
Similar content being viewed by others


Facts
-
Reactive oxygen species (ROS) production and thiol redox state imbalance are induced immediately upon nutrient deprivation and represent important mediators of autophagy.
-
ROS and reactive nitrogen species (RNS) irreversibly oxidize DNA and cellular biomolecules, thereby representing the primary source of damage in biological systems.
-
Autophagy contributes to clearing the cells of all irreversibly oxidized biomolecules (proteins, DNA and lipids), this is all the more reason why it could be included in the antioxidant and DNA damage repair systems.
Open Questions
-
How do ROS and oxidative stress affect autophagy?
-
Which are the main ROS able to signal autophagy being activated and going on?
-
Does nitric oxide act as a real inhibitor of autophagy?
-
How does autophagy sense DNA damage?
-
How can autophagy contribute to DNA damage repair?
In the 1950s, Christian de Duve,1,2 contextually with the discovery of glucagon, clarified the intracellular localization of several enzymes by setting up centrifugation-based tissue fractionation of rat liver homogenates.3 During his work, he discovered and coined the names of many organelles, whose purification, characterization and distribution contributed to earning him the Nobel Prize for Physiology and Medicine in 1974. In his studies on carbohydrate metabolism and insulin action, he described for the first time the lysosomes as the intracellular granules containing the enzymes glucose-6-phosphatase and acid phosphatase, in addition to a set of hydrolases that were deputed to digest, recycle and remove intracellular material,4 such as worn-out or damaged organelles, and engulfed pathogens, by means of a process that he named autophagy.
More than 10 years later, in 1966, he also defined the structure and composition of microbodies:5 the cellular districts in which hydrogen peroxide is endogenously produced to a high extent as a side effect of the reactions catalyzed by many oxidases involved in amino acid, purines and fatty acid metabolism, and for this reason named peroxisomes. Although the toxicity of hydrogen peroxide had been reported many years before,6 only in the late 1950s its real implications in biology were coming up. Progress in the field of metallobiology and the fine characterization of metalloenzyme-mediated catalysis provided compelling evidence for an endogenous and physiological production of partially reduced oxygen species (nowadays usually referred to as reactive oxygen species (ROS)), such as superoxide anion (O ), hydrogen peroxide (H2O2) and hydroxyl radical (·OH). Their being highly reactive towards lipids, proteins and DNA,7, 8, 9 and severely harmful for cell survival when present at very high concentrations, both led to the concept of oxidative stress as detrimental condition occurring in all living systems and arising from the imbalance between oxidants species and antioxidant defence. It is not a coincidence that in the same years, Denham Harman10 postulated the ‘free radical theory of ageing’ in which he stated that free radicals were the primary cause of massive damage to DNA and all cellular macromolecules, culminating in cancer and in a diffuse cell dysfunction distinctive of ageing.
), hydrogen peroxide (H2O2) and hydroxyl radical (·OH). Their being highly reactive towards lipids, proteins and DNA,7, 8, 9 and severely harmful for cell survival when present at very high concentrations, both led to the concept of oxidative stress as detrimental condition occurring in all living systems and arising from the imbalance between oxidants species and antioxidant defence. It is not a coincidence that in the same years, Denham Harman10 postulated the ‘free radical theory of ageing’ in which he stated that free radicals were the primary cause of massive damage to DNA and all cellular macromolecules, culminating in cancer and in a diffuse cell dysfunction distinctive of ageing.
When de Duve characterized the peroxisomes and found out that they were the organelles in which the antioxidant enzyme catalase resides, he probably did not realize that all his findings could be basically interconnected by a finely organized signalling system, where primary/primitive stimuli (e.g., nutrient availability and oxidative insults) differently impinge on the maintenance of biomolecule integrity and cell viability through the intermediate activity of homeostatic processes (mainly based on repair and degradation), the most complex and versatile of which was the very same autophagy he discovered 10 years before.
Autophagy: Converging Point of Different Stimuli
There are three main types of autophagy culminating to lysosome-mediated degradation: (1) macroautophagy (hereafter referred to as autophagy) that involves the formation of a double-membrane vesicle (autophagosome) deputed to sequester damaged organelles and biomolecules; (2) microautophagy, by which the cytosolic material is directly engulfed by the lysosome; and (3) chaperone-mediated autophagy. It is now well established that autophagy is a very sensitive process underlying cell response induced by almost every stressful condition affecting cellular homeostasis.11 Through autophagy, cells coordinate energy and building blocks demanded for vital processes (e.g., growth and proliferation) with the extracellular stimuli and carbon source availability, such as amino acids and glucose. If they are not sufficient to maintain the rate of protein synthesis, or to provide the required amount of ATP needed to sustain metabolic reactions, then cells activate autophagy in order to rapidly degrade the old or burned-out components and reuse the generated pool of biomolecules.
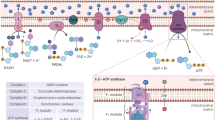
Both glucose and amino acids signals converge on a unique molecular transducer of cellular needs, the mammalian target of rapamycin complex 1 (mTORC1) (Figure 1).12 Active mTORC1 controls the activity of translation eukaryotic initiation factors (eIFs) and eukariotic elongation factors (eEFs), namely eIF2, -3 and -4 and eEF2, by direct phosphorylation of two key protein targets, EIF4E-binding protein 1 (4E-BP1) and protein S6 kinase (p70S6K).13 Both are required for a correct and efficient protein synthesis, as they regulate the interactions between the mRNA 5′ cap, the poly(A)-tail and the 40S and 60S ribosomal subunits (Figure 1).14 Concomitantly, active mTORC1 prevents autophagy by phospho-inhibiting the UNC51-like kinase 1 (ULK1) at Ser757 and its interacting partner, the autophagy related gene 13 (Atg13) that, together with the FAK-family interacting protein of 200 kDa (FIP200), form the so-called ULK1 complex (Figure 1).12,15 Upon autophagic stimuli, mTORC1 is inhibited, thus leading to the activation of ULK1. Once activated, ULK1 is able to phosphorylate Atg13 and FIP200 inducing to the following activation of the class III phosphoinositide 3-kinase (PI3K) complex via the activating molecule in Beclin1-regulated autophagy 1 (Ambra1).15,16 The formation of PI3K complex is required to initiate phagophore nucleation that represents the first step leading to autophagosome formation (Figure 1).12,15,16

Main molecular pathways activated in the presence or absence of nutrients. (a) The synergic import of leucine (Leu) and glutamine (Gln) (top left) results in mTORC1 recruitment to the lysosomal membrane and its subsequent activation by at least two distinct pathways. The first one proposes that cytosolic amino acids enter the lysosome and signal their presence to RAG-A and RAG-C (or RAG-B and RAG-D, not shown in the figure) through the lysosome-located proton pump v-ATPase and the multimolecular complex called Ragulator. The second pathway provides for the double deamination of Gln catalysed by the enzymes glutaminase (GLN) and glutamate dehydrogenase (GDH). This sequence of reactions subsequently generates glutamate (Glu) and α-ketoglutarate (αKG) that, by acting as co-substrate for prolyl hydroxylases (PHD), finally leads to RAG activation. GTP-bound RAG-A (or B) and GDP-bound RAG-C (or D) can then recruit mTORC1 to the lysosome membrane where it is activated by RHEB (middle left). Once activated, mTORC1 activates protein synthesis by phosphorylating 4EBP1 and p70S6K, and concomitantly inhibits autophagy by phosphorylating ULK1 complex at the level of ULK1 and Atg13 (bottom left). Glucose is taken up through specific transporters (GLUTs) and phosphorylated to glucose-6-phosphate (G6P) by hexokinase (the only mitochondrial isoform II, HKII, is shown in the top right side of the figure). G6P is then isomerized to fructose-6-phosphate (F6P), oxidized through the glycolytic pathway to generate pyruvate (Pyr) and acetyl-CoA that fuels the mitochondrial TCA cycle and the respiratory chain for the production of ATP (middle right) through the oxidative phosphorylation (OXPHOS). G6P can also undergo oxidation via the glucose-6-phosphate dehydrogenase (G6PDH)-mediated catalysis along the pentose phosphate pathway (middle right). In this way, electrons required for NADP+-to-NADPH reduction, and sugars needed for DNA de novo synthesis (e.g., ribulose-5-phosphate, R5P) are also provided (bottom right). (b) Upon amino acid deprivation, RAGs exchange nucleotides located in their binding sites (GTP with GDP or vice versa), thus leading to mTORC1 release from the lysosome membrane (top left). These 2 events are associated with the inhibitory binding of mTORC1 to HKII that takes place upon glucose deficiency and G6P level decrease (top right). This condition leads to a decrease of NADPH and ATP levels that finally result in a reduced antioxidant capacity of the cell (especially in regenerating the reduced thiol pool) (middle right) and in energetic stress that the cell attempts to counteract by the adenylate kinase (AK)-mediated conversion of ADP into ATP and AMP (centre). AMP increase induces to the activation of AMPK that inhibits protein synthesis by phosphorylating TSC2 and mTORC1 and activates autophagy by phospho-activating ULK1. Once activated, ULK1 phosphorylates its interactors (Atg13 and FIP200) and recruits microtubule-associated PI3K complex by means of an AMBRA1-mediated process to initiate the nucleation phase of autophagic vesicles from the endoplasmic reticulum (or mitochondria, not shown). Many other factors, such as Atg proteins coming from Golgi apparatus (e.g., Atg9) contribute to phagophore elongation and autophagosome formation (bottom)
Amino acid signal
Among the amino acids that are able to signal their presence to the cell, and then let autophagy be induced in case of any deficiency, leucine and glutamine play the most important roles because of their essentiality and their tight interdependence in the mechanism regulating their uptake.17,18 Leucine is an essential amino acid indispensible for cell survival as it is nonsynthesizable de novo through alternative pathways, such as by transformation of other amino acids or by transamination of carboxylates coming up from glycolysis and the tricarboxylic acid (TCA) cycle. Glutamine, instead, though nonessential, represents the most abundant amino acid in the human body and one of the main substrates of anaplerotic reactions fuelling the TCA cycle. It has been estimated that hypercatabolism or other stressful conditions (e.g., infection and severe injuries) are accompanied by a significant decrease of glutamine in skeletal muscle cells,19 thus arguing for it being a reliable marker of both amino acid and energetic status.
At the molecular level, the presence of an adequate amount of amino acids induces members of the RAS-related GTP-binding protein (RAG) family of small GTPases (i.e., RAG A–D) to bind guanine nucleotides (GTP or GDP in dependence of the member), leading to their activation and, subsequently, to the recruitment of mTORC1 on the lysosome membrane (Figure 1).20,21 Here, mTORC1 can be targeted by RAS homologue enriched in brain (RHEB) that, upon binding to GTP, acts as positive regulator of mTORC1.22 In agreement with this model, RAGs are the primary sensors that, by means of at least two distinct mechanisms, signal amino acid availability to mTORC1 and modulate its activation state.23 One mechanism suggests that RAGs sense the amino acid pool (mainly leucine) present within the lumen of the lysosome by the vacuolar H+-ATPase (v-ATPase) and a molecular complex called Ragulator (Figure 1).24,25 This mechanism could underlie the negative feedback acting on autophagy (because of mTOR reactivation) once amino acid levels have been successfully restored. Alternatively, it has been proposed that RAGs are activated by glutamine, specifically by α-ketoglutarate generated upon double deamination occurring in the glutaminolytic pathway.26 Although mTOR activation through this mechanism needs to be still clarified, it seems to require prolyl hydroxylase (PHD) activity that, in fact, is positively regulated by α-ketoglutarate (Figure 1). As above reported, this mechanism has been shown to be responsive to leucine also, as it binds to and activates glutamate dehydrogenase, the enzyme catalysing the last deamination step leading to α-ketoglutarate production.27 In line with these pieces of evidence, compartmentalization of mTORC1 at the level of the lysosomes could provide some explanations regarding mTOR negative regulation on autophagy.13
Glucose signal
Glucose is the primary carbon source that, upon sequential oxidation steps taking place during glycolysis and TCA cycle, provides the electrons (energy) coming from the breakdown of its chemical bonds, required for ATP production. The maintenance of endergonic processes strictly depends on the maintenance of ATP levels, and for this reason, cells (1) actively synthesize ATP and (2) have evolved sophisticated mechanisms to face up energetic stress.28
AMP-dependent protein kinase (AMPK) is the genuine sensor of the energetic state of the cell, and directly responds to the so-called adenylate energy charge as the enzyme is activated by very low increases of AMP levels (and, to certain extent, of ADP), and deactivated by ATP.28, 29, 30 In order to restore the correct adenylate energy charge, phospho-active AMPK concertedly stimulates catabolic pathways (e.g., glycolysis and fatty acid oxidation), inhibits the rate of anabolic reactions (protein and fatty acid synthesis)28 and activates autophagy.
At the molecular level, active AMPK stimulates autophagy by means at least three distinct mechanisms. These include (1) phosphorylation of the mTORC1 inhibitor, tuberous sclerosis 2 (TSC2) at Ser1387,31 which induces RHEB GTPase activity; (2) phosphorylation of the mTORC1 component Raptor at Ser722 and Ser792,32 which is preparatory for its binding to 14–3–3; and (3) phosphorylation of ULK1 at Ser317 and Ser777 (Figure 1).33 From a mechanistic point of view, the first two phosphorylations catalysed by AMPK inhibit mTORC1 and reduce its inhibitory effects on ULK1. ULK1 is then free to interact with and to be phosphoactivated by AMPK (Figure 1).
Fascinatingly, it has very recently been reported that glucose sensing by the cells does not only depend on AMPK, as indirect transducer of the intracellular energy state, but also relies upon more direct mechanisms, thereby making the system redundant and controlled at multiple levels. Roberts et al.34 demonstrated that hexokinase II (HKII), the mitochondria-located enzyme responsible for the first step of glycolysis, binds to and inhibits mTORC1, and that this interaction is enhanced by glucose deprivation, namely by a decrease in glucose-6-phosphate levels (Figure 1). As proposed by the authors, this new mechanism could contribute to the modulation of cell metabolism in circumstances of glucose deficiency,34 but could also have deep implications in redox homeostasis. Indeed, besides many indications arguing for a direct role of HKII in preventing ROS generation from mitochondria,35, 36, 37 it should be also taken into account that glucose-6-phosphate, via the pentose phosphate pathway, is also the primary source of electrons for NADP+-to-NADPH reduction (Figure 1). NADPH directly participates in bioreductive synthesis and provides the electrons required for thiol redox homeostasis (Figure 1). In particular, NADPH acts as a co-substrate of glutathione reductase – the enzyme responsible for the reduction of the disulphide (GSSG) to the sulphhydryl (GSH) form of glutathione – as well as of many other reductases deeply implicated in sulphhydryl regeneration and, in turn, in the defence against oxidative stress.
Oxidative Stress
Living cells are always subjected to the hazardous effects of exogenously or endogenously produced highly reactive oxidizing molecules. These can be radicals and nonradicals (e.g., H2O2), but have in common the ability to easily take electrons from (oxidize) molecules with which they remain in contact, such as all cellular biomolecules, generating chain reactions and ultimately leading to cell structure damage. Among these classes of molecules, those deriving from ROS and reactive nitrogen species (RNS) have the main biological impact because they are endogenously produced at the highest concentration, and for this reason the concept of oxidative stress can be widened so as to nitro-oxidative stress.
Oxidative damage
It is commonly accepted that the principal source of ROS in the cell is the mitochondrial respiratory chain. Indeed, mitochondrial complexes (mainly complexes I and III) can leak electrons, leading to the partial reduction of oxygen to O that spontaneously, or by the superoxide dismutase (SOD)-mediated catalysis, very rapidly disproportionates into H2O2. It has been estimated that ROS produced by mitochondria are ∼1–2% of the total rate of oxygen consumption.38 This at first glance could appear very low; yet, if one considers that the average rate of oxygen utilization in each single cell of human body is ∼2.5 × 10-18 mol/s (that means 2.2 × 1010 molecules everyday),39 the amount of ROS daily generated intracellularly reaches ∼1 billion molecules. Multiplying this value for the number of cells in human body (∼50 trillion) gives an idea of the intensity of ROS flux to which we are exposed physiologically. Moreover, considering that in some circumstances, the electron flux through the mitochondrial respiratory chain is intensified (e.g., upon increased energetic demand), or that mitochondrial efficiency might decrease (e.g., during ageing), along with the fact that other exogenous (e.g., UV radiation) and endogenous sources of ROS (e.g., oxidases and oxygenases) can operate as well, it becomes evident that a highly efficient antioxidant response has probably been selected by evolution to protect and preserve, as far as possible, the cellular components.40
that spontaneously, or by the superoxide dismutase (SOD)-mediated catalysis, very rapidly disproportionates into H2O2. It has been estimated that ROS produced by mitochondria are ∼1–2% of the total rate of oxygen consumption.38 This at first glance could appear very low; yet, if one considers that the average rate of oxygen utilization in each single cell of human body is ∼2.5 × 10-18 mol/s (that means 2.2 × 1010 molecules everyday),39 the amount of ROS daily generated intracellularly reaches ∼1 billion molecules. Multiplying this value for the number of cells in human body (∼50 trillion) gives an idea of the intensity of ROS flux to which we are exposed physiologically. Moreover, considering that in some circumstances, the electron flux through the mitochondrial respiratory chain is intensified (e.g., upon increased energetic demand), or that mitochondrial efficiency might decrease (e.g., during ageing), along with the fact that other exogenous (e.g., UV radiation) and endogenous sources of ROS (e.g., oxidases and oxygenases) can operate as well, it becomes evident that a highly efficient antioxidant response has probably been selected by evolution to protect and preserve, as far as possible, the cellular components.40
The antioxidant enzymes SOD, catalase and glutathione peroxidases (GSH-Px or GPx) are those responsible for removal of O , H2O2 and peroxides in general. They are present in all cellular districts and act in concert with other proteins, such as peroxiredoxins, thioredoxins (Trx) and glutaredoxins (Grx), as well as low-molecular-weight antioxidants (e.g., GSH, tocopherols and ascorbate) to fully scavenge ROS and restore the reduced protein and lipid pool.41, 42, 43 The efficiency of the antioxidant defence is also important to modulate the levels of RNS, a class of molecules deriving from peroxynitrite (ONOO-), a very dangerous compound generated by reaction between O2.- and nitric oxide (NO).44,45 Nitric oxide is a highly reactive gaseous radical, soluble in water and diffusible across cell membranes.45,46 It is endogenously produced by NO synthases (NOS1-3), a family of constitutive or inducible enzymes with different tissue distribution and that use arginine and NADPH as substrates for reaction. As already described for ROS, NO-derived oxidant species contribute to establishing oxidative conditions as well, resulting in irreversible damage to biomolecules when produced at an extent high enough to overcome the antioxidant response.47
, H2O2 and peroxides in general. They are present in all cellular districts and act in concert with other proteins, such as peroxiredoxins, thioredoxins (Trx) and glutaredoxins (Grx), as well as low-molecular-weight antioxidants (e.g., GSH, tocopherols and ascorbate) to fully scavenge ROS and restore the reduced protein and lipid pool.41, 42, 43 The efficiency of the antioxidant defence is also important to modulate the levels of RNS, a class of molecules deriving from peroxynitrite (ONOO-), a very dangerous compound generated by reaction between O2.- and nitric oxide (NO).44,45 Nitric oxide is a highly reactive gaseous radical, soluble in water and diffusible across cell membranes.45,46 It is endogenously produced by NO synthases (NOS1-3), a family of constitutive or inducible enzymes with different tissue distribution and that use arginine and NADPH as substrates for reaction. As already described for ROS, NO-derived oxidant species contribute to establishing oxidative conditions as well, resulting in irreversible damage to biomolecules when produced at an extent high enough to overcome the antioxidant response.47
Redox signal
In the late 1990s, a new ‘radical free’ concept for free radicals began to take root,48 and a new signalling role for ROS and RNS emerged. Many lines of evidence were accumulating, indicating that ROS and RNS were able to modify proteins in a reversible manner at the level of the sulphur-containing residues, cysteine and methionine, thus providing evidence for the existence of a redox-based signal.40,49 In particular, reactive cysteine thiol groups (SH) of a growing number of proteins were revealed to be able to rapidly undergo reactions with H2O2 and NO in biological systems, thus forming the S-hydroxylated (S-OH) and S-nitrosylated (S-NO) derivatives, respectively. Upon reaction with other cysteines (e.g., those belonging to GSH or other protein thiols), both these adducts usually covert to disulphide (S-S), and are finally reduced back to sulphhydryl at the expense of the reducing equivalents provided by NADPH through the Trx/Trx reductase (or Grx/Grx reductase) system.40,41 Oxidative modifications of reactive cysteines cause changes in protein structure and function; they affect localization and physical interactions, as well as the capability to undergo further posttranslational modifications (e.g., phosphorylation).40 This is the reason that reactive cysteines are deemed to be the primary molecular switches that are able to transduce a redox signal.48
Autophagy and Oxidative Stress
ROS have been copiously reported as early inducers of autophagy upon nutrient deprivation.50 However, to date, it is still unclear as to which species exactly drives the process. A detailed work from Chen et al.51 proposes that O2.- is the primary ROS involved in autophagy induced by glucose, glutamine, pyruvate or serum deprivation. Further lines of evidence indicate, instead, that H2O2 is the molecule produced immediately after starvation,52,53 whereas many others just hypothesize that ROS are crucial for autophagy execution as treatment with antioxidants partially or completely reverts the process.54
Mitochondria as main source of ROS in autophagy signalling
Although the question is still far from being solved, there are at least other two issues that deserve to be considered. The first is ‘where ROS are so rapidly produced’. It would be actually more logical that a stimulus coming from the outside of the cell is transduced by a ROS-producing system located at, or nearby, the plasma membrane, such as the NADPH oxidase (NOX) complexes. Nevertheless, although attractive, this hypothesis has been verified only in macrophages upon bacterial infection, where ROS generated by NOX2 are indispensable for the recruitment of the microtubule-associated protein light chain 3 (LC3) on phagosomes that, thus modified, are degraded by autophagy to prevent pathogen escape.55 A large amount of data, instead, converge to state that the mitochondria represent the principal source of ROS required for autophagy induction,56,57 although they are not in close proximity to the plasma membrane. A possible explanation for this unexpected evidence is that nutrient deprivation suddenly results in energetic stress that, in turn, increases ATP demand and causes mitochondrial overburden to face up adverse conditions. As a consequence, electron leakage and ROS production also increase. Another hypothesis is that a still uncharacterized factor could act as transducer, linking the upstream autophagic signal with mitochondria. A good candidate could be HKII that, by sequestering mTORC1, could loosen its inhibition on permeability transition pore (PTP) and its ability to decrease ROS.34,35 In support of this hypothesis, it has been reported that the two protein kinases positively regulating HKII activity, Akt and myotonic dystrophy protein kinase (DMPK), are negative modulators of autophagy.58,59
ROS and mitophagy
As principal sites of ROS production, mitochondria are the organelles that are able to turn on and tune autophagy. However, upon chronic impairment of mitochondrial function, ROS can be generated at high extent, thus shifting their role from bulk autophagy inducers into a self-removal signal for mitochondria through a selective process called mitophagy. This represents a fine mechanism of negative feedback regulation by which autophagy eliminates the source of oxidative stress and protects the cell from oxidative damage (see below).
Although necessary, mitophagy represents an ‘extreme decision’ for a cell subjected to nutrient deprivation because of at least two main reasons. The first reason is that mitochondria underpin ATP production that is fundamental upon carbon source limitation. The second reason lies in the fact that mitochondria are relatively large organelles that require being beforehand fragmented in order to be properly recognized and engulfed within the autophagosomes.60 Both these issues contribute to explain why mitochondria are in general refractory to undergo mitophagy, unless they are severely damaged. Recently, it has been proposed that under nutrient deprivation, mitochondria attempt to protect themselves from autophagic removal by promoting fusion and inhibiting fission events.61,62 The combination of these two inputs results in mitochondrial elongation that further impedes organelle engulfment within the autophagosomes and, concomitantly, allows to maximize ATP production.62 Only upon prolonged starvation, mitochondria depolarize and become fragmented in order to assist their removal by mitophagy.
So far, at least two different molecular mechanisms underlying mitophagy have been described and characterized. The first one is mediated by NIX/Bnip3L (Bcl-2/adenovirus E1B 19-kDa-interacting protein 3, long form),63,64 an atypical BH3 protein that is able to directly recognize the autophagosome-sited GABA receptor-associated protein (GABARAP), a functional homologue of LC3 and, in turn, allow mitochondria to be removed. This is a ‘programmed’ event, independent on any damage response that is required, for example, in mitochondrial elimination during erythrocyte differentiation.65,66 The second mechanism is activated for the selective dismissal of impaired or dysfunctional mitochondria. It is responsive to mitochondrial depolarization and is regulated by the PTEN-induced putative kinase 1 (PINK1) and Parkin,67 a ubiquitin E3 ligase whose mutations have been associated with familial form of Parkinson’s disease. PINK1 is a Ser/Thr kinase that translocates on the outer mitochondrial membrane where it is stabilized by low mitochondrial transmembrane potential, thereby acting as real sensor of mitochondrial depolarization.68 Here, PINK1 recruits Parkin68 that ubiquitylates a number of outer mitochondrial membrane-located proteins, for example, the voltage-dependent anion channel 1 (VDAC1).69 Once ubiquitylated, these proteins are recognized by p62/sequestosome 1 (p62/SQSTM1, or simply p62),69,70 a ubiquitin-binding protein acting as a scaffold for several protein aggregates and triggering their degradation through the proteasome, or the lysosome pathway via autophagy.71,72 p62 contains an LC3 interacting region (LIR)73 that has been indicated being fundamental for p62 to mediating mitophagy. Indeed, by means of this motif, p62 can bridge autophagy-targeted mitochondria to LC3 located on the autophagosomes surface, thereby driving their degradation.
Interestingly, our group has recently identified a role for Ambra1 in mitophagy, driven by its selective interaction with LC3 and independent from Parkin and p62.74
Redox signalling in autophagy
Another issue to be considered is ‘how oxidative stress can crosstalk with autophagic machinery’. As previously mentioned, antioxidant treatment prevents autophagy, suggesting that redox imbalance has a pivotal role in driving the process. The very fast induction of autophagy upon ROS production from mitochondria argues for a rapid (ON/OFF) response mediated by redox-sensitive proteins, among which AMPK could be a good candidate. Indeed, AMPK has been proposed as being activated upon H2O2 exposure, particularly through S-glutathionylation (formation of a mixed disulphide with GSH) of reactive cysteines located at the α- (Cys299 and Cys304) and β-subunits (still not identified) (Figure 2).30,75 Although the role of redox regulation in AMPK activation is still a matter of debate, these results are in line with the recent observations indicating that, once deprived of nutrients, cells actively extrude GSH by the drug efflux pump, multidrug resistance protein 1 (MRP1) in order to shift intracellular redox environment towards more oxidizing conditions and prime redox-sensitive proteins to be oxidatively modified (Figure 2).76 The evidence that the sole chemically induced oxidation of GSH is able to induce autophagy even in the absence of any autophagic stimulus76 underlines the importance of thiol redox homeostasis in autophagy commitment. This assumption is in line with the evidence indicating that a number of proteins involved in both induction and execution of autophagy act by means of Cys residues. Among them, the two ubiquitin-like systems Atg7-Atg3 and Atg7-Atg10, some members of Rab GTPase (e.g., Rab33b), and the phosphatase and tensin homologue deleted (PTEN)50 are included. Along this line, it is worthwhile to note that p62 contains a zinc-finger motif (ZZ) rich in cysteine residues that are necessary for metal binding and that could be redox regulated. Although no evidence has been provided yet about a possible redox sensitivity of p62, it could be conceived that, similar to other ZZ-containing proteins,77 p62 could also undergo oxidation and structural alterations that are able to modify/regulate its role in autophagy.

Crosstalk between autophagy and oxidative stress. Superoxide (O ) and H2O2 are the main ROS produced by mitochondria upon nutrient deprivation. They positively regulate autophagy by means of at least three different mechanisms, including: (1) S-glutathionylation (SH → S-SG) of cysteines located in the α and β subunits of AMPK (top right); (2) oxidation of Cys81 (SH → Sox) of Atg4 that in turn leads to the inactivation of its ‘delipidating’ activity on LC3 and to the accumulation of the pro-autophagic LC3-II isoform (top centre); and (3) wide alteration of thiol redox state (e.g., decrease of GSH/GSSG ratio and general increase of oxidized thiols, Sox) that is facilitated by the release of reduced glutathione (GSH) to the extracellular milieu through the multidrug resistance protein 1 (MRP1) (top left). In a redox-independent manner, it has also been demonstrated that p62, when bound to ubiquitylated protein aggregates, can undergo phosphorylation on Ser351, thereby sequestering Keap1 and leading to its detachment from Nrf2 (bottom left). Consequently, Nrf2 is no longer degraded by the ubiquitin-3 proteasome system, but translocates in the nucleus, binds to antioxidant-responsive elements (AREs) located in the promoter regions of antioxidant genes and activates their transcription (bottom right)
) and H2O2 are the main ROS produced by mitochondria upon nutrient deprivation. They positively regulate autophagy by means of at least three different mechanisms, including: (1) S-glutathionylation (SH → S-SG) of cysteines located in the α and β subunits of AMPK (top right); (2) oxidation of Cys81 (SH → Sox) of Atg4 that in turn leads to the inactivation of its ‘delipidating’ activity on LC3 and to the accumulation of the pro-autophagic LC3-II isoform (top centre); and (3) wide alteration of thiol redox state (e.g., decrease of GSH/GSSG ratio and general increase of oxidized thiols, Sox) that is facilitated by the release of reduced glutathione (GSH) to the extracellular milieu through the multidrug resistance protein 1 (MRP1) (top left). In a redox-independent manner, it has also been demonstrated that p62, when bound to ubiquitylated protein aggregates, can undergo phosphorylation on Ser351, thereby sequestering Keap1 and leading to its detachment from Nrf2 (bottom left). Consequently, Nrf2 is no longer degraded by the ubiquitin-3 proteasome system, but translocates in the nucleus, binds to antioxidant-responsive elements (AREs) located in the promoter regions of antioxidant genes and activates their transcription (bottom right)
Notwithstanding the large amount of data supporting the hypothesis of a redox regulation of autophagic signalling, so far the only redox-based mechanism demonstrated to be able to regulate an autophagic protein goes back to 2007, when Scherz-Shouval et al.78 proved that H2O2-mediated oxidation of Atg4 at Cys was required for inactivating its hydrolyzing (delipidating) activity on LC3, thus allowing autophagosome to be correctly elongated (Figure 2).52 No further protein has been added to the list since then, suggesting that redox modifications of proteins – although reasonably proposed as likely modulators of autophagic signal transduction50 – are not the main mechanism linking ROS and autophagy.
Conflicting role of NO and nitrosative stress in autophagy
Results emerging in the past 5 years suggest that NO, by means of S-nitrosylation mechanisms, has also a role in modulating autophagy. However, rather than a positive effector of the process, it seems that it could act as an inhibitory molecule. This assumption is completely in contrast with that described above for ROS, and contributes to making the functional relationship between oxidative stress and autophagy even more complex. Sarkar et al.,79 indeed, demonstrated that treatment with NO donors or enhancement of NOS activity in HeLa cells results in autophagy prevention because of S-nitrosylation, and subsequent inhibition of the c-Jun-N-terminal kinase 1 (JNK1) and IκB kinase β (IKKβ) that regulate Beclin1 detachment from Bcl2 and AMPK activation, respectively. This is in line with results reporting that S-nitrosylation of TSC2 prevents its inhibitory activity on mTOR,80 thereby preventing autophagy and inducing proliferation of melanoma cells. However, these data are in contrast with the well-documented role of NO and nitrosative stress in the activation of AMPK–TSC2 pathway via Ataxia telangiectasia mutated (ATM) in response to DNA damage (see below).81 This discrepancy is likely because of the fact that the biological effects of NO range from prosurvival to apoptotic depending on the real concentrations used (reason why NO has been identified as Janus-faced molecule) that frequently are completely unknown (e.g., upon treatment with NO donors). In support to this, very recently it has been reported that genetic ablation of the main denitrosylating enzyme, S-nitrosoglutathione reductase (GSNOR) – which is the elective experimental approach to study the effects of S-nitrosylation in biological contexts – does not absolutely affect bulk autophagy (Figure 3, unpublished results from Filomeni’s laboratory), but exclusively results in an impairment of the sole mitochondrial autophagy (mitophagy) in skeletal muscle models.78

Autophagy is not affected by S-nitrosylation. (a) Western blot analyses of S-nitrosothiols in total homogenates of skeletal muscles obtained from GSNOR-KO (KO) and wild-type (WT) mice, subjected to biotin-switch assay and revealed by HRP-conjugated streptavidin. Lactate dehydrogenase (LDH) was selected as loading control. Results show that even in the absence of any treatment with NO-delivering drugs, GSNOR ablation induces a significant increase of S-nitrosylated proteins. (b) Representative fluorescence microscopy images of satellite cell-derived myotubes isolated from KO and WT mice expressing LC3-conjugated green fluorescent protein (GFP-LC3) in heterozygosis. Cells were then subjected to two different autophagic stimuli: (1) they were treated for 6 h with 5 μM of the proteasome inhibitor MG132 or, alternatively, (2) allowed to grow for 6 h in a nutrient-deprived cell medium (stv). Images are representative of three independent experiments that gave similar results. Both genotypes displayed a significant and similar increase of fluorescent dots, plausibly representing autophagosomes, thereby indicating that autophagy is not impaired by S-nitrosylation
Altogether, these observations indicate that a straightforward idea about how oxidative stress functions in autophagy is still lacking, despite abundant evidence corroborating its implication in each phase of this process.
p62/Keap1/Nrf2 system: how autophagy couples with redox response
In the past few years, autophagy and oxidative stress have been shown to be interconnected in a more intimate and coordinated maner than by a simple ON/OFF signal. Particularly, in 2010, it was discovered that p62 activates the antioxidant transcription factor Nrf2 (nuclear factor erythroid 2-related factor 2) by a ‘non-canonical’ pathway (Figure 2). The underlying mechanism is completely redox independent, and involves the recruitment of Kelch-like ECH- associated protein 1 (Keap1) that functions as an adapter protein of the Cul3-ubiquitin E3 ligase complex responsible for degrading Nrf2.82,83 In agreement with this model, p62 binds to aggregates of ubiquitylated proteins and increases its affinity for Keap1 when phosphorylated at Ser351 (Figure 2).84 This event induces Keap1 degradation via autophagy85 and leaves Nrf2 free to accumulate and translocate in the nucleus. Here, Nrf2 binds to the antioxidant-responsive elements (ARE) in the promoter regions of antioxidant and detoxifying genes,86 as well as genes involved in DNA damage response, such as 8-oxoguanine glycosylase (OGG1)87 and p53 binding protein 1 (53BP1),88 inducing their transcription (Figure 2). It has also been suggested that Nrf2 activation by this pathway is sustained by Sestrins,89 a highly conserved family of small ‘antioxidant-like’ proteins transcriptionally induced by p53 upon stressful conditions, which are involved in autophagy since function as AMPK activators and mTORC1 inhibitors.90,91
Antioxidant Role of Autophagy: Focus on Nucleus and DNA Damage
On the basis of what has been reported so far, antioxidant response and autophagy are mechanisms simultaneously induced by oxidative stress conditions in order to concomitantly decrease ROS and RNS concentration (upstream causes) and reduce the oxidative damage to biomolecules and organelles (downstream effect). This finely orchestrated repair system perfectly fits the needs of a cell attempting to find a new homeostatic state. By responding very rapidly to oxidative stress, and by decreasing the toxicity of oxidized molecules and organelles through their selective removal, autophagy can be in principle encompassed in the large family of antioxidant processes. However, at variance with proteins and organelles that are present in several copies inside the cell (e.g., mitochondria and ribosomes), autophagy cannot mediate nucleus degradation, even though it is severely damaged, because this could lead to the complete loss of genetic information. Genomic DNA cannot be destroyed, de novo synthesised or entirely replaced like the other biomolecules. Its integrity should be prevented and maintained, and any damage accurately repaired.
An exception to the rule is a highly selective and unusual nuclear DNA degradation by means of the so-called piecemeal microautophagy (PMN). Indeed, in a way resembling nucleophagy occurring in fungi and nematodes,92,93 mammalian cells can specifically remove part of nuclei containing damaged DNA.94,95 In particular, this phenomenon has been observed in nuclear envelopathies94 where the presence of perinuclear autophagosomes or autophagolysosomes containing DNA clearly represented an operative autophagy. It has been reported that micronuclei containing chromosomes, or parts of them, that are not properly incorporated in the daughter nuclei during cell division can be removed by autophagy as well,95 thus providing this process with a direct role in cleaning up damaged content in the nucleus and in maintaining genomic stability.
However, besides PMN, a number of observations indicate that autophagy is deeply involved in DNA damage repair, although without any direct degradative activity on DNA. This phenomenon, mainly occurring upon ROS and RNS-mediated damage to DNA, represents an issue that deserves discussion so as to comprehend how autophagy acts as a preventive and reparative process upon genotoxic stress. Understanding the underlying molecular mediators and the mechanisms would make it possible to clarify once and for all the antioxidant activity of autophagy.
Oxidative Stress and DNA Damage
ROS and RNS are one of the major sources of DNA damage96 as they could directly modify the DNA or indirectly generate different lesions, both affecting cell viability. Among ROS, ·OH can directly attack the DNA backbone by generating five classes of oxidative damage: oxidized bases, abasic sites, DNA–DNA intrastrand adducts, single-strand break (SSB), double-strand break (DSB) and DNA–protein crosslinks.97,98 Among the nucleobases, guanine is the most susceptible to ROS because of its low redox potential, and the main products of its oxidation are 8-hydroxyguanine and 8-hydroxydeoxyguanosine (8-OHG and 8-OHdG). Both are highly mutagenic and carcinogenic as they can match with both cytosine and adenine, thus leading to GC-to-AT transversions.99,100 The 8-OHG and 8-OHdG are the most commonly used markers of DNA damage as they easily accumulate, thus being measured as good index of oxidative lesions to DNA. Nitric oxide and RNS (i.e., NO2·, ONOO-, N2O3 and HNO2) can cause DNA damage as well, and are considered potentially mutagenic96 as they can induce nitration, nitrosylation and deamination of DNA bases.
ROS and RNS are also harmful for mitochondrial DNA (mtDNA) integrity. This feature can deeply affect the transcription of mtDNA-coded proteins and RNAs that underlie the synthesis of a number of subunits belonging to the complexes of the mitochondrial respiratory chain (except Complex II). A vicious cycle is then established in which mitochondria, with oxidized mtDNA, become dysfunctional and produce a high rate of ROS, leading to further mitochondrial impairment. This condition can ultimately result in severe nuclear DNA damage and cell death.101
DNA Damage and Autophagy: A Complex Crosstalk
When the DNA is damaged by ROS and RNS, cells activate a number of pathways in order to maintain genomic integrity, these being associated to the DNA damage response (DDR). Different classes of proteins are implicated in DDR, among which the sensors specifically recognize the lesions to DNA, whereas the mediators and the effectors transduce the signal from the nucleus to the cytosol where several processes are contextually activated in order to better face up to adverse conditions.102,103 For instance, cell cycle checkpoints are soon activated to block proliferation until lesions are repaired. However, if DNA is severely damaged or unrepaired, cells remain quiescent or undergo cell death.104 Autophagy is considered both a pro-survival mechanism and a type of cell death. Therefore, once induced by DNA injury, it makes a crucial contribution in regulating cell fate.105,106 For example, many lines of evidence argue that autophagy can delay apoptotic cell death upon DNA damage by sustaining the energy demand required to support DNA repair processes,107,108 a phenomenon that concurs to the development of chemoresistance mechanisms in cancer cells.109 Conversely, in cells where DNA is unrepaired and apoptosis is defective, DNA damage-induced autophagy has been reported to contribute to cell death, thereby acting as a tumour suppressor process.110
It is worthwhile noting that cases where autophagy impairment results in DNA damage have been reported as well, leading to the assumption that the interplay might be broader, and suggesting that many molecular players could exist to biunivocally link the two processes. In particular, it has been demonstrated that the deficiency of autophagy genes, such as Beclin 1, UV irradiation resistance-associated gene (UVRAG), Atg5 and Atg7, leads to DNA damage accumulation and promotes tumourigenesis.111, 112, 113, 114, 115 In line with this, the suppression of the ULK1-interacting protein FIP200 has been reported to impair DDR, thus triggering cell death upon ionizing radiation-induced oxidative stress.116
Direct and indirect roles of autophagy in the DDR
All this evidence strongly suggests that autophagy participates, directly or indirectly, in the DDR to ROS and RNS-mediated genotoxic stress. However, how this occurs is still a matter of debate. In yeast, for example, it has been demonstrated the selective degradation of specific proteins, mostly through the so-called ‘cytoplasm to vacuole targeting’ (Cvt),117 is directly involved in: (1) inducing cell cycle arrest in G2/M phase118 by degrading proteins involved in cell cycle progression (e.g., Psd1 and Esp1); (2) optimizing dNTP production and DNA synthesis by targeting the subunit 1 of the ribonucleotide reductase complex;119 and (3) regulating the dynamics of homologous recombination by degrading the endonuclease Sae2 once catalysed the resection of DNA ends.120
In higher eukaryotes, no Cvt has ever been identified, nor has any orthologue of proteins belonging to this pathway been revealed. One of the most accredited hypotheses explaining a role of autophagy in supporting the DDR is that by degrading damaged mitochondria (mitophagy) and toxic aggregates, autophagy eliminates putative sources of ROS, reduces their levels and, if only indirectly, decreases DNA damage accumulation.114,121,122 This assumption provides a general explanation of the important role of autophagy in maintaining genomic stability that is strictly related to its tumour suppressor function.106,113,114 However, evidence supporting a direct role for autophagy in the DDR, at least in mammals, is still lacking.
Sensor proteins transduce the signal of DNA damage to autophagy
A number of works in recent years indicate that once ROS and RNS damage the DNA, the event is transduced in order to activate the DDR, and concomitantly is signalled to the autophagic pathway in order to orchestrate the response. PolyADP-ribose polymerase 1 (PARP1) is among the proteins directly linking the DDR and autophagy (Figure 4).123,124 It is a nuclear enzyme that catalyses poly-ribosylation of nuclear proteins by converting NAD+ into polymers of polyADP-ribose (PAR), and deeply participates in SSB repair, thereby regulating nuclear homeostasis. PARP1 is hyperactivated upon ROS-induced DNA damage; this leads to NAD+ consumption and ATP depletion (Figure 4). Such energetic imbalance results in the activation of autophagy via AMPK pathway (Figure 4)123,124 in order to recycle metabolic precursors for ATP and to provide energy for the DDR.

Implication of autophagy in DNA damage repair. Endogenous (e.g., dysfunctional mitochondria, top right) or exogenous (e.g., radiations or genotoxic stimuli, bottom left) sources of ROS and RNS induce DNA damage, whose primary sensors are PARP1 and ATM. Once activated by DNA breaks, PARP1 catalyses poly-ADP ribosylation of itself, as well as of other nuclear proteins, thereby leading to a massive decrease of NAD+ and to a subsequent energetic stress (bottom left). Upon DNA damage, ATM can activate p53-mediated transcription of autophagic genes (bottom right). Alternatively, cytosolic pool of ATM could be directly activated by ROS through a still unidentified mechanism and it directly induces the activation of LKB1 (centre). The issue of whether cytosolic and nuclear pool of ATM are interconnected still waits to be demonstrated. Both PARP1 and ATM signalling pathways converge on AMPK, whose activation induces the autophagic machinery to remove the main source of DNA damage and contribute to its repair through a negative feedback loop (top)
Another DNA repair protein linking the DDR to autophagy is ATM (Figure 4), a DNA damage sensor orchestrating the cell cycle with damage response checkpoints and DNA repair to safeguard the integrity of the DNA.125 It has been demonstrated that under ROS-induced cellular damage, cytosolic ATM, through the LKB1/AMPK pathway, can activate TSC2 tumour suppressor to inhibit mTORC1 and induce autophagy (Figure 4).126 These new findings integrate different stress response pathways occurring in different cellular compartments. From this perspective, ATM would be required to both initiate (nucleus) and mediate (cytosol) the DDR.
The principal regulator of DDR, however, remains p53 that, together with the other members of its family, p63 and p73, has been demonstrated to modulate many autophagic genes (Figure 4).105,127, 128, 129 In particular, p53 is very rapidly activated when DNA damage occurs, and transcriptionally activates both DNA repair and cell cycle checkpoints proteins to allow repair of DNA lesions.104 It has been recently demonstrated that the strength of the stimulus and the dynamics of the binding to DNA underlies the genes transactivated by p53. For instance, in case of inefficient repair, p53 can shift transcription from cell cycle modulators (e.g., p21waf) to genes regulating cell senescence and apoptosis (e.g., PUMA and BAX).104 In addition, autophagy genes are also induced and their transcription finely orchestrated by p53 (Figure 4). Targets of p53 are, indeed, both upstream regulators of autophagy (e.g,. PTEN, TSC2, β1, β2 and γ subunits of AMPK) and proteins directly involved in autophagosome formation (e.g., ULK1, UVRAG, ATG2, 4, 7, 10).129 It is worth noting that p63 and p73 also share some autophagic genes as transcriptional targets, such as ATG 4, 7, 10, ULK1 and UVRAG, hence suggesting a redundancy with p53.128 In fact, p53 has also been shown to function in an opposite way depending on its subcellular localization, with the cytosolic pool of p53 inhibiting, rather than activating, autophagy.130
The dual role of p53 and p53-related members, as well as of many other proteins that have been demonstrated having a role in regulating both autophagy and apoptosis, is an issue that deserves to be deeply investigated in the future. Indeed, several lines of evidence clearly indicate that the molecular pathways – from the upstream stimuli (e.g., mitochondrial ROS or genotoxic stress) through the mediators (e.g., BH3 proteins or transcription factors like p53) and the effectors of the signal (e.g., Bnip3 and AMPK) – are shared between autophagy and apoptosis.131 How can they be differently induced? Which are the signals, or how much high should be the threshold level to allow cell response being switched from stress adaptation and survival (autophagy) to cell dismantling (apoptosis)? These issues are still debated. Speculations and hypotheses based on the ‘strength’ of the signal have been reasonably done, but no direct evidence of the causative event underlying the decision by the cell to activate autophagy or apoptosis has been provided as yet.
Concluding Remarks
Several lines of evidence indicate that ROS and RNS are the upstream modulators of autophagy, likely acting at multiple levels in the process. In line with this assumption, ROS and RNS would act as the intracellular ‘alarm molecules’ of the extracellular availability of nutrients (primitive stimuli) by signalling their presence to the autophagic machinery. Through a negative feedback regulation, autophagy could be then induced to provide energy and building blocks in order to restore homeostasis and, concomitantly, remove oxidative damage. From this perspective, autophagy is required for the cell to simultaneously overcome starvation and oxidative stress conditions. Therefore, any dysfunction in this regard has been found to be involved in the onset of pathological states where a primary role for oxidative stress and/or alterations in metabolic demand (such as cancer) has also been reported. Accordingly, genetic defects of autophagic genes lead to an increased production of ROS and accumulation of damaged organelles and DNA that in turn promote metabolic reprogramming and induce tumourigenesis.
Although a number of possible mechanisms underlying the intimate interplay between oxidative stress and autophagy have been postulated, to date only a few of them have been shown to have a role in tuning autophagy. Understanding the fine molecular regulation of autophagy by ROS and RNS, as well as the tight relationship between metabolism and redox state, could therefore provide valuable information that could be useful in the future to improve anticancer treatment and develop new selective therapies.
Abbreviations
- 4E-BP1:
-
EIF4E-binding protein 1
- 53BP1:
-
p53 binding protein 1
- 8-OHdG:
-
8-hydroxydeoxyguanosine
- 8-OHG:
-
8-hydroxyguanine
- Ambra1:
-
activating molecule in Beclin1-regulated autophagy
- AMPK:
-
AMP-dependent protein kinase
- ARE:
-
antioxidant-responsive element
- Atg4:
-
autophagy related gene 4
- Atg5:
-
autophagy related gene 5
- Atg7:
-
autophagy related gene 7
- Atg13:
-
autophagy related gene 13
- ATM:
-
Ataxia telangiectasia mutated
- Bnip3:
-
Bcl-2/adenovirus E1B 19-kDa-interacting protein 3
- Cvt:
-
cytoplasm-to-vacuole targeting
- DDR:
-
DNA damage response
- DMPK:
-
myotonic dystrophy protein kinase
- DSB:
-
double-strand break
- eEF:
-
eukariotic elongation factor
- eIF:
-
eukaryotic initiation factor
- Esp1:
-
extra spindle pole bodies homologue 1
- FIP200:
-
FAK-family interacting protein of 200 kDa
- GABARAP:
-
GABA receptor-associated protein
- GSH-Px or GPx:
-
glutathione peroxidise
- Grx:
-
glutaredoxin
- GSH:
-
reduced glutathione
- GSNOR:
-
S-nitrosoglutathione reductase
- GSSG:
-
disulphide glutathione
- HKII:
-
hexokinase II
- IKKβ:
-
IκB kinase β
- JNK1:
-
c-Jun-N-terminal kinase 1
- Keap1:
-
Kelch-like ECH- associated protein 1
- LC3:
-
light chain 3
- LKB1:
-
liver kinase B1
- MRP1:
-
multidrug resistance protein 1
- mtDNA:
-
mitochondrial DNA
- mTORC1:
-
mammalian target of rapamycin complex 1
- NO:
-
nitric oxide
- NOX:
-
NADPH oxidase
- Nrf2:
-
nuclear factor erythroid 2-related factor 2
- OGG1:
-
8-oxoguanine glycosylase
- p70S6K:
-
p70 ribosomal protein S6 kinase
- PAR:
-
polyADP-ribose
- PARP1:
-
polyADP-ribose polymerase 1
- PHD:
-
prolyl hydroxylase
- PI3K:
-
phosphoinositide 3-kinase
- PINK1:
-
PTEN-induced putative kinase 1
- PMN:
-
piecemeal microautophagy
- Psd1:
-
pleckstrin and Sec7 domain containing 1
- PTEN:
-
phosphatase and tensin homologue
- PTP:
-
permeability transition pore
- RAG:
-
RAS-related GTP-binding protein
- RHEB:
-
RAS homologue enriched in brain
- RNS:
-
reactive nitrogen species
- ROS:
-
reactive oxygen species
- Sae2:
-
sporulation in the absence of SPO11 protein 2
- SOD:
-
superoxide dismutase
- SQSTM1:
-
sequestosome 1
- SSB:
-
single-strand break
- TCA:
-
tricarboxylic acid
- Trx:
-
thioredoxin
- TSC2:
-
tuberous sclerosis 2
- ULK1:
-
upstream kinase UNC51-like kinase 1
- UVRAG:
-
UV irradiation resistance-associated gene
- v-ATPase:
-
vacuolar H+-ATPase
- VPS34:
-
vacuolar protein sorting 34
References
Baudhuin P, Berleur AN, De Duve C, Wattiaux R . Tissue fractionation studies. VIII. Cellular localization of bound enzymes. Biochem J 1956; 63: 608–612.
De Duve C, Gianetto R, Appelmans F, Wattiaux R . Enzymic content of the mitochondria fraction. Nature 1953; 172: 1143–1144.
De Duve C, Berthet J . Reproducibility of differential centrifugation experiments in tissue fractionation. Nature 1953; 172: 1142.
De Duve C, Wattiaux R . Functions of lysosomes. Annu Rev Physiol 1966; 28: 435–492.
De Duve C, Baudhuin P . Peroxisomes (microbodies and related particles). Physiol Rev 1966; 46: 323–357.
Mann PJ, Quastel JH . Toxic effects of oxygen and of hydrogen peroxide on brain metabolism. Biochem J 1946; 40: 139–144.
Davies KJ . Protein damage and degradation by oxygen radicals. I. general aspects. J Biol Chem 1987; 262: 9895–9901.
Halliwell B . Biochemical mechanisms accounting for the toxic action of oxygen on living organisms: the key role of superoxide dismutase. Cell Biol Int Rep 1978; 2: 113–128.
Imlay JA, Linn S . DNA damage and oxygen radical toxicity. Science 1988; 240: 1302–1309.
Harman D . Aging: a theory based on free radical and radiation chemistry. J Gerontol 1956; 11: 298–300.
Kroemer G, Marino G, Levine B . Autophagy and the integrated stress response. Mol Cell 2010; 40: 280–293.
Yang Z, Klionsky DJ . Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol 2010; 22: 124–131.
Neufeld TP . Autophagy and cell growth—the yin and yang of nutrient responses. J Cell Sci 2012; 125: 2359–2368.
Zoncu R, Efeyan A, Sabatini DM . mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol 2011; 12: 21–35.
Nazio F, Cecconi F . mTOR AMBRA1, and autophagy: an intricate relationship. Cell Cycle 2013; 12: 2524–2525.
Fimia GM, Di Bartolomeo S, Piacentini M, Cecconi F . Unleashing the Ambra1-Beclin 1 complex from dynein chains: Ulk1 sets Ambra1 free to induce autophagy. Autophagy 2011; 7: 115–117.
Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 2009; 136: 521–534.
Cohen A, Hall MN . An amino acid shuffle activates mTORC1. Cell 2009; 136: 399–400.
Furst P, Stehle P . What are the essential elements needed for the determination of amino acid requirements in humans? J Nutr 2004; 134: 1558S–1565S.
Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L et al. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 2008; 320: 1496–1501.
Kim E, Goraksha-Hicks P, Li L, Neufeld TP, Guan KL . Regulation of TORC1 by Rag GTPases in nutrient response. Nat Cell Biol 2008; 10: 935–945.
Long X, Lin Y, Ortiz-Vega S, Yonezawa K, Avruch J . Rheb binds and regulates the mTOR kinase. Curr Biol 2005; 15: 702–713.
Shimobayashi M, Hall MN . Making new contacts: the mTOR network in metabolism and signalling crosstalk. Nat Rev Mol Cell Biol 2014; 15: 155–162.
Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM . mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science 2011; 334: 678–683.
Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM . Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010; 141: 290–303.
Duran RV, Oppliger W, Robitaille AM, Heiserich L, Skendaj R, Gottlieb E et al. Glutaminolysis activates Rag-mTORC1 signaling. Mol Cell 2012; 47: 349–358.
Duran RV, MacKenzie ED, Boulahbel H, Frezza C, Heiserich L, Tardito S et al. HIF-independent role of prolyl hydroxylases in the cellular response to amino acids. Oncogene 2013; 32: 4549–4556.
Hardie DG . AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes Dev 2011; 25: 1895–1908.
Oakhill JS, Steel R, Chen ZP, Scott JW, Ling N, Tam S et al. AMPK is a direct adenylate charge-regulated protein kinase. Science 2011; 332: 1433–1435.
Cardaci S, Filomeni G, Ciriolo MR . Redox implications of AMPK-mediated signal transduction beyond energetic clues. J Cell Sci 2012; 125: 2115–2125.
Inoki K, Ouyang H, Zhu T, Lindvall C, Wang Y, Zhang X et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell 2006; 126: 955–968.
Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell 2008; 30: 214–226.
Kim J, Kundu M, Viollet B, Guan KL . AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011; 13: 132–141.
Roberts DJ, Tan-Sah VP, Ding EY, Smith JM, Miyamoto S . Hexokinase-II positively regulates glucose starvation-induced autophagy through TORC1 inhibition. Mol Cell 2014; 53: 521–533.
da-Silva WS, Gomez-Puyou A, de Gomez-Puyou MT, Moreno-Sanchez R, De Felice FG, de Meis L et al. Mitochondrial bound hexokinase activity as a preventive antioxidant defense: steady-state ADP formation as a regulatory mechanism of membrane potential and reactive oxygen species generation in mitochondria. J Biol Chem 2004; 279: 39846–39855.
Sun L, Shukair S, Naik TJ, Moazed F, Ardehali H . Glucose phosphorylation and mitochondrial binding are required for the protective effects of hexokinases I and II. Mol Cell Biol 2008; 28: 1007–1017.
Wu R, Wyatt E, Chawla K, Tran M, Ghanefar M, Laakso M et al. Hexokinase II knockdown results in exaggerated cardiac hypertrophy via increased ROS production. EMBO Mol Med 2012; 4: 633–646.
Chance B, Sies H, Boveris A . Hydroperoxide metabolism in mammalian organs. Physiol Rev 1979; 59: 527–605.
Wagner BA, Venkataraman S, Buettner GR . The rate of oxygen utilization by cells. Free Radic Biol Med 2011; 51: 700–712.
Filomeni G, Rotilio G, Ciriolo MR . Disulfide relays and phosphorylative cascades: partners in redox-mediated signaling pathways. Cell Death Differ 2005; 12: 1555–1563.
Filomeni G, Rotilio G, Ciriolo MR . Cell signalling and the glutathione redox system. Biochem Pharmacol 2002; 64: 1057–1064.
Flohe L . Changing paradigms in thiology from antioxidant defense toward redox regulation. Methods Enzymol 2010; 473: 1–39.
Bindoli A, Fukuto JM, Forman HJ . Thiol chemistry in peroxidase catalysis and redox signaling. Antioxid Redox Signal 2008; 10: 1549–1564.
Beckman JS, Koppenol WH . Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am J Physiol 1996; 271: C1424–C1437.
Di Giacomo G, Rizza S, Montagna C, Filomeni G . Established principles and emerging concepts on the interplay between mitochondrial physiology and S-(De)nitrosylation: implications in cancer and neurodegeneration. Int J Cell Biol 2012; 2012: 361872.
Stamler JS, Singel DJ, Loscalzo J . Biochemistry of nitric oxide and its redox-activated forms. Science 1992; 258: 1898–1902.
Allen BW, Demchenko IT, Piantadosi CA . Two faces of nitric oxide: implications for cellular mechanisms of oxygen toxicity. J Appl Physiol (1985) 2009; 106: 662–667.
Jones DP . Radical-free biology of oxidative stress. Am J Physiol Cell Physiol 2008; 295: C849–C868.
Holmstrom KM, Finkel T . Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat Rev Mol Cell Biol 2014; 15: 411–421.
Filomeni G, Desideri E, Cardaci S, Rotilio G, Ciriolo MR . Under the ROS. thiol network is the principal suspect for autophagy commitment. Autophagy 2010; 6: 999–1005.
Chen Y, Azad MB, Gibson SB . Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ 2009; 16: 1040–1052.
Scherz-Shouval R, Shvets E, Elazar Z . Oxidation as a post-translational modification that regulates autophagy. Autophagy 2007; 3: 371–373.
Zhang C, Yang L, Wang XB, Wang JS, Geng YD, Yang CS et al. Calyxin Y induces hydrogen peroxide-dependent autophagy and apoptosis via JNK activation in human non-small cell lung cancer NCI-H460 cells. Cancer Lett 2013; 340: 51–62.
Levonen AL, Hill BG, Kansanen E, Zhang J, Darley-Usmar VM . Redox regulation of antioxidants, autophagy, and the response to stress: Implications for electrophile therapeutics. Free Radic Biol Med 2014; 71C: 196–207.
Huang J, Canadien V, Lam GY, Steinberg BE, Dinauer MC, Magalhaes MA et al. Activation of antibacterial autophagy by NADPH oxidases. Proc Natl Acad Sci USA 2009; 106: 6226–6231.
Murphy MP . How mitochondria produce reactive oxygen species. Biochem J 2009; 417: 1–13.
Scherz-Shouval R, Z. Elazar . ROS, mitochondria and the regulation of autophagy. Trends Cell Biol 2007; 17: 422–427.
Wang RC, Wei Y, An Z, Zou Z, Xiao G, Bhagat G et al. Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science 2012; 338: 956–959.
Oude Ophuis RJ, Wijers M, Bennink MB, van de Loo FA, Fransen JA, Wieringa B et al. A tail-anchored myotonic dystrophy protein kinase isoform induces perinuclear clustering of mitochondria, autophagy, and apoptosis. PLoS One 2009; 4: e8024.
Campello S, Strappazzon F, Cecconi F . Mitochondrial dismissal in mammals, from protein degradation to mitophagy. Biochim Biophys Acta 2014; 1837: 451–460.
Rambold AS, Kostelecky B, Elia N, Lippincott-Schwartz J . Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc Natl Acad Sci USA 2011; 108: 10190–10195.
Gomes LC, Di Benedetto G, Scorrano L . During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol 2011; 13: 589–598.
Zhang J, Ney PA . Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ 2009; 16: 939–946.
Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep 2010; 11: 45–51.
Schweers RL, Zhang J, Randall MS, Loyd MR, Li W, Dorsey FC et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci USA 2007; 104: 19500–19505.
Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M et al. Essential role for Nix in autophagic maturation of erythroid cells. Nature 2008; 454: 232–235.
Youle RJ, Narendra DP . Mechanisms of mitophagy. Nat Rev Mol Cell Biol 2011; 12: 9–14.
Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol 2010; 189: 211–221.
Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 2010; 12: 119–131.
Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ . p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 2010; 6: 1090–1106.
Vadlamudi RK, Joung I, Strominger JL, Shin J . p62, a phosphotyrosine-independent ligand of the SH2 domain of p56lck, belongs to a new class of ubiquitin-binding proteins. J Biol Chem 1996; 271: 20235–20237.
Bjorkoy G, Lamark T, Johansen T . p62/SQSTM1: a missing link between protein aggregates and the autophagy machinery. Autophagy 2006; 2: 138–139.
Ichimura Y, Kumanomidou T, Sou YS, Mizushima T, Ezaki J, Ueno T et al. Structural basis for sorting mechanism of p62 in selective autophagy. J Biol Chem 2008; 283: 22847–22857.
Strappazzon F, Nazio F, Corrado M, Cianfanelli V, Romagnoli A, Fimia GM et al. AMBRA1 is able to induce mitophagy via LC3 binding, regardless of PARKIN and p62/SQSTM1. Cell Death Differ 2014; e-pub ahead of print 12 September 2014; doi:10.1038/cdd.2014.139.
Zmijewski JW, Banerjee S, Bae H, Friggeri A, Lazarowski ER, Abraham E . Exposure to hydrogen peroxide induces oxidation and activation of AMP-activated protein kinase. J Biol Chem 2010; 285: 33154–33164.
Desideri E, Filomeni G, Ciriolo MR . Glutathione participates in the modulation of starvation-induced autophagy in carcinoma cells. Autophagy 2012; 8: 1769–1781.
Giles NM, Gutowski NJ, Giles GI, Jacob C . Redox catalysts as sensitisers towards oxidative stress. FEBS Lett 2003; 535: 179–182.
Montagna C, Di Giacomo G, Rizza S, Cardaci S, Ferraro E, Grumati P et al. S-nitrosoglutathione reductase deficiency-induced S-nitrosylation results in neuromuscular dysfunction. Antioxid Redox Signal. 2014; 21 (4): 570–587.
Sarkar S, Korolchuk VI, Renna M, Imarisio S, Fleming A, Williams A et al. Complex inhibitory effects of nitric oxide on autophagy. Mol Cell 2011; 43: 19–32.
Lopez-Rivera E, Jayaraman P, Parikh F, Davies MA, Ekmekcioglu S, Izadmehr S et al. Inducible nitric oxide synthase drives mTOR pathway activation and proliferation of human melanoma by reversible nitrosylation of TSC2. Cancer Res 2014; 74: 1067–1078.
Tripathi DN, Chowdhury R, Trudel LJ, Tee AR, Slack RS, Walker CL et al. Reactive nitrogen species regulate autophagy through ATM-AMPK-TSC2-mediated suppression of mTORC1. Proc Natl Acad Sci USA 2013; 110: E2950–E2957.
Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol 2010; 12: 213–223.
Lau A, Wang XJ, Zhao F, Villeneuve NF, Wu T, Jiang T et al. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: direct interaction between Keap1 and p62. Mol Cell Biol 2010; 30: 3275–3285.
Ichimura Y, Waguri S, Sou YS, Kageyama S, Hasegawa J, Ishimura R et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol Cell 2013; 51: 618–631.
Taguchi K, Fujikawa N, Komatsu M, Ishii T, Unno M, Akaike T et al. Keap1 degradation by autophagy for the maintenance of redox homeostasis. Proc Natl Acad Sci USA 2012; 109: 13561–13566.
Ma Q . Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol 2013; 53: 401–426.
Singh B, Chatterjee A, Ronghe AM, Bhat NK, Bhat HK . Antioxidant-mediated up-regulation of OGG1 via NRF2 induction is associated with inhibition of oxidative DNA damage in estrogen-induced breast cancer. BMC Cancer 2013; 13: 253.
Kim SB, Pandita RK, Eskiocak U, Ly P, Kaisani A, Kumar R et al. Targeting of Nrf2 induces DNA damage signaling and protects colonic epithelial cells from ionizing radiation. Proc Natl Acad Sci USA 2012; 109: E2949–E2955.
Bae SH, Sung SH, Oh SY, Lim JM, Lee SK, Park YN et al. Sestrins activate Nrf2 by promoting p62-dependent autophagic degradation of Keap1 and prevent oxidative liver damage. Cell Metab 2013; 17: 73–84.
Lee JH, Budanov AV, Park EJ, Birse R, Kim TE, Perkins GA et al. Sestrin as a feedback inhibitor of TOR that prevents age-related pathologies. Science 2010; 327: 1223–1228.
Lee JH, Budanov AV, Talukdar S, Park EJ, Park HL, Park HW et al. Maintenance of metabolic homeostasis by Sestrin2 and Sestrin3. Cell Metab 2012; 16: 311–321.
Shoji JY, Kikuma T, Arioka M, Kitamoto K . Macroautophagy-mediated degradation of whole nuclei in the filamentous fungus Aspergillus oryzae. PLoS One 2010; 5: e15650.
McGee MD, Weber D, Day N, Vitelli C, Crippen D, Herndon LA et al. Loss of intestinal nuclei and intestinal integrity in aging C. elegans. Aging Cell 2011; 10: 699–710.
Krick R, Muhe Y, Prick T, Bredschneider M, Bremer S, Wenzel D et al. Piecemeal microautophagy of the nucleus: genetic and morphological traits. Autophagy 2009; 5: 270–272.
Rello-Varona S, Lissa D, Shen S, Niso-Santano M, Senovilla L, Marino G et al. Autophagic removal of micronuclei. Cell Cycle 2012; 11: 170–176.
Wiseman H, Halliwell B . Damage to DNA by reactive oxygen and nitrogen species: role in inflammatory disease and progression to cancer. Biochem J 1996; 313 (Pt 1): 17–29.
Cooke MS, Evans MD, Dizdaroglu M, Lunec J . Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J 2003; 17: 1195–1214.
Cadet J, Delatour T, Douki T, Gasparutto D, Pouget JP, Ravanat JL et al. Hydroxyl radicals and DNA base damage. Mutat Res 1999; 424: 9–21.
Neeley WL, Essigmann JM . Mechanisms of formation, genotoxicity, and mutation of guanine oxidation products. Chem Res Toxicol 2006; 19: 491–505.
Iida T, Furuta A, Nakabeppu Y, Iwaki T . Defense mechanism to oxidative DNA damage in glial cells. Neuropathology 2004; 24: 125–130.
Lenaz G . Mitochondria and reactive oxygen species. Which role in physiology and pathology? Adv Exp Med Biol 2012; 942: 93–136.
Ciccia A, Elledge SJ . The DNA damage response: making it safe to play with knives. Mol Cell 2010; 40: 179–204.
Roos WP, Kaina B . DNA damage-induced cell death: from specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett 2013; 332: 237–248.
De Zio D, Cianfanelli V, Cecconi F . New insights into the link between DNA damage and apoptosis. Antioxid Redox Signal 2013; 19: 559–571.
Rodriguez-Rocha H, Garcia-Garcia A, Panayiotidis MI, Franco R . DNA damage and autophagy. Mutat Res 2011; 711: 158–166.
Vessoni AT, Filippi-Chiela EC, Menck CF, Lenz G . Autophagy and genomic integrity. Cell Death Differ 2013; 20: 1444–1454.
Yoon JH, Ahn SG, Lee BH, Jung SH, Oh SH . Role of autophagy in chemoresistance: regulation of the ATM-mediated DNA-damage signaling pathway through activation of DNA-PKcs and PARP-1. Biochem Pharmacol 2012; 83: 747–757.
Abedin MJ, Wang D, McDonnell MA, Lehmann U, Kelekar A . Autophagy delays apoptotic death in breast cancer cells following DNA damage. Cell Death Differ 2007; 14: 500–510.
Bordin DL, Lima M, Lenz G, Saffi J, Meira LB, Mesange P et al. DNA alkylation damage and autophagy induction. Mutat Res 2013; 753: 91–99.
Maiuri MC, Zalckvar E, Kimchi A, Kroemer G . Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol 2007; 8: 741–752.
Yue Z, Jin S, Yang C, Levine AJ, Heintz N . Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci USA 2003; 100: 15077–15082.
Liang C, Feng P, Ku B, Dotan I, Canaani D, Oh BH et al. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat Cell Biol. 2006; 8: 688–699.
Karantza-Wadsworth V, Patel S, Kravchuk O, Chen G, Mathew R, Jin S et al. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev 2007; 21: 1621–1635.
Mathew R, Kongara S, Beaudoin B, Karp CM, Bray K, Degenhardt K et al. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev 2007; 21: 1367–1381.
Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S et al. Autophagy-deficient mice develop multiple liver tumors. Genes Dev 2011; 25: 795–800.
Bae H, Guan JL . Suppression of autophagy by FIP200 deletion impairs DNA damage repair and increases cell death upon treatments with anticancer agents. Mol Cancer Res 2011; 9: 1232–1241.
Lynch-Day MA, Klionsky DJ . The Cvt pathway as a model for selective autophagy. FEBS Lett 2010; 584: 1359–1366.
Dotiwala F, Eapen VV, Harrison JC, Arbel-Eden A, Ranade V, Yoshida S et al. DNA damage checkpoint triggers autophagy to regulate the initiation of anaphase. Proc Natl Acad Sci USA 2013; 110: E41–E49.
Dyavaiah M, Rooney JP, Chittur SV, Lin Q, Begley TJ . Autophagy-dependent regulation of the DNA damage response protein ribonucleotide reductase 1. Mol Cancer Res 2011; 9: 462–475.
Robert T, Vanoli F, Chiolo I, Shubassi G, Bernstein KA, Rothstein R et al. HDACs link the DNA damage response, processing of double-strand breaks and autophagy. Nature 2011; 471: 74–79.
Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006; 10: 51–64.
Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell 2009; 137: 1062–1075.
Munoz-Gamez JA, Rodriguez-Vargas JM, Quiles-Perez R, Aguilar-Quesada R, Martin-Oliva D, de Murcia G et al. PARP-1 is involved in autophagy induced by DNA damage. Autophagy 2009; 5: 61–74.
Rodriguez-Vargas JM, Ruiz-Magana MJ, Ruiz-Ruiz C, Majuelos-Melguizo J, Peralta-Leal A, Rodriguez MI et al. ROS-induced DNA damage and PARP-1 are required for optimal induction of starvation-induced autophagy. Cell Res 2012; 22: 1181–1198.
Hurley PJ, F. Bunz . ATM and ATR: components of an integrated circuit. Cell Cycle 2007; 6: 414–417.
Alexander A, Cai SL, Kim J, Nanez A, Sahin M, MacLean KH et al. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc Natl Acad Sci USA 2010; 107: 4153–4158.
Pietrocola F, Izzo V, Niso-Santano M, Vacchelli E, Galluzzi L, Maiuri MC et al. Regulation of autophagy by stress-responsive transcription factors. Semin Cancer Biol 2013; 23: 310–322.
Kenzelmann Broz D, Spano Mello S, Bieging KT, Jiang D, Dusek RL, Brady CA et al. Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev 2013; 27: 1016–1031.
Fullgrabe J, Klionsky DJ, Joseph B . The return of the nucleus: transcriptional and epigenetic control of autophagy. Nat Rev Mol Cell Biol 2014; 15: 65–74.
Maiuri MC, Galluzzi L, Morselli E, Kepp O, Malik SA, Kroemer G . Autophagy regulation by p53. Curr Opin Cell Biol 2010; 22: 181–185.
Marino G, Niso-Santano M, Baehrecke EH, Kroemer G . Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol 2014; 15: 81–89.
Acknowledgements
We thank M Acuña Villa and MW Bennett for editorial and secretarial work. We are also grateful to Costanza Montagna and Giuseppina Di Giacomo for having provided part of unpublished results obtained on GSNOR-KO mice. The Unit of Cell Stress and Survival is supported by grants from the Danish Cancer Society (KBVU R72-A4408 to FC and KBVU R72-A4647 to GF); Lundbeck Foundation (nn. R167-2013-16100) and NovoNordisk (nn. 7559). We are also grateful to AIRC (IG2010 to FC and MFAG2011 to GF); Telethon Foundation (GGP10225); the Italian Ministry of University and Research (PRIN 2009 and FIRB Accordi di Programma 2011); and the Italian Ministry of Health (RF 2009 to FC and GR 2008).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Filomeni, G., De Zio, D. & Cecconi, F. Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ 22, 377–388 (2015). https://doi.org/10.1038/cdd.2014.150
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2014.150
This article is cited by
-
Selective autophagy in cancer: mechanisms, therapeutic implications, and future perspectives
Molecular Cancer (2024)
-
The therapeutic potential of probucol and probucol analogues in neurodegenerative diseases
Translational Neurodegeneration (2024)
-
Targeting DGAT1 inhibits prostate cancer cells growth by inducing autophagy flux blockage via oxidative stress
Oncogene (2024)
-
STUB1 exacerbates calcium oxalate-induced kidney injury by modulating reactive oxygen species-mediated cellular autophagy via regulating CFTR ubiquitination
Urolithiasis (2024)
-
Advances in the study of autophagy in breast cancer
Breast Cancer (2024)
