
Abstract
The ubiquitin proteasome pathway plays a critical role in regulating many processes in the cell which are important for tumour cell growth and survival. Inhibition of proteasome function has emerged as a powerful strategy for anti-cancer therapy. Clinical validation of the proteasome as a therapeutic target was achieved with bortezomib and has prompted the development of a second generation of proteasome inhibitors with improved pharmacological properties. This review summarises the main mechanisms of action of proteasome inhibitors in cancer, the development of proteasome inhibitors as therapeutic agents and the properties and progress of next generation proteasome inhibitors in the clinic.
Similar content being viewed by others

The ubiquitin proteasome pathway
The degradation of cellular proteins is a highly complex and tightly regulated process that plays a central role in regulating cellular function and maintaining homeostasis. The ubiquitin proteasome pathway (UPP) represents the major pathway for intracellular protein degradation. More than 80% of cellular proteins are degraded through this pathway including those involved in a broad array of processes such as cell cycle, apoptosis, transcription, DNA repair, protein quality control and antigen presentation. It has become increasingly clear that defects within this pathway are associated with a number of diseases, including cancer.
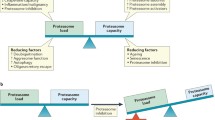
Degradation of a protein via the UPP involves two distinct and successive pathways (Fig. 1). Proteins destined for proteolysis are initially tagged by a covalently linked polyubiquitin chain in a multistep process involving the coordinated action of three enzymes—E1 (ubiquitin-activating enzyme), E2 (ubiquitin-conjugating enzyme) and E3 (ubiquitin ligase). The process begins with activation of ubiquitin by formation of a thiol ester bond between E1 and ubiquitin, in an ATP-dependent manner (McGrath et al. 1991). The activated ubiquitin moiety is then transferred to an active site residue within an E2 enzyme which shuttles ubiquitin either directly or in concert with an E3 enzyme to a lysine residue in the target protein. There are more than 30 different E2 and over 500 E3 enzymes, which work in conjunction to confer exquisite substrate specificity to the UPP (Nagy and Dikic 2010). The successive conjugation of ubiquitin moieties generates a polyubiquitin chain that acts as a signal to target the protein for degradation by the 26S proteasome. Ubiquitin conjugation can occur through linkage on one of ubiquitin’s seven acceptor lysines, resulting in ubiquitin chains of various lengths, types and functions. Polyubiquitination mediated by lysine 48 is required for substrate degradation via the proteasome whereas polyubiquitination at lysine 63 plays a role in cellular signalling.

The Ubiquitin Proteasome Pathway. Stage 1—Ubiquitin Tagging: An E1 ubiquitin-activating enzyme binds ubiquitin (Ub), which is then transferred to an E2 ubiquitin-conjugating enzyme. An E3 ubiquitin ligase subsequently recruits the target protein and mediates the transfer of ubiquitin to the protein. Stage 2—Proteolytic Degradation: Polyubiquitin chains target the protein for degradation by the 26S proteasome. Ubiquitin molecules are removed and the protein is unfolded and fed into the inner catalytic chamber of the 20S proteasome, where the protein is cleaved into small peptides using 3 main catalytic activities; chymotrypsin-like (CT-L), trypsin-like (T-L) and caspase-like (C-L)
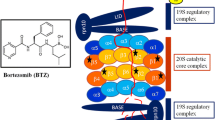
The 26S proteasome
The 26S or constitutive proteasome is found in the nucleus and cytoplasm of all eukaryotic cells. It is composed of a core 20S particle capped with two 19S structures. The 20S catalytic core is made up of 28 subunits arranged into four stacked rings, creating a central chamber where proteolysis occurs. The two outer rings are composed of 7 different α subunits, which are predominantly structural and the two inner rings are composed of 7 different β subunits, at least three of which contain catalytic sites (Groll et al. 1997). Catalytic activities of the proteasome are classified into three major categories, based upon preference to cleave a peptide bond after a particular amino acid residue. These activities are referred to as chymotrypsin-like (CT-L), trypsin-like (T-L) and caspase-like (C-L) and are associated with β5, β2 and β1 subunits respectively. The CT-L activity cleaves after hydrophobic residues, the T-L activity cleaves after basic residues and the C-L activity cleaves after acidic residues (Groll et al. 1999; Heinemeyer et al. 1997). Substrates gain access to the proteolytic chamber by binding to the 19S regulatory particle at either end of the 20S proteasome. Polyubiquitin-tagged proteins are recognised by the 19S particle, where ubiquitin is cleaved off and recycled and the target protein is unfolded and fed into the 20S catalytic chamber (Groll et al. 2000a, b; Navon and Goldberg 2001).
An alternative proteasome isoform known as the immunoproteasome can be formed in response to cytokine signalling. Interferon-gamma and tumour necrosis factor-alpha induce the expression of a different set of catalytic β-subunits and a different regulatory cap to form the immunoproteasome. Subunits β1i (LMP2), β2i (MECL1) and β5i (LMP7) replace constitutive subunits β1, β2 and β5 and the 19S regulatory cap is replaced with an 11S regulatory structure. These modifications allow the immunoproteasome to generate antigenic peptides for presentation by the major histocombatability (MHC) class 1 mediated immune response (Rock and Goldberg 1999). The expression of the immunoproteasome appears to be tissue-specific and is particularly abundant in immune-related cells.
The proteasome as a drug target
Proteasome inhibitors were initially synthesized as in vitro probes to investigate the function of the proteasome’s catalytic activity. As the essential role of the proteasome in cell function was unravelled, the possibility that proteasome inhibitors may have potential as therapeutic agents was considered. Early studies showed that proteasome inhibitors induced apoptosis in leukaemic cell lines (Imajoh-Ohmi et al. 1995; Shinohara et al. 1996; Drexler 1997) and were active in an in vivo model of Burkitt’s lymphoma (Orlowski et al. 1998). Further in vitro investigations demonstrated that proteasome inhibitors displayed a broad spectrum anti-proliferative and pro-apoptotic activity against haematological and solid tumours. While these studies established the potential of proteasome inhibitors as anti-cancer agents, many of the compounds available were limited to laboratory studies due to a relative lack of potency, specificity or stability. This led to the design of new inhibitors with more potent and selective activity.
Critical targets for proteasome inhibitors in malignant cells
Pre-clinical studies have demonstrated that malignant cells are more susceptible to the cytotoxic effects of proteasome inhibition than normal cells. The mechanisms behind the higher sensitivity of malignant cells are unclear, however, it is likely that they exploit the proteasome to regulate proliferation and anti-apoptotic pathways. Most tumour cells are highly proliferative and have an increased requirement for protein synthesis which would make them more vulnerable to proteasome inhibition. We have previously demonstrated that increased proteasome activity in leukaemic cell lines is correlated with an increased sensitivity to proteasome inhibitors (Crawford et al. 2009). In accordance with this, Nawrocki et al. (2008), have shown a direct correlation between proteasome inhibitor sensitivity and rates of translation in multiple myeloma cells. Nonetheless, proteasome inhibitors demonstrate better efficacies in certain malignancies than others and there are clearly other determinants that account for this. It is likely that the relative importance of the mechanisms depends on the tumour type. Inhibition of NFκB activity, altered degradation of cell cycle related proteins, altered pro-apoptotic and anti-apoptotic protein balance, endoplasmic reticulum stress and inhibition of angiogenesis and DNA repair have all been reported to contribute to the apoptotic affect of proteasome inhibitors in tumour cells. These mechanisms are summarised below and in Fig. 2.

Critical targets for proteasome inhibitors in malignant cells
NFκB
One of the first mechanisms of action attributed to proteasome inhibitors was inhibition of the inflammation-associated transcription factor NFκB, through stabilization of its inhibitor IκB (Traenckner et al. 1994). NFκB regulates various immune and inflammatory responses, but also plays an important role in tumourigenesis by inducing angiogenesis, proliferation, migration and suppression of apoptosis. NFκB is bound to its inhibitor IκB in the cytoplasm and is activated by proteasomal degradation of IκB. Inhibition of proteasome activity prevents degradation of IκB and subsequent activation and translocation of NFκB to the nucleus to activate downstream pathways. NFκB is constitutively active in a large proportion of advanced cancers (reviewed in Luqman and Pezzuto 2010) and has been shown to play a role in resistance to chemotherapeutic agents. It has therefore been of interest as a potential therapeutic target for some time. Proteasome inhibition was demonstrated to induce accumulation of IκB and pre-clinical and clinical studies with bortezomib showed down-regulation of transcriptional targets of NFκB (eg IL-6). However, studies are now challenging the concept that proteasome inhibitors inhibit NFκB activation. Dolcet et al. (2006) first reported that proteasome inhibitors actually activate NFκB in endometrial carcinoma cells. This work was supported by a study in MM cells demonstrating that bortezomib activates two upstream NFκB activating kinases (RIP2 and IKKβ), promotes non-proteasomal degradation of IκB and increases NFκB DNA binding (Hideshima et al. 2009). The discrepancy between the original concepts and recent discoveries of the effect of proteasome inhibition on NFκB may be in part accounted for by the fact that earlier studies demonstrated that proteasome inhibitors blocked inducible NFκB activity but did not investigate the effect of proteasome inhibition on basal NFκB activity. The role of NFκB in mediating the effects of proteasome inhibition remains controversial.
Cell cycle
Progression through the cell cycle occurs through tightly controlled interplay between cyclins and cyclin dependent kinases (CDKs). Loss of cell cycle control is a critical step in oncogenesis. The ubiquitin proteasome pathway mediates the degradation of many cell cycle regulatory proteins. There are a number of ways in which proteasome inhibitors may induce cell cycle arrest by interfering with the degradation of cyclins and cell cycle regulatory proteins in malignant cells. For example, the tumour suppressor molecule p27 is a CDK inhibitor that inhibits both cyclin D and cyclin E to negatively regulate progression through the G1/S phase of the cell cycle (Sherr and Roberts 1999). Degradation of p27 therefore promotes progression through the cell cycle and cellular levels of p27 are controlled by the ubiquitin proteasome pathway. Low p27 expression is reported to be associated with tumour progression and poor prognosis in various malignancies including lymphoma, breast, lung, colon, prostate, ovarian and brain cancer (Chu et al. 2008). The ubiquitin ligase S-phase kinase protein 2 (Skp-2) targets p27 for degradation by the proteasome. High expression of Skp-2 has been reported in some cancers including non-small cell lung carcinoma (Inui et al. 2003) and it is thought to contribute to enhanced degradation of p27. Proteasome inhibition has been shown to result in a downregulation of Skp-2 and accumulation of p27 resulting in cell cycle arrest (Hussain et al. 2009).
Regulation of apoptosis
Apoptosis is regulated by the opposing activities of pro-apoptotic and anti-apoptotic molecules. Cancer cells often have disregulated apoptotic signalling pathways which give malignant cells a survival advantage and can confer resistance to chemotherapeutic agents. The proteasome is involved in the control of apoptosis by modulating the levels of pro- and anti-apoptotic factors. Inhibition of proteasome activity results in an upregulation of pro-apoptotic factors such as p53, Bax and NOXA, while reducing levels of anti-apoptotic proteins such as Bcl-2 and IAP (inhibitor of apoptosis) proteins (McConkey and Zhu 2008). Proteasome inhibitors have been demonstrated to induce apoptosis in numerous malignant cell types when used as a single agent and induce sensitivity to other chemotherapeutic agents in combination.
p53
The tumour suppressor p53 is a critical regulator of apoptosis induced by DNA damage and transforming oncogenes. It is often inactivated in malignant cells, leading to tumour progression and drug resistance. Hyperactivation of MDM2, an E3 ligase for p53, and subsequent proteasomal degradation is a common mechanism for downregulation of p53 activity (Momand et al. 1998). Proteasome inhibition results in accumulation of p53 and has been shown to activate p53 downstream target genes such as p21, Fas ligand, PUMA and Bax (Williams and McConkey 2003). Proteasome inhibitors have been demonstrated to induce p53-dependent apoptosis in malignancies such as renal cell carcinoma cell lines (Vaziri et al. 2009), colon cancer (Ding et al. 2007), melanoma and multiple myeloma (Qin et al. 2005). However, this appears to be cell type dependent as bortezomib has been shown to act independently of p53 in B-cell lymphoma (Strauss et al. 2007) and glioma cells (Yin et al. 2005).
Endoplasmic reticulum stress
The endoplasmic reticulum (ER) plays an important role in protein folding and maturation. Unfolded or misfolded proteins are directed to the proteasome for degradation. Proteasome inhibition results in the accumulation and aggregation of misfolded proteins in the ER resulting in ER stress, which in turn elicits the unfolded protein response (UPR). The UPR is primarily a pro-survival response to reduce the accumulation of unfolded proteins and restore ER function. However, if protein accumulation is persistent, as in the case of proteasome inhibition, signalling switches from pro-survival to pro-apoptotic. Malignant cells generally have higher protein synthesis rates than their normal counterparts, thus making them more prone to protein aggregation and perhaps more sensitive to proteasome-inhibitor induced apoptosis. For example, multiple myeloma cells constitutively express ER stress survival factors to function as antibody-secreting cells. Inhibition of proteasome activity has been demonstrated to induce pro-apoptotic ER stress in numerous cancer cells including, multiple myeloma (Obeng et al. 2006), pancreatic (Nawrocki et al. 2005), head and neck cancer (Fribley and Wang 2006) and non small cell lung carcinoma (Morgillo et al. 2010).
Inhibition of angiogenesis
The success of proteasome inhibitors in multiple myeloma has been attributed not only to direct effects on myeloma cells but also the effects of proteasome inhibitors on the tumour microenvironment, including anti-angiogenic effects. Proteasome inhibitors were initially shown to have an indirect effect on angiogenesis by decreasing the secretion of vascular endothelial growth factor (VEGF) (Hideshima et al. 2001; Nawrocki et al. 2002). Subsequently, direct anti-proliferative effects of bortezomib on vascular endothelial cells were demonstrated using a range of functional assays including chemotaxis, adhesion to fibronectin and capillary formation (Roccaro et al. 2006). More recently, Tamura et al. (2010), have shown that bortezomib potently inhibits cell growth of vascular endothelial cells by suppressing the G2/M transition of the cell cycle and increasing the permeability, thus displaying a unique mechanism of action as a vascular targeting drug.
FOXM1
The oncogenic transcription factor Forkhead Box M1 (FoxM1) has recently emerged as a new target for proteasome inhibitors. FoxM1 induces the expression of genes involved in cell cycle progression and is overexpressed in many cancers including non small cell lung carcinoma (Gialmanidis et al. 2009), breast cancer (Bektas et al. 2008), colorectal cancer (Yoshida et al. 2007), glioblastomas (Liu et al. 2006), pancreatic carcinomas (Wang et al. 2007) and squamous cell carcinomas (Gemenetzidis et al. 2009). Conversely, FoxM1 is lowly expressed in normal non-dividing cells and for this reason it provides an attractive target for anticancer drugs. Gartel and colleagues demonstrated that proteasome inhibitors suppress FoxM1 transcriptional activity and expression in a number of malignant cell lines (Bhat et al. 2009; Gartel 2010) and may contribute to the anti-cancer effect of proteasome inhibitors.
DNA repair
Recent research has demonstrated a role for the UPP in regulating DNA repair through mechanisms such as nucleotide excision repair, post-replication repair and homologous recombination (reviewed in Motegi et al. 2009). Proteasome inhibitors may impact these pathways through the depletion of available nuclear ubiquitin; inhibition of proteasome activity results in an accumulation of nondegraded polyubiquitinated proteins leading to a reduction in the amount of free ubiquitin in the cell. This depletion of free ubiquitin results in a loss of monoubiquitinated histones in the nucleus and consequently impairs DNA-damage responses. Proteasome inhibitors have been shown to sensitize tumour cells to various anti-tumour therapies such as radiation, camptothecin and topoisomerase inhibitors, all of which induce DNA damage.
Proteasome inhibitors in clinical development
Bortezomib, a peptide boronate inhibitor, was the first proteasome inhibitor to enter clinical practice. Growing evidence from translational research and clinical trials with bortezomib established the proteasome as a novel and legitimate therapeutic target. However, there are restrictions to the use of bortezomib including dose limiting toxicity, particularly peripheral neuropathy, limited activity in solid tumours, resistance and intravenous administration. This prompted the development of a new generation of structurally distinct proteasome inhibitors. In addition to bortezomib, there are currently five proteasome inhibitors in clinical development, representing three different structural classes—peptide boronic acids, peptide epoxyketones and β-lactones (Fig. 3). These inhibitors bind either reversibly or irreversibly to catalytic sites within the proteasome. An overview of bortezomib along with second generation proteasome inhibitors currently in clinical development is presented below (Table 1).

Chemical structures of proteasome inhibitors in clinical development
Bortezomib
Bortezomib is a reversible inhibitor primarily acting on the CT-L activity of the proteasome. This compound was chosen from a panel of boronic acid analogues that were screened against the National Cancer Institute’s (NCI’s) panel of 60 cancer cell lines, on the basis of its potency and cytotoxicity (Adams et al. 1999). Bortezomib was further investigated in vitro and in vivo in various tumour types and showed early indications of activity in non-small cell lung cancer, prostate cancer, multiple myeloma and mantle cell and follicular non-Hodgkin’s lymphoma. Bortezomib proved to be particularly active against multiple myeloma and Phase I through to Phase III clinical trials quickly confirmed its efficacy in this disease (Orlowski et al. 2002; Richardson et al. 2003; Jagannath et al. 2004; Richardson et al. 2005). Bortezomib was approved for third-line treatment of multiple myeloma by the FDA in 2003 (Kane et al. 2006) and expanded to first-line treatment in 2008; approval for use in mantle cell lymphoma came in 2006 (Kane et al. 2007). While bortezomib exhibits considerable activity as a single agent, its main use is as a means to overcome resistance and induce sensitivity to a variety of other chemotherapeutic agents. Bortezomib has been combined with doxorubicin, thalidomide, melphalan, dexamethasone, and lenalidomide, among others and has generally been successfully combined with other agents without increased toxicity. There are currently over 200 active clinical trials involving bortezomib, the majority of which are investigating novel combination therapy for haematological malignancies, particularly multiple myeloma and lymphoma. There are also trials involving a wide variety of advanced solid tumours, most notably non small cell lung carcinomas, renal cell carcinoma, and breast cancer; further information on these trials can be found at www.clinicaltrials.gov. Although bortezomib exhibited anti-tumour activity in multiple malignancies in pre-clinical studies, clinical trials in solid tumours have proved disappointing to date. The reasons for this are unclear but it is postulated that the dosing regimes may be sub-optimal for the treatment of solid tumours (Bennett and Kirk 2008) and has prompted interest in the possibility that second-generation proteasome inhibitors may have a broader clinical efficacy.
Carfilzomib
Epoxomicin, a member of the epoxyketone family of natural peptide proteasome inhibitors, inhibits proteasome activity through a unique mechanism, by binding to both the hydroxyl and amino groups of the catalytic site threonine residue (Groll et al. 2000a, b). Carfilzomib (formerly PR-171) is an epoxomicin-based proteasome inhibitor, with improved pharmaceutical properties. Unlike bortezomib, carfilzomib binds irreversibly to the CT-L subunit, leading to more sustained proteasome inhibition. In preclinical studies carfilzomib was shown to exhibit equal potency but greater selectivity than bortezomib for the CT-L activity in vitro and in vivo studies demonstrated anti-tumour activity, tolerability and dosing flexibility in several xenograft models (Kuhn et al. 2007; Demo et al. 2007). Carfilzomib has also been shown to act synergistically with histone deacetylase inhibitors in vitro in lymphoma and leukaemia (Fuchs et al. 2009; Dasmahapatra et al. 2010). Results from Phase I studies in patients with haematological malignancies demonstrated that it was well tolerated and may exhibit less peripheral neuropathy than bortezomib (O’Connor et al. 2009). Carfilzomib is currently in Phase III trials in multiple myeloma and Phase I trials for acute myeloid leukaemia, acute lymphoblastic leukaemia, chronic lymphocytic leukaemia and solid tumours.
NPI-0052
NPI-0052, also known as Salinosporamide A, is a β-lactone compound derived from the marine bacterium Salinospora tropica (Macherla et al. 2005) and is structurally related to the lactacystin-derived proteasome inhibitor Omuralide. In contrast to bortezomib which is a slowly reversible inhibitor, NPI-0052 binds irreversibly to all three catalytic activities of the proteasome. While bortezomib is administered intravenously, NPI-0052 has the advantage of being orally bioactive. Initial in vitro studies established the effectiveness of this compound in multiple myeloma cell lines, including those that were resistant to bortezomib (Chauhan et al. 2005). Pre-clinical studies have also shown activity of NPI-0052 in Waldenstrom’s macroglobulinemia (Roccaro et al. 2008), acute leukaemias (Miller et al. 2007, 2009), chronic lymphocytic leukaemia (Ruiz et al. 2006) and prostate (Baritaki et al. 2009), pancreatic (Sloss et al. 2008) and colon cancer (Cusack et al. 2006). Animal tumour model studies demonstrated reduced tumour growth without significant toxicity (Chauhan et al. 2005; Singh et al. 2010a). Phase I trials of NPI-0052 in advanced solid tumours, refractory lymphoma and non small cell lung carcinoma are currently ongoing.
MLN9708
MLN9708 like bortezomib is also a boron containing peptide proteasome inhibitor and was selected from a panel of inhibitors based on having a biochemical profile distinct from that of bortezomib. MLN9708 hydrolyses immediately in plasma to its biologically active form MLN2238. MLN2238 displays similar potency and selectivity for the CT-L proteasome subunit, however, it has a substantially shorter half-life than bortezomib which may improve tissue distribution. Cell viability studies revealed a strong anti-proliferative effect on a variety of tumour cell lines and in vivo studies have demonstrated efficacy in human prostate xenograft, colon cancer and lymphoma models where both intravenous and oral dosing were effective (Kupperman et al. 2010). This compound is currently being evaluated in Phase I studies in patients with lymphoma and non-haematological malignancies and in Phase I/II trials for multiple myeloma.
CEP-18770
CEP-18770 is a next-generation boronic acid-based proteasome inhibitor and in common with bortezomib it is a reversible inhibitor, primarily of the CT-L activity. CEP-18770 was demonstrated to induce apoptosis in multiple myeloma cell lines and primary myeloma cells, while displaying a favourable cytotoxicity profile towards normal cells (Piva et al. 2008; Dorsey et al. 2008). Its anti-tumour activity was demonstrated in several animal tumour models and it has been shown to demonstrate marked anti-myeloma effects in combination with Bortezomib and melphalan (Sanchez et al. 2010). CEP-18770 has completed early Phase I trials for solid tumours and non-Hodgkin’s and is currently being evaluated in Phase I/II trials for multiple myeloma.
ONX0912
ONX0912 (formerly PR-047) is a novel orally available analogue of the proteasome inhibitor carfilzomib. Carfilzomib, in common with bortezomib, is administered intravenously, however, proteasome inhibitor therapy requires twice weekly dosing and therefore an orally available inhibitor would be advantageous. ONX0912 has demonstrated similar anti-tumour activity to carfilzomib in vitro in cell lines and primary cells and enhanced the anti-myeloma activity of bortezomib, lenolidomide and histone deacetylase inhibitors; animal models of multiple myeloma, non-Hodgkin’s lymphoma and colorectal cancer demonstrated reduced tumour progression and prolonged survival (Zhou et al. 2009; Roccaro et al. 2010; Chauhan et al. 2010). A Phase I trial of ONX0912 in advanced solid tumours is currently recruiting.
Immunoproteasome inhibitors
A novel approach that is looking promising is the use of proteasome inhibitors to specifically inhibit catalytic activities of the immunoproteasome. Immunoproteasomes are constitutively expressed in immune tissues and expressed at a much lower level in other cell types. Thus targeting immunoproteasomes confers a certain amount of specificity and provides an opportunity to overcome toxicities associated with proteasome inhibition, such as peripheral neuropathy and gastrointestinal effects. A number of immunoproteasome specific inhibitors have recently been described and exhibit encouraging pre-clinical activity in haematological malignancies. PR-924 is a tripeptide epoxyketone related to carfilzomib. It exhibits 100-fold greater selectivity for β5i than carfilzomib and was demonstrated to inhibit the growth of multiple myeloma cell lines and primary tumour cells and inhibited tumour growth in animal models without significant toxicity (Singh et al. 2010b). The immunoproteasome inhibitor ISPI-101 is a peptide aldehyde which preferentially inhibits the β1i subunit. ISPI-101 induced accumulation of polyubiquitinated proteins and pro-apoptotic proteins and inhibited proliferation in in vitro models of haematological malignancies (Kuhn et al. 2009). At the time of writing this review there are no clinical trials of immunoproteasome inhibitors in progress, however, it is likely that the encouraging pre-clinical data on PR-924 and ISPS-101 will form the basis for future clinical evaluation of these compounds.
Summary
The UPP is now widely appreciated for its critical role in regulating diverse cellular processes and the clinical efficacy of bortezomib has established the proteasome as a therapeutic target. Although the precise mechanisms of action of proteasome inhibitors are not yet fully defined, there are a number of pathways that appear to be important in the selectivity for malignant cells. While bortezomib treatment results in impressive response rates in multiple myeloma and other haematological malignancies, its success in solid tumours has been disappointing. Whether this limited activity in solid tumours is specific to bortezomib or whether it extends to proteasome inhibitors as a class is as yet unknown. A number of chemically distinct next generation proteasome inhibitors have been developed which display unique mechanisms of action against the proteasome. The clinical development of these proteasome inhibitors along with the development of novel drug combinations should help to address some of the key issues with bortezomib and offer possibilities for future anti-cancer therapies.
References
Adams J, Palombella VJ, Sausville EA, Johnson J, Destree A, Lazarus DD, Maas J, Pien CS, Prakash S, Elliott PJ (1999) Proteasome inhibitors: a novel class of potent and effective antitumor agents. Cancer Res 59:2615–2622
Baritaki S, Chapman A, Yeung K, Spandidos DA, PAlladino M, Bonavida B (2009) Inhibition of epithelial to mesenchymal transition in metastatic prostate cancer cells by the novel proteasome inhibitor NPI-0052: pivitol roles of Snail repression and RKIP induction. Oncogene 28:3573–3585
Bektas N, Haaf A, Veeck J, Wild PJ, Luscher-Firzlaff J, Hartmann A, Knuchel R, Dahl E (2008) Tight correlation between expression of the Forkhead transcription factor FoxM1 and Her2 in human breast cancer. BMC Cancer 8:42
Bennett MK, Kirk CJ (2008) Development of proteasome inhibitors in oncology and autoimmune diseases. Curr Opin Drug Discov Devel 11:616–625
Bhat UG, Halasi M, Gartel AL (2009) FoxM1 is a general target for proteasome inhibitors. PLoS One 4e6593
Chauhan D, Catley L, LiG PK, Hideshime T, Velanker M, Mitsiades C, Mitsiades N, Yasui H, Letai A, Ovaa H, Berkers C, Chao TH, Neuteboom ST, Richardson O, Palladino MA, Anderson KC (2005) A novel orally active proteasome inhibitor induces apoptosis in multiple myeloma cells with mechanisms distinct from bortezomib. Cancer Cell 8:407–419
Chauhan D, Singh AV, Aujay M, Kirk CJ, Bandi M, Ciccarelli B, Raje N, Richardson P, Anderson KC (2010) A novel orally active proteasome inhibitor ONX 0912 triggers in vitro and in vivo cytotoxicity in multiple myeloma. Blood 116:4906–4915
Chu IM, Hengst L, Joyce M (2008) The CDK inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. Nat Rev Cancer 8:253–267
Crawford LJ, Windrum P, Magill L, Melo JV, McCallum L, McMullin MF, Ovaa H, Walker B, Irvine AE (2009) Proteasome proteolytic profile is linked to Bcr-Abl expression. Exp Hematol 37:357–366
Cusack JC, Liu R, Xia L, Chao TH, Pien C, Niu W, Palombella VJ, Neuteboom ST, Palladino MA (2006) NPI-0052 enhances tumoricidal response to conventional cancer therapy in a colon cancer model. Clin Cancer Res 12:6758–6764
Dasmahapatra G, Lembersky D, Kramer L, Fisher RI, Friedberg J, Dent P, Grant S (2010) The pan-HDAC inhibitor vorinostat potentiates the activity of the proteasome inhibitor carfilzomib in human DLBCL cells in vitro and in vivo. Blood 115:4478–4487
Demo SD, Kirk CJ, Aujay MA, Buchholz TJ, Dajee M, Ho MN, Jiang J, Laidig GJ, Lewis ER, Parlati F, Shenk KD, Smyth MS, Sun CM, Vallona MK, Woo TM, Molineaux CJ, Bennett MK (2007) Antitumor activity of PR-171, a novel irreversible inhibitor of the proteasome. Cancer Res 67:6383–6391
Ding WX, Ni HM, Chen X, Yu J, Zhang L, Yin XM (2007) A coordinated action of Bax, PUMA and p53 promotes MG-132—induced mitochondria activation and apoptosis in colon cancer cells. Mol Cancer Ther 6:1062–1069
Dolcet X, Llobet D, Encinas M, Pallares J, Cabero A, Schoenenberger JA, Comella JX, Matias-Guiu X (2006) Proteasome inhibitors induce death but activate NF-kappaB on endometrial carcinoma cell lines and primary culture explants. J Biol Chem 281:22118–22130
Dorsey BD, Iqbal M, Chatterjee S, Menta E, Bernardini R, Bernareggi A, Cassara PG, D’Arasmo G, Ferretti E, De Munari S, Oliva A, Pezzoni G, Allievi C, Strepponi I, Ruggeri B, Ator MA, Williams M, Mallamo JP (2008) Discovery of a potent, selective and orally active proteasome inhibitor for the treatment of cancer. J Med Cham 51:1068–1072
Drexler HC (1997) Activation of the cell death program by inhibition of proteasome function. Proc Nat Acad Sci 94:855–860
Fribley A, Wang CU (2006) Proteasome inhibitor induces apoptosis through induction of endoplasmic reticulum stress. Cancer Biol Ther 5:745–748
Fuchs O, Provaznikova D, Marinov I, Kuzelova K, Spika I (2009) Antiproliferative and proapoptotic effects of proteasome inhibitors and their combination with histone deacetylase inhibitors on leukemia cells. Cardiovasc Hematol Disord Drug Targets 9:62–77
Gartel AL (2010) A new target for proteasome inhibitors: FoxM1. Expert Opin Investig Drugs 19:235–242
Gemenetzidas E, Bose A, Riaz AM, Chaplin T, Young BD, Ali M, Sugden D, THurlow JK, Cheong SC, Teo SH, Wan H, Waseem A, PArkinason EK, Fortune F, Teh MT (2009) FoxM1 upregulation is an early event in human squamous cell carcinoma and it is enhanced by nicotine during malignant transformation. PLoS One 4:e4849
Gialmanidas IP, Bravou V, Amanetopoulou SG, Varakis J, Kourea H, Papadaki H (2009) OVerexpression of hedgehog pathway molecules and FOXM1 in non-small cell lung carcinomas. Lung Cancer 66:64–74
Groll M, Dtizel L, Lowe J, Stock D, Bochtler M, Wolf DH, Huber R (1997) Structure of the 20S proteasome from yeast at 2.4 A resolution. Nature 386:463–471
Groll M, Heinemeyer W, Jager S, Ulrich T, Bochtler M, Wolf DH, Huber R (1999) The catalytic sites of 20S proteasomes and their role in subunit maturation: A mutational and crystallographic study. Proc Nat Acad Sci 96:10975–10983
Groll M, Bajorek M, Kohler A, Moroder L, Rubin DM, Huber R, Glickman MH, Finley D (2000a) A gated channel into the proteasome core particle. Nat Struct Biol 7:1062–1067
Groll M, Kim KB, Kairies N, Huber R, Crews CM (2000b) Crystal structure of epoxomicin: 20S proteasome reveals a basis for selectivity of alpha, beta—epoxyketone proteasome inhibitors. J Am Chem Soc 122:1237–1238
Heinemeyer W, Fischer M, Krimmer T, Stachon U, Wolf DH (1997) The active sites of the eukaryotic 20S proteasome and their involvement in subunit precursor processing. J Biol Chem 272:25200–25209
Hideshima T, Chauhan D, Podar K, Schlossman RL, Richardson P, Anderson KC (2001) Novel therapies targeting the myeloma cell and its bone marrow microenvironment. Semin Oncol 28:607–612
Hideshima T, Ikeda H, Chauahan D, Okawa Y, Raje N, Podar K, Mitsiades C, Munshi NC, Richardson PG, Carrasco RD, Anderson KC (2009) Bortezomib induces canonical nuclear factor-kappaB activation in multiple myeloma cells. Blood 114:1046–1052
Hussain AR, Ahmed M, Ahmed SO, Al-Thari S, Khan AS, Razack S, Platanias LC, Al-Kuraya KS, Uddin S (2009) Proteasome inhibitor MG-132 mediated expression of p27Kip1 via S-phase kinase protein 2 degradation induces cell cycle couple apoptosis in primary effusion lymphoma cells. Leuk Lymphoma 50:1204–1213
Imajoh-Ohmi S, Kawaguchi T, Sugiyama S, Tanaka K, Omura S, Kikuchi H (1995) Lactacystin, a specific inhibitor of the proteasome, induces apoptosis in human monoblast U937 cells. Biochem Biophys Res Cummun 217:1070–1077
Inui N, Kitagawa K, Miwa S, Hattori T, Chida K, Nakamura H, Kitagawa M (2003) High expression of Cks1 in human non-small cell lung carcinoma. Biochem Biophys Res Commun 303:978–984
Jagannath S, Barlogie B, Berenson J, Siegel D, Irwin D, Richardson PG, Niesvizky R, Alexanian R, Limentani SA, Alsine M, Adams J, Kauffman M, Esseltine DL, Schenkein DP, Anderson KC (2004) A Phase 2 study of two doses of bortezomib in relapsed or refractory myeloma. Br J Haematol 127:165–172
Kane RC, Farell AT, Sridhara R, Pazdur R (2006) United States Food and Drug Administration approval summary: bortezomib for the treatment of progressive multiple myeloma after one prior therapy. Clin Cancer Res 12:2955–2960
Kane RC, Dagher R, Farell A, Ko CW, Sridhara R, Justice R, Pazdur R (2007) Bortezomib for the treatment of mantle cell lymphoma. Clin Cancer Res 13:5291–5294
Kuhn DJ, Chen Q, Voorhees PM, Strader JS, Shenk KD, Sun CM, Demo SD, Bennett MK, van Leeuwen FW, Chanan-Khan AA, Orlowski RZ (2007) Potent activity of carfilzomib, a novel, irreversible inhibitor of the ubiquitin proteasome pathway against preclinical models of multiple myeloma. Blood 110:3281–3280
Kuhn DJ, Hunsucker SA, Chen Q, Voorhees PM, Orlowski M, Orlowski RZ (2009) Targeted inhibition of the immunoproteasome is a potent strategy against models of multiple myeloma that overcomes resistance to conventional drugs and nonspecific proteasome inhibitors. Blood 113:4667–4776
Kupperman E, Lee EC, Cao Y, Bannerman B, Fitzgerald M, Berger A, Yu J, Yang Y, Hales P, Bruzzese F, Liu J, Blank J, Garcia K, Tsu C, Dick L, Fleming P, Yu L, Manfredi M, Rolfe M, Bolen J (2010) Evaluation of the proteasome inhibitor MLN9708 in preclinical models of human cancer. Cancer Res 70:1970–1980
Liu M, Dai B, Kang SH, Ban K, Huang FJ, Lang FF, Aldape KD, Xie TX, Pelloski CE, Xie K, Sawaya R, Huang S (2006) FoxM1B is overexpressed in human glioblastomas and critically regulates the tumorigenicity of glioma cells. Cancer Res 66:3593–3602
Luqman S, Pezzuto JM (2010) NFkappaB: a promising target for natural products in cancer chemoprevention. Phytother Res 24:949–963
Macherla VR, Mitchell SS, Manam RR, Reed KA, Chao TH, Nicholson B, Deyanat-Yazdi G, Mai B, Jensen PR, Fenical WF, Neuteboom ST, Lam KS, Palladino MA, Potts BC (2005) Structure-activity relationaship studies of salinosporamide A (NPI-0052), a novel marine-derived proteasome inhibitor. J Med Chem 48:3684–3687
McConkey DJ, Zhu K (2008) Mechanisms of proteasome inhibitor action and resistance in cancer. Drug Resist Update 11:164–179
McGrath P, Jentsch S, Varshavsky A (1991) UBA 1: an essential yeast gene encoding ubiquitin-activating enzyme. EMBO J 10:227–236
Miller CP, Ban K, McConkey DJ, Munsell M, Palladino M, Chandra J (2007) NPI-0052, a novel proteasome inhibitor, induces caspase-8 and ROS-dependent apoptosis alone and in combination with HDAC inhibitors in leukemia cells. Blood 110:267–277
Miller CP, Rudra S, Keating MJ, Wierda WG, Palladino M, Chandra J (2009) Caspase-8 dependent histone acetylation by a novel proteasome inhibitor, NPI-0052: a mechanism for synergy in leukemia cells. Blood 113:4289–4299
Momand J, Jung D, Wilczynski S, Niland J (1998) The MDM2 gene amplification database. Nucleic Acids Res 26:3452–3459
Morgillo F, D’Aiuto E, Troiani T, MArtinelli E, Cascone T, De Palma R, Orditura M, De Vita F, Ciardiello F (2010) Antitumor activity of bortezomib in human cancer cells with acquired resistance to anti-epidermal growth factor receptor tyrosine kinase inhibitors. Lung Cancer Epub ahead of print
Motegi A, Murakawa Y, Takeda S (2009) The vital link between the ubiquitin-proteasome pathway and DNA repair: impacy on cancer therapy. Cancer Lett 283:1–9
Nagy V, Dikic I (2010) Ubiquitin ligase complexes: from substrate selection to conjugational specificity. Biol Chem 391:163–169
Navon A, Goldberg AL (2001) Proteins are unfolded on the surface of the ATPase ring before transport into the proteasome. Mol Cell 8:1339–1349
Nawrocki ST, Bruns CJ, Harbison MT, Bold RJ, Gotsch BS, Abbruzzese JL, Elliott P, Adams J, McConkey DJ (2002) Effects of the proteasome inhibitor PS-341 on apoptosis and angiogenesis in orthotopic human pancreatic tumor xenografts. Mol Cancer Ther 1:1243–1253
Nawrocki ST, Carwe JS, Pino MS, Highshaw RA, Dunner K, Huang P, Abbruzzese JL, McConkey DJ (2005) Bortezomib sensitizes pancreatic cells to endoplasmic reticulum stress-mediated apoptosis. Cancer Res 65:11658–11666
Nawrocki ST, Carew JS, Maclean KH, Courage JF, Huang P, Houghton JA, Cleveland JA, Giles FJ, McConkey DJ (2008) Myc regulates aggresome formation, the induction of Noxa, and apoptosis in response to the combination of bortezomib and SAHA. Blood 112:2917–2926
O’Connor OA, Stewart AK, Vallone M, Molineaux CJ, Kunkel LA, Gerecitano JF, Orlowski RZ (2009) A phase 1 dose escalation study of the safety and pharmacokinetics of the novel proteasome inhibitors carfilzomib (PR-171) in patients with hematologic malignancies. Clin Cancer Res 15:7085–7091
Obeng EA, Carlson LM, Gutman DM, Harrington WJ, Lee KP, Boise LH (2006) Proteasome inhibitors inude a terminal unfolded protein response in multiple myeloma cells. Blood 107:4907–4916
Orlowski RZ, Eswara JR, Lafond-Walker A, Grever MR, Orlowski M, Dang CV (1998) Tumor growth inhibition induced by a murine model of human Burkitt’s lymphoma by a proteasome inhibitor. Cancer Res 59:2615–2622
Orlowski RZ, Stinchcombe TE, Mitchell BS, Shea TC, Baldwin AS, Stahl S, Adams J, Esseltine DL, Elliott PJ, Pien CS, Guerciolini R, Anderson JK, Depcit-Smaith ND, Bhagat R, Lehman MJ, Novick SC, O’Connor OA, Soignet SL (2002) Phase I trial of the proteasome inhibitor PS341 in patients with refractory haematological malignancies. J Clin Oncol 20:4420–4427
Piva R, Ruggeri B, Williams M, Costa G, Tamagno I, Ferrero D, Giai V, Coscia M, Peola S, Massaia M, Pezzoni G, Allievi C, Pescalli N, Cassin M, di Giovine S, Nicoli P, de Feudis P, Strepponi I, Roato I, Ferracini R, Bussolati B, Camussi G, Jones-Bolin S, Hunter K, Zhao H, Neri A, Palumbo A, Berkers C, Ovaa H, Bernareggi A, Inghirami G (2008) CEP-18770: A novel, orally active proteasome inhibitor with a tumor-selective pharmacological profile competitive with bortezomib. Blood 111:2765–2775
Qin JZ, Ziffra J, Stennett L, Bodner B, Bonish BK, Chaturvedi V, Bennett F, Pollock PM, Trent JM, Hendrix MJ, Rizzo P, Miele L, Nickoloff BJ (2005) Proteasome inhibitors trigger NOXA-mediated apoptosis in melanoma and myeloma cells. Cancer Res 65:6282–6293
Richardson PG, Barlogie B, Berenson J, Singhal S, Jaganath S, Irwin D, Rajkumar SV, Srkalovic G, Alsina M, Alexanian R, Siegel D, Orlowski RZ, Kuter D, Limentani SA, Lee S, Hideshima T, Esseltine DL, Kauffman M, Adams J, Schenkein DP, Anderson KC (2003) A phase 2 study of bortezomib in relapsed, refractory myeloma. N Engl J Med 348:2609–2617
Richardson PG, Sonneyeld P, Schuster MW, Irwin D, Stadtmauer EA, Facon T, Harousseau JL, Ben-Yehuda D, Lonial S, Goldschmidt H, Reece D, San-Miguel JF, Blade J, Boccadoro M, Kavenagh J, Dalton WS, Boral AL, Esseltine DL, Porter JB, Schenkein D, Anderson KC (2005) Assessment of Proteasome Inhibition for Extending Remission (APEX) Investigators: bortezomib or high dose dexamethasone for relapsed multiple myeloma. N Engl J Med 352:2487–2498
Roccaro AM, Hideshima T, Raje N, Kumar S, Ishitsuka K, Yasui H, Shiraishi N, Ribatti D, Nico B, Vacca A, Dammacco F, Richardson PG, Anderson KC (2006) Bortezomib mediates antiangiogenesis in multiple myeloma via direct and indirect effects on endothelial cells. Cancer Res 66:184–191
Roccaro AM, Leleu X, Sacco A, Jia X, Melham M, Moreau AS, Ngo HT, Runnels J, Azab A, Azab F, Burwick N, Farag M, Treon SP, Palladino MA, Hideshima T, Chauhan D, Anderson KC, Ghobrial IM (2008) Dual targeting of the proteasome regulates survival and homing in Waldenstrom macroglobulinemia. Blood 111:4752–4763
Roccaro AM, Sacco A, Aujay M, Ngo HT, Azab AK, Quang P, Maiso P, Runnels J, Anderson KC, Demo S, Ghobrial IM (2010) Selective inhibition of chymotrypsin-like activity of the immunoproteasome and constitutive proteasome in Waldenstrom macroglobulinemia. Blood 115:4051–4060
Rock KL, Goldberg AL (1999) Degradation of cell proteins and the generation of MHC class 1-presented peptides. Annu Rev Immunol 17:739–799
Ruiz S, Krupnik Y, Keating M, Chandra J, Pallidino M, McConkey D (2006) The proteasome inhibitor NPI-0052 is a more effective inducer of apoptosis than bortezomib in lymphocytes from patients with chronic lymphocytic leukemia. Mol Cancer Ther 5:1836–1843
Sanchez E, Li M, Steinberg JA, Wang C, Shen J, Bonavida B, Li ZW, Chen H, Berenson JR (2010) The proteasome inhibitor CEP-18770 enhances the anti-myeloma activity of bortezomib and melphelan. Br J Haematol 148:569–581
Sherr CJ, Roberts JM (1999) CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev 13:1501–1512
Shinohara K, Tomoika M, Nakano H, Tone S, Ito H, Kawahima S (1996) Apoptosis induction resulting from proteasome inhibition. Biochem J 317:385–388
Singh AV, Palladino MA, Lloyd GK, Potts BC, Chauhan D, Anderson KC (2010a) Pharmacodynamic and efficacy studies of the novel proteasome inhibitor NPI-0052 (marizomib) in a human plasmacytoma xenograft murine model. Br J Haematol 149:550–559
Singh AV, Bandi M, Aujay MA, Kirk CJ, Hark DE, Raje N, Chauhan D, Anderson KC (2010b) PR-924, a selective inhibitor of the immunoproteasome subunit LMP-7, blocks multiple myeloma cell growth both in vitro and in vivo. Br J Haematol 152:155–163
Sloss CM, Wang F, Liu R, Xia L, Houston M, Ljungman D, Palladino MA, Cusack CJ (2008) Proteasome inhibition activates epidermal growth factor receptor (EGFR) and EGFR-independent mitogenic kinase signaling pathways in pancreatic cancer cells. Clin Cancer Res 14:5116–5123
Strauss SJ, Higginbottom K, Juliger S, Maharaj L, Allen P, Schenkein D, Lister TA, Joel SP (2007) The proteasome inhibitor bortezomib acts independently of p53 and induces cell death via apoptosis and mitotic catastrophe in B-cell lymphoma cell lines. Cancer Res 67:2783–2790
Tamura D, Arao T, Tanaka K, Kaneda H, Matsumoto K, Kudo K, Aomatsu K, Fujita Y, Watanabe T, Saijo N, Kotani Y, NishimuraY NK (2010) Bortezomib potentially inhibits cellular growth of vascular endothelial cells through suppression of G2/M transition. Cancer Sci 101:1403–1408
Traenckner EB, Wilk S, Baeuerle PA (1994) A proteasome inhibitor prevents activation of NF-kappa B and stabilizes a newly phosphorylated form of i-kappa B-alpha that is still bound to NF-kappa B. EMBO J 15:5433–5441
Vaziri SA, Grabowski DR, Hill J, Rybicki LR, Burk R, Bukowski RM, Ganapathi MK, Gamapathi R (2009) Inhibition of proteasome activity by bortezomib in renal cancer cells is p53 and VHL independent. Anticancer Res 29:2961–2969
Wang Z, Banerjee S, Kong D, Li Y, Sarker FH (2007) Downregulation of Forkhead Box M1 transcription factor leads to the inhibition of invasion and angiogenesis of pancreatic cancer cells. Cancer Res 67:8293–8300
Williams SA, McConkey DJ (2003) The proteasome inhibitor bortezomib stabilizes a novel active form of p53 in human LNCaP-Pro5 cancer cells. Cancer Res 63:7338–7344
Yin D, Zhou H, Kumagai T, Liu G, Ong JM, Black KL, Koeffler HP (2005) Proteasome inhibitor PS-341 causes cell growth arrest and apoptosis in human glioblastoma multiforme (GBM). Oncogene 24:344–354
Yoshida Y, Wang IC, Yoder HM, Davidson NO, Costa RH (2007) The forkhead box M1 transcription factor contributes to the development and growth of mouse colorectal cancer. Gastroenterology 132:1420–1431
Zhou HJ, Aujay MA, Bennett MK, Dajee M, Demo SD, Fang Y, Ho MN, Jiang J, Kirk CJ, Laidig GJ, Lewis ER, Lu Y, Muchamuel T, Parlati F, Ring E, Shenk KD, Shields J, Shwonek PJ, Stanton T, Sun CM, Sylvain C, Woo TM, Yang J (2009) Design and synthesis of an orally bioavailable and selective peptide epoxyketone proteasome inhibitor (PR-047). J Med Chem 52:3028–3038
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Crawford, L.J., Walker, B. & Irvine, A.E. Proteasome inhibitors in cancer therapy. J. Cell Commun. Signal. 5, 101–110 (2011). https://doi.org/10.1007/s12079-011-0121-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12079-011-0121-7