

Abstract
Background/Aim: Breast cancer is the most common type of cancer among women worldwide, and about 57,000 new cases are expected for the Brazilian population in 2015. Elucidation of protein expression and modification is essential for the biological understanding, early diagnosis and therapeutics of breast cancer. The main objectives of the study are comparison between the proteome of tumor and paired non-tumor breast cancer tissues, describing all identified proteins, highlighting the ones most differentially expressed and comparing the data with existing literature. Materials and Methods: The five paired samples from patients with invasive ductal carcinoma were analyzed by 2-DE and MS. Results: We collected 161 identified spots corresponding to 110 distinct proteins. Forty-three differentially-expressed spots were common to at least two samples, and the ten proteins with the highest-fold changes were CASPE, ENOG, TPM1, CAPG, VIME, TPM3, TRFE, PDIA6, WDR61 and PDIA3. Metabolic enzymes and proteins with binding functions were the most representative functional classes of proteins with increased and decreased expression in tumor tissue respectively. Conclusion: Taking the fold change as a parameter, we point to future targets to be studied by functional methods in a search for biomarkers for initiation and progress of breast cancer.
- Breast cancer
- proteome
- differential expression
- biomarker identification
Abbreviations: 2DE: Two-dimensional electrophoresis; MS: mass spectrometry; IDC: invasive ductal carcinoma; MALDI: matrix-assisted laser desorption/ionization; TOF: time of flight; PMF: peptide mass fingerprinting; BCT: breast cancer tissue; NBT: non-tumor breast tissue; PAGE: polyacrylamide gel electrophoresis; IEF: isoelectric focusing; pI: isoelectric point; MM: molecular mass; ER: estrogen receptor.
Breast cancer is the most common type of cancer in women worldwide. Survival rates vary according to world regions ranging from around 80% in North America, Sweden and Japan, 60% in middle-income countries and up to 40% in low-income countries (1). In Brazil, about 57,120 new cases are expected in 2015 (2). Despite all efforts to control the disease, incidence is still rising in most countries and it is expected to keep rising in the next 20 years (3). Invasive ductal carcinoma (IDC) is the most representative (65-80%) breast cancer type (4).
Proteins are the major conductors of genetic information and the molecules that can better-reflect the functional status of the cell. That is why the elucidation of protein expression and modification is essential in breast cancer biology understanding, also for cancer risk predictors, early diagnosis biomarkers and therapeutic targets identification (5). Proteomics, working together with genomics, might refine current breast cancer classifications and management protocols (6).
Despite the development of alternative techniques, two-dimensional electrophoresis (2-DE) continues to be widely employed for differential expression studies, so the number of published articles using 2-DE continues to be steadily high, confirming its status as a central technique in proteomics. Through its high resolving power, 2-D gels allow for separation of different isoelectic point (pI) and molecular mass (MM) protein. Coupled with mass spectrometry (MS), 2-DE remains the mature technology and sometimes thegold-standard depending on the study goal (7, 8). The MALDI-TOF/MS profiling techniques are commonly used to determine different markers and mechanisms involved in cancer development. MALDI-TOF/MS peptide mass fingerprinting (PMF) is a fast and cheap protein identification method (9). This method has been used in several recently published biomarker determination studies on breast cancer (10-12).
Patient's clinicopathological data.
The main objective of this study is the comparison between the proteome of tumor and non-tumor breast cancer tissue, describing all identified proteins, highlighting the ones most differentially expressed and comparing data with the current literature.
Materials and Methods
Sample collection and clinical evaluation. Matched pairs of sporadic breast cancer tissue (BCT) and non-tumor breast (NTB) tissue were obtained from five female patients (average age=58.6±12.3 years) diagnosed with IDC without any neoadjuvant therapy. Samples were collected during surgical intervention at the Hospital Nossa Senhora das Graças in Curitiba, Brazil, and immediately stored at −80°C. Non-tumor tissues were removed from the opposite quadrant of tumor area and then used after confirmation of normality by a pathologist. The study was approved by the Ethics Committee and patients signed an informed consent to participate in this piece of research. Table I shows the patients' information.
Protein extraction and quantification. Tissue fragments were solubilized in lysis buffer containing 7M urea, 2M thiourea, 4% CHAPS, 40 mM Tris and 0.2% PMSF and the cells were homogenized by means of an electric tissue disruptor. The total lysate was centrifuged at 15,300 × g for 5 min to remove debris. The protein concentration was determined by the Bradford assay (13).
2D-PAGE. This step was made as previously described (14). Briefly, 1mg total protein was solubilized in the rehydration buffer. Passive rehydration of 13-cm linear IPG (immobilized pH gel) strips (pH 4-7) (GE Helthcare, Milwaukee, USA) occurred at room temperature (RT) for 16h. Isoelectric focusing (IEF) was performed according to the program suggested by the manufacturer. After IEF, strips were equilibrated for 15min with DTT and then for another 15min with iodoacetamide. SDS-PAGE was performed with 10% gels using Hoefer SE 600 Ruby (GE Healthcare,Milwaukee, USA) at 11°C for 30min at 15mA and 4.5h at 30mA. Gels were fixed for 1h and stained with Coomassie G-250 for 16h. Gels were produced in triplicate for each sample. No depletion method was used for removing plasma proteins.
Image analysis. Stained gels were scanned with ImageScanner™ II (GE Healthcare, Milwaukee, USA) and analyzed with ImageMaster™ 2D Platinum v6.0 (GE Healthcare, Milwaukee, USA). The parameters used to detect spots by the software were: area min - 5; smooth - 3; and saliency - 25. Triplicates were cropped to frame the same cluster of spots across samples and one representative gel was used to create a match-set. Logarithmic ratios of spots with precise matching were considered for normalization at software analysis. Only spots with expression levels above 2 folds were considered for statistical analysis. The ImageMaster™ software was also used to perform Student's t-test to select differential spots (p<0.05).
Mass Spectrometry and protein identification. The spots were manually excised from the gels and were de-stained in 50% acetonitrile and 25 mM ammonium bicarbonate. Dehydration was performed in two rounds of 100 μl of acetonitrile for 5 min. The supernatant was discarded and gels were dried at room temperature. Afterwards, the gel pieces were rehydrated in 20 μl of solution containing 40 mM ammonium bicarbonate, 10% acetonitrile and 15 ng/μl trypsin (Sequencing Grade Modified Trypsin; Promega, Fitchburg, Wisconsin, USA) for 30 min on ice bath. The digestion occurred at 37°C for 16-20h. To improve tryptic peptides removal, supernatant was removed to a 0.5 ml tube and gel fragment was submerged in 20 μl of trifluoroacetic acid (TFA) 5% and acetonitrile 50% solution for 30 min under agitation. The supernatant was added to the same 0.5ml tube and submitted to SPD1010 Integrated SpeedVac™ (Thermo Scientific,Waltham, USA) for 30 min (RT) for peptide concentration. Concentrated peptide extracts were dissolved (1:1) in a matrix solution (50% acetonitrile, 0.1% trifluoroacetic acid and saturated α-cyano-4-hydroxycinnamic acid) and spotted onto scout MTP MALDI ion source 384 target (Bruker Daltonics, Billerica, Massachusetts, USA). Tryptic peptide masses were obtained from MALDI-TOF/TOF/MS/MS AutoflexII (Bruker Daltonics, Billerica, Massachusetts, USA) in positive reflector mode; 20kV acceleration voltage; 150ns interval between the laser pulse and voltage application; and acquisition range of 800-3,200Da. External calibration was performed with a mix containing ACTH1-17; ACTH 1-24; ACTH 18-39; angiotensin I and II and somatostatin. Mass specters were analyzed through FlexControl 2.0 software (Bruker Daltonics, Billerica, Massachusetts, USA) using trypsin autolysis picks (842.50 Da and 2211.10 Da) for internal calibration. The PMF and/or MS/MS data were compared against thetheoretical molecular masses and isoelectric point from UniProtKB/Swiss-Prot annotation, using the Matrix Science (MASCOT) database. For protein identification, the taxonomic category was restricted to Homo sapiens, maximum 200 ppm of mass tolerance and one missed enzymatic cleavage for trypsin. A number of fixed (carbamidomethylation of cysteine residues) and variable modifications (methionine oxidation) were included as search parameters. The threshold value for p<0.05 was 56.
Results
Comparative analysis of breast cancer tissues and matched non-tumor breast tissue samples. The paired samples obtained from five patients diagnosed with IDC were compared in order to observe individual differences in expression profile (Figure 1). The 30 gels from non-tumor tissues showed a mean of 630±100.6 spots while the 30 ones from tumors presented a mean of 970±285.9 spots. The average of differentially spots detected and proteins identified in the five paired samples was 85.2 and 53.4 (62.7%), respectively. The results showed 161 identified spots corresponding to 110 distinct proteins: 33 spots (24 distinct proteins) with decreased expression and 128 spots (91 distinct proteins) with increased expression in tumor breast tissue (Supplementary data). Five proteins, Vimentin (VIME), Peroxirredoxin-2 (PRDX2), Glutathione S-transferase P (GSTP1), Alpha-soluble NSF attachment protein (SNAA) and Alpha-1-antitrypsin (A1AT) were identified with increased expression in both tissues but not at the same corresponding spots, reducing the total number to 110 proteins.
From this number, we selected the seven differentially expressed spots that were common to all five samples, 9 present in four samples, 10 found in three samples and 17 present in two samples, amounting to 43 protein spots (Figure 2 and Table II).
Table III shows the ten proteins that displaying the highest fold change mean all over the study (CAPG appears twice), the regulation in breast cancer tissue (if up- or down-regulated) related to non-tumor breast tissue, the number of samples they were identified and the functional class of each one.
Table IV shows a comparison between our data and the current literature, taking into account the main proteins discussed by other authors.
Functional classification. Proteins were classified according to their biological functions in 12 classes, according to Pucci-Minafra et al. (15, 16): (1) cytoskeleton and associated proteins; (2) metabolic enzymes; (3) molecular chaperones/heat shock proteins; (4) proteolysis regulation; (5) de-toxification and redox proteins; (6) cell growth and proliferation regulators; (7) proteins with binding functions; (8) membrane-associated proteins with multiple activities; (9) proteins with extracellular activity; (10) protein biosynthesis; (11) nucleotide biosynthesis; (12) other functions (Figure 3).
Discussion
In the present study we first aimed of all to describe the differentially expressed proteins by comparing matched samples of IDC and their normal counterpart (tissue collected in the opposite side of the tumor and confirmed as normal by a pathologist) from five patients. Table II shows all proteins identified according to the number of samples they are found in (two to five samples). Through this approach, we noticed that only five proteins were identified in the five samples. This was expected considering the high heterogeneity of breast cancer and the use of a broad histological classification (IDC), without considering the immunohistochemical markers ER, PR, HER2, Ki-67 and others for sub-classification. After this observation, we tried to extract the main group considering the difference in expression. Using the fold change as a parameter, the 10 proteins with the highest-fold change (Table IV) are discussed focusing on their role in BC and their potential as diagnostic biomarkers when compared to normal tissue.
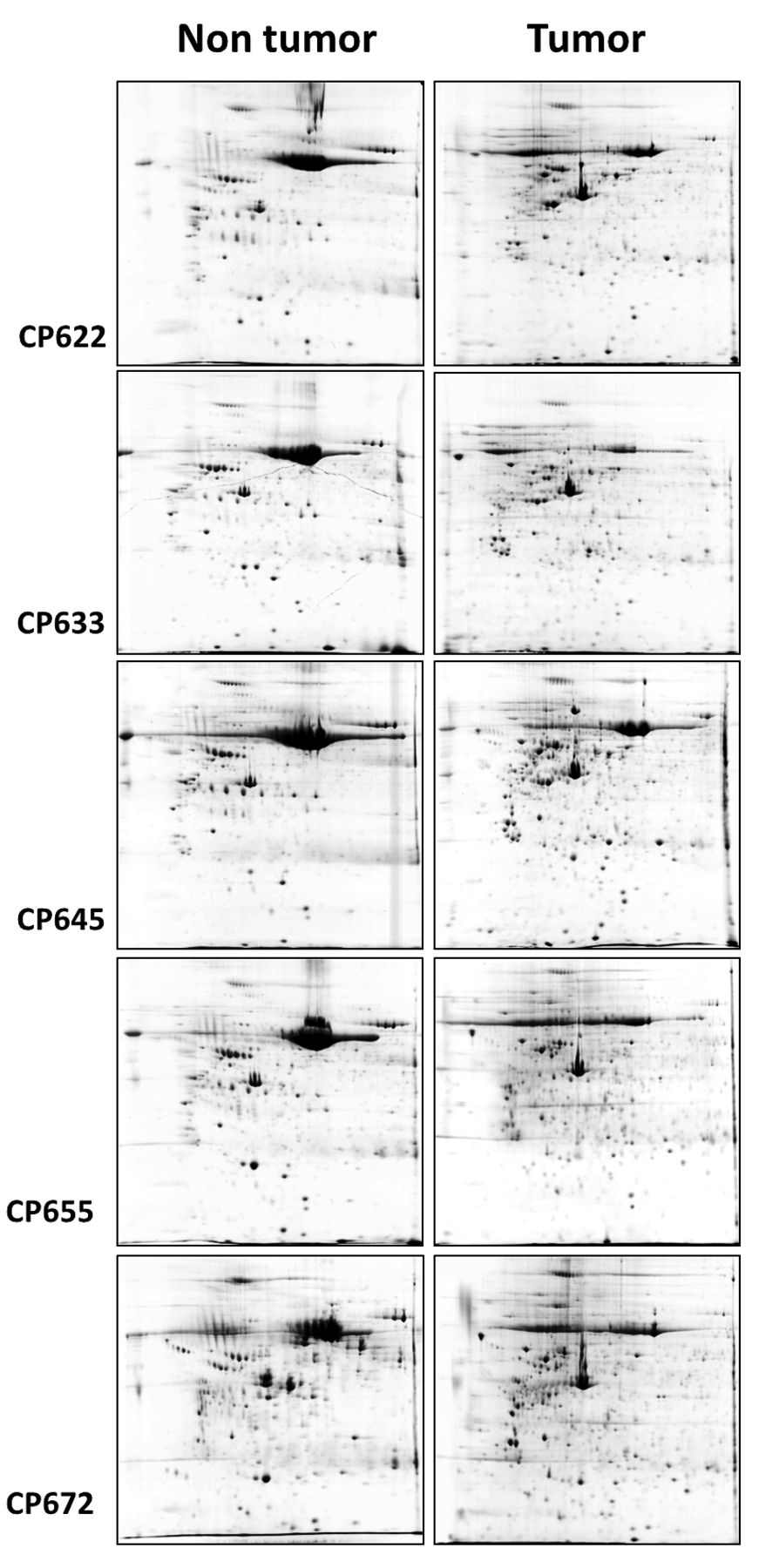
Reference 2-DE gels of non-tumor and tumor tissues from each sample.
Caspase-14 (CASPE) is a non-apoptotic caspase involved in epithelial differentiation and highly expressed in embryonic tissues. Caspase-14 is unique among the caspase family proteins and it may have a different cellular role than related proteins. A 2005 study associated a higher CASPE expression to high-grade tumors and it seems to be a very early alteration in the pathogenesis of breast cancer (53). CASPE overexpression in breast cancer was also found in two other studies (22, 54). In 2011, a study identified CASP14 as a probable transcriptional target of Gata-3, and the overexpression of CASPE in human breast cancer cells has the same effect of GATA3 overexpression, which significantly delayed tumor growth (55).
Differentially expressed proteins according to the number of samples they were identified.
Enolase molecules are dimers composed by three distinct subunits (α, β and γ). When used in cancer characterization and diagnosis, γγ and αγ-enolase are referred to as neuron-specific enolases. Several immunostaining studies reported a high proportion of γ-enolase (ENOG) staining cells detected in some breast carcinomas while no positive staining was observed in non-tumor breast tissue (59-61). ENOG high expression is described as a characteristic of neuroendocrine breast cancer, but this study showed an increased ENOG expression in IDC in contrast to NTB.

CP645 reference 2-DE gel from (A) non tumor and (B) tumor tissue.
The CAPG is a gelsolin-related actin-binding protein that is involved in the control of actin-based cell motility and phagocytosis (70, 71), once it is involved in cytoplasm. When it is found in cell nucleus its function is unknown, but CAPG is the only member of this family that accumulates at this location (63). We found two spots identified as CAPG, both overexpressed in BCT. CAPG has an increased expression in breast cancer, especially in metastasizing ones, than in normal breast epithelium (63). Kang et al. (2010) also found a higher CAPG expression in breast tumor tissue (35).
Vimentin (VIME) is a type-III intermediate filament that maintains cell and tissue integrity (72) and is highly expressed in high-grade ductal breast carcinoma or in tumors with low ER levels (50). Overexpression of vimentin is interpreted as a sign of epithelial-mesenchymal transition, related to tumor cell dedifferentiation, growth, invasion and metastasis in numerous types of cancer, including breast cancer (51). Proteomic studies have reported several vimentin spots at the same sample gel (15, 52), probably due to post-translational modifications. Our study found vimentin spots with an increased expression and also, in some cases, with a decrease in BCT. Despite its correlation to malignancy, the role of VIME in cancer regulation is still unclear.
Increased expression of TRFE in breast tumor patients was observed by immunohistochemistry of BCT (17) and by nipple aspirate fluid and serum analysis (73). The primary role of TRFE is to transport iron, derived from dietary absorption and from macrophage recycling, safely around the body and deliver it to growing cells (74). Free iron can be toxic, promoting free radical formation that results in oxidative damage to tissues (75) and also causes lipid peroxidation by converting hydroperoxides into reactive peroxyl and alkoxyl radicals (76). The decreased expression in BCT found in this study may be due to the balance between cell and serum proteins. Considering that normal tissue presents low cell amount, serum proteins may be more evident when comparing to the high cell amount found in breast cancer tissue (15).
WDR61 is a member of the Paf1 complex, which is well-known for promoting RNA polymerase II transcription elongation and transcription-coupled histone modifications. Paf1 complex also plays a role in gene expression and silencing, RNA maturation, DNA repair, cell cycle progression and prevention of disease states in higher eukaryotes (77). A recent study reported that WDR5, another WD repeat-containing protein, is required for MYC to broadly associate with target genes in vivo and to drive tumorigenesis (78). There is a lack of information about its expression in breast cancer and in cancer in general. Nevertheless, due to its participation in several cellular processes, WDR61 should be observed with caution.
Representative proteins based on their fold change.
The last two groups of proteins, tropomyosins (TPMs) and disulfide isomerases (PDIs), were studied by our group to validate the preliminary data of this study (26, 38). Quantitative real-time PCR using a Sybr green protocol was performed in order to analyze the mRNA expression level. Both studies used a very similar sample of the present study, collected at the same center and comparing breast cancer and normal breast tissue. Related to TPMs, a reduced mRNA expression of TPM1, increased expression of TPM3 and no difference in TPM4 expression in BCT were seen, corroborating the proteomic analysis just for TPM3, since our data pointed to an increase in TPM1, TPM3 and TPM4 expression in BCT, as well as found in other proteomic studies (15, 25). Changes in the tropomyosin expression contribute to the re-arrangement of microfilaments, morphological alterations and cell motility (79). Frequently, a decreased expression is associated with tumor development (23, 24). As TPMs have several isoforms (high molecular weight and low molecular weight) generated by alternative splicing, more studies are necessary regarding their regulation levels, e.g. TPM1 transcript was shown as a potential target of mir-21 in a breast cancer cell line study (80).
The PDIs fully corroborated the proteomic data, showing a higher expression of both genes and an association with lymph node metastasis and tumor grade for PDIA3, suggesting their potential use as an aggressiveness marker (38). PDIs act in disulfide bond formation and isomerization. They also play a role as chaperone, by binding polypeptide chains and assisting in the correct protein folding as well as inhibiting unfolding substrates aggregation (81). These proteins are associated to several types of cancers (82-84).
Functional classes. According to functional classification, the main classes overexpressed in BCT were metabolic enzymes (21%) and cytoskeleton and associated proteins (20%), while proteins with binding functions (33%) and cytoskeleton and associated proteins (17%) were the major classes of proteins with decreased expression in BCT. We noticed that proteins related to cytoskeleton have great changes in expression levels between tumor and non-tumor tissues and it is known that they play an important role in tumor invasiveness. Proteins with binding functions was the most expressive class with decreased expression in BCT, suggesting that proteins with these functions may be essential in tumorigenesis. These data are difficult to compare against those of other authors since the methods used to classify them are not homogeneous. Despite the lack of regularity, it is important to describe the functional classes in an effort to cluster the big amount of proteins identified by high throughput methods.
Comparison between the data of the present study and current literature.

{kind=link}
{kind=link}
{kind=link}
Identified proteins distributed according to their biological function and increased (a) or decreased (b) expression in analyzed tumor samples.
Comparison with literature data. Table IV shows a comparison between our data and the data from literature. Several proteins share the same type of regulation expression, the majority displaying an increased expression in BCT. On the contrary, there were some proteins with conflicting data, without sufficient information or in disagreement with the information described by other authors, as discussed below.
In two spots, fibrinogen beta chain expression was around four folds decreased in BCT. Although observed in a different spot location, FIBB was shown as increased in breast tumor (18). The participation of fibrinogen, fibrin and their degradation products is described in blood clotting, inflammation, angiogenesis and metastasis (19, 85), although their role is not sufficiently known yet.
IPYR was found to have an increased expression in samples of gastric cancer showing a relation with cell migration but not with invasion, and it may be a useful poor prognosis marker for gastric cancers (20). Increased expression was also observed in prostate cancer (21). In a comparative study using breast cancer and healthy mammary cell lines, no significant differential expression was observed (22). A recent study showed that knockdown of PPA1 (IPYR coding gene) decreased colony formation and viability of MCF7 cells (86).
ATPB was found overexpressed in our study but it has conflicting data about expression in breast cancer. There are evidences of overexpression (27, 28) and decreased expression in BCT (29) as well as no expression change observed in a cell-line study (22).
Alpha-1 antitrypsin was found overexpressed while haptoglobin showed decreased expression in BCT in the present study. Increased expression of acute-phase protein A1AT in cancer patients' sera may be due to a non-specific inflammatory host response to tumor (87). A serum proteomic analysis suggested that A1T1 may be a useful serum biomarker for early-stage breast cancer screening and diagnosis due to overexpression in patients' sera (11), but two comparative proteomic studies found A1AT decreased in contrast to NTB (18, 30). Hamrita et al. found two A1AT and four HPT isoforms in the sera of patients with infiltrating ductal carcinoma (87).
IF4A1 and HNRPF were identified at the same spot. IF4A1 is a subunit from translation regulator eIF4F complex and it has a helicase function (88). Recent studies pointed IF4A1 as a promising therapeutic target in ER-negative breast cancer (65, 66) Heterogeneous nuclear ribonucleoprotein F (HNRPF) plays a role in regulation of alternative splicing events by participating in hnRNP complexes. Other members of these complexes were found overexpressed (29, 34) but also decreased (89) in BCT. There is a lack of information on this specific protein expression in breast cancer. A colon cancer study also reported overexpression in contrast to adjacent non-tumor tissue (67). Probably because the two proteins showed up together at the same spot, this was recognized as overexpressed in our analysis.
Cytochrome b-c1 complex subunit 1 has been associated with the generation of reactive oxygen species (ROS) and dysregulation may cause several problems including cancer (90). Hepatocellular cancer showed increased QCR1 expression (68).
Two proteasome subunit alpha types were identified as overexpressed in BCT, PSA3 and PSA5. These alpha subunits are involved in 20S proteasome composition that, together with the 19S, forms the 26S proteasome, which is involved in several biological processes, including cell cycle progression, apoptosis and DNA repair (64). Despite their importance as proteasome subunits, there is little information about expression changes of the two subunits found in this study.
The function of TCPE in cancer is unknown, but this protein subunit participates in the TCP1 ring complex (TRiC). Evidences strongly suggest that TRiC plays a key role in cell cycle progression and that it could be implicated in tumor development (91-93). A recent study also showed overexpression in nitric oxid-stimulated NIH/3T3 cells (62).
Conclusion
We conclude that 2DE coupled to MALDI-TOF/MS is a very useful proteomic approach to discriminate proteins with differential expression in breast cancer. With the description of the most important ten proteins, taking the fold change as a parameter, we point to future targets to be studied by several validation and functional methods in search of good biomarkers in early stages and progress of breast cancer.
Acknowledgments
The Authors would like to acknowledge Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) and Fundação Araucária for the financial support. Theyalsorecognizethe Universidade Federal do Paraná, Hospital Nossa Senhora das Graças, Curitiba/BR, for providing structure and professional assistance.
- Received June 9, 2015.
- Revision received July 26, 2015.
- Accepted August 7, 2015.
- Copyright© 2015, International Institute of Anticancer Research (Dr. John G. Delinasios), All rights reserved