


Abstract
Background/Aim: There exists considerably large interpatient variability in pharmacokinetic exposure of high dose melphalan in multiple myeloma patients with hematopoietic stem-cell transplantation. In this study, we aimed to evaluate the potential impacts of CYP3A4*1B (rs2940574) and CYP3A5*3 (rs776746) variations on pharmacokinetic properties of melphalan and clinical outcomes in multiple myeloma (MM) patients. Patients and Methods: Genotypes of CYP3A4*1B (rs2940574) and CYP3A5*3 (rs776746) were determined by validated gene-specific real-time PCR (RT-PCR) assays using DNA samples from 108 MM patients; plasma concentrations of melphalan at different time points were quantified using liquid chromatography-tandem mass spectrometry (LC-MS/MS). Results: CYP3A4*1B/*1B and CYP3A5*3/*3 carriers appeared to have a short median progression-free survival time and a higher maximum melphalan plasma concentration than non-carriers [792 vs. over 950 days, p=0.08; 9.91 (2.67, 34.03) vs. 8.66 (4.46, 17.61) mg/l, p=0.039]. Conclusion: CYP3A4*1B/*1B and CYP3A5*3/*3 variations might influence melphalan therapy in MM patients through yet-to-be-identified mechanisms.
Multiple myeloma (MM) accounts for 1% of all cancers and is the most common hematologic malignancy, only secondary to lymphoma (1). MM is characterized by a pattern of recurrent relapses and remains incurable due to resistance and relapse in almost all patients, of which over 50% die within 5 years of diagnosis (1, 2). Despite the development of novel proteasome inhibitors, immunomodulatory drugs and epigenetic agents, hematopoietic autologous stem cell transplant (ASCT) with high dose intravenous melphalan (HDM) remains the current standard of care and the most effective treatment for transplant-eligible patients (3-6). Melphalan is a bifunctional alkylating agent that forms interstrand, intrastrand, or DNA-protein crosslinks (7). It has been used for the treatment of a variety of malignancies including MM and ovarian carcinoma for more than 60 years. Notably, the treatment outcome of autoHSCT-HDM varies considerably among MM patients, and about 20% of patients even have melphalan resistant myeloma as demonstrated by a progression-free survival (PFS) of less than 12 months, much shorter than the PFS of most MM patients (8, 9). It is arguably believed that variability in melphalan pharmacokinetics (PK) at least partially contributes to both excessive adverse effects (such as oral mucositis, gastrointestinal toxicity, and infection) and inadequate dosing, thus resulting in treatment failure (10-14). Previous studies have shown that PK of melphalan appears to be dose-independent and there is up to 10-fold variation in plasma melphalan area under the concentration-time curve (AUC) between patients receiving the standard 200 mg/m2 of melphalan (15),
Defining the PK behaviors of melphalan remains very challenging to date. Melphalan undergoes spontaneous hydrolysis in the plasma, with “inactive” monohydroxyl and dihydroxyl metabolites appearing quickly after melphalan administration, suggesting that melphalan does not require metabolic activation (16-19). Interestingly, melphalan is found to extensively bind to plasma proteins (mainly serum albumin), which decreases the hydrolysis of melphalan (8,19). As such, only 5% of melphalan is eliminated through hydrolysis (18). It is known that a considerable portion of melphalan (up to 40%) is excreted intact in urine, indicative of a potential association between renal function with PK of melphalan (12, 15). Such association is further supported by previous observations that the toxicity and perturbation in PK properties of melphalan, such as the half-life time and AUC, increased when it was administered at high doses to MM patients with renal insufficiency (20). However, it has been reported that together with lean body weight and hematocrit, renal function (i.e., creatinine clearance) only accounts for 20% of interpatient variability in melphalan PK, indicating that there could be other clinical factors contributing to the vast interpatient variability in its PK properties (18, 21). For example, previous studies have identified at least three metabolites of melphalan other than monohydroxyl and dihydroxyl melphalan (the products of hydrolysis) in the plasma of mice and dogs, implying that there exist certain metabolic pathways in these animals, even though these pathways may not be the major routes of melphalan elimination (22, 23).
CYP3A4 and CYP3A5 are cytochrome P450 enzymes involved in phase I metabolism of many chemotherapeutic agents including alkylating agents, which may have direct or indirect impacts on the PK properties of these agents (24-26). For example, it has been reported that patients carrying CYP3A5*3/*3 (CYP3A5 non-expressers, with 6986A→G polymorphisms) exhibited significantly higher dose-corrected trough concentrations of tacrolimus (24, 25). In a prospective randomized clinical trial (HOVON-24), MM patients carrying CYP3A5*3/*3 and CYP3A4*1B/*1B (290A→G; a variant in a close linkage with the CYP3A5*3 allele) had significantly poorer progression-free survival (PFS) and overall survival (OS) after high-dose melphalan and VAD therapies than non-carriers of CYP3A5*3/*3 and CYP3A4*1B/*1B, as demonstrated in univariate survival analyses (26, 27). Additionally, CYP3A4*1B SNP was found to be statistically significantly associated with OS and PFS in a cohort of MM patients with ASCT and bortezomib-cyclophosphamine-dexamethasone therapy (28).
Taken together, these findings demonstrate that even though high doses of melphalan have been used in the context of autologous HCT for plasma cell myeloma for 30 years, the interpatient variability in its PK properties (as well as its underlying mechanisms) remains to be further elucidated. In this study, we evaluated the potential impacts of CYP3A4*1B (rs2940574) and CYP3A5*3 (rs776746) variations on PFS, 90-days response, and oral mucositis in a cohort of 108 MM patients with HSCT-HDM therapy. We also investigated the associations of these genetic variations with the PK properties of melphalan, namely, area under the plasma concentration-time curve extrapolated to infinity (AUCinf), the maximum plasma concentration (Cmax), time to reach the maximum plasma concentration (Tmax), and creatinine clearance (CrCL).
Patients and Methods
Blood samples and pharmacokinetic (PK) parameter determination. Blood samples were collected from 119 MM patients who underwent melphalan-based autologous stem cell transplant (prior to melphalan treatment) with approval from the Cancer Institutional Research Board’s guidance (IRB#2011C0080, NCT01653106). Venous blood samples were collected in heparin tubes prior to melphalan administration (time 0) and then at different time points after completion of melphalan infusion: 5, 30, 45, 60, 180 and 360 min. The concentration of melphalan in each plasma was assessed using liquid chromatography-tandem mass spectrometry (LC-MS/MS) following a well-validated procedure. Non-compartmental PK parameters, such as AUCinf (the area under the plasma concentration-time curve extrapolated to infinity), Cmax (the maximum plasma concentration), Tmax (time to reach the maximum plasma concentration), were determined using Phoenix WinNonlin (v6.3, Pharsight, Mountain View, CA, USA). NONMEM 7, version 7.1.2 (ICON Development Solutions, Ellicott City, MD, USA) was used to build the basic two-compartment population pharmacokinetic model and the covariate model for melphalan as previously described (21).
DNA preparation and genotyping. After PK analysis, PBMCs were successfully separated from the remaining blood samples of 108 patients. DNA and RNA were purified using a Blood DNA/RNA Purification kit (Qiagen, Valencia, CA, USA) following the manufacturer’s instructions. Genotypes of CYP3A5 *3 and CYP3A4 *1B SNPs were determined on a QuantStudio™ 7 Flex system (Thermo Fisher Scientific, Waltham, MA, USA) using the following Taqman® pre-validated genotyping kits (Life Technologies, Carlsbad, CA, USA): C__26201809_30 for CYP3A5 *3 (rs776746) and C__1837671_50 for CYP3A4 *1B (rs2740574). Assays were conducted in duplicate (6).
Statistical analyses. R3.4 (R Foundation for Statistical Computing, Vienna, Austria; https://CRAN.R-project.org) was used in all statistical procedures in this study. For each SNP, the consistency between its distribution and the Hardy Weinberg equilibrium (HWE) principle was analyzed using the chi-square test. Count data were analyzed using Fisher’s exact test or chi-square test, where appropriate. Potential non-random allele associations between selected SNPs were investigated using the “genetics” package of R.
Time to relapse was determined as the time from transplant until the earliest of the following time points: progressive disease, clinical relapse, or relapse from CR (complete response) as determined by the International Myeloma Working Group (IMWG) (28). Patients without known progression were censored at the date of last follow up. Time to relapse was calculated from Kaplan-Meier curves with the difference between the curves analyzed using the log-rank test. Multivariate analysis was conducted using the proportional hazard regression model of Cox (Cox PH regression), initially including all factors with a p-Value of less than 0.20 in the univariate analyses and using a stepwise backward approach for model reduction. Regarding the sample size, the final multivariate model contained covariates with p-values ≤0.10 (29).
Univariate and multivariate logistic regression analyses were used to investigate the potential associations between mucositis (no: mucositis grade 0 and 1; yes: mucositis grade 2 and 3) and SNPs as well as other demographic cand clinical covariates. Similarly, the relationship between response at day 90 after transplant (no: negative, minor response and partial response; yes: very good partial response, complete response, and stringent complete response) and SNPs as well as other covariates were evaluated using logistic regression.
p-Values were two-sided, and unless specified, the significance level was α=0.05.
Results and Discussion
CYP3A4*1B and CYP3A5*3 variations in MM patients. The human CYP3 subfamily, located on chromosome 7, comprises four genes, namely, CYP3A4, CYP3A5, CYP3A7 and CYP3A43, whose protein products are involved in the oxidative metabolism of about 50% of all prescribed drugs, such as HIV protease inhibitors, calcium channel blockers, antineoplastic drugs and immunosuppressants (24, 30, 31). Out of these CYP3A enzymes, CYP3A4 is the predominant isoform that metabolizes macrolide antibiotics, statins, opioids, antidepressants, immunosuppressants, and some anticancer drugs (32). CYP3A5 shares up to 85% homology in protein sequence with CYP3A4 and the substrate specificities of these two enzymes overlap considerably (27). It has been reported that some SNPs in CYP3A4 and CYP3A5 genes down-regulate their enzymatic activities, which impact the metabolisms of some drugs, leading to different clinical outcomes (33). For example, two closely linked SNPs, CYP3A4 rs2940574 and CYP3A5 rs776746, have been found to be associated with the metabolism of tacrolimus in kidney transplant patients (34, 35). CYP3A5 rs776746 (CYP3A5*3) causes a splicing defect in the CYP3A5 transcript, resulting in significantly lower expression of active CYP3A5 enzyme. Consequently, patients who harbored homozygous CYP3A5*3 alleles had significantly higher dose-corrected trough concentrations of tacrolimus (36-38). The effect of CYP3A4 rs2940574 on tacrolimus metabolism is thought to be caused by its linkage to active CYP3A5 allele (i.e., CYP3A5*1), rather than modulating CYP3A4 itself. While hydrolysis and renal exertion are regarded as major route in melphalan metabolism in MM patients, a previous study showed that CYP3A4*1B and CYP3A5*3 variations tended to be associated with the survival of MM patients to certain extent, suggesting that the potential association of CYP3A4*1B and CYP3A5*3 variations and melphalan therapy might need to be evaluated (27).
We first determined the genotypes of CYP3A4*1B (rs2940574) and CYP3A5*3 (rs776746) variations in a cohort of 108 MM patients with HSCT-HDM therapy. The results are summarized in Table I. While the MAF (minor allele frequency) of CYP3A4*1B was about 14 %, the MAF of CYP3A5*3 was over 90%, which is consistent with the fact that most of these MM patients were white (about 85%) (38). It has been demonstrated that most of Caucasians (white) are CYP3A5*3/*3 carriers, and about 70% of the African population are CYP3A5 expressers harboring one of two CYP3A5*1 alleles (39).
Genotypes of CYP3A4 *1B and CYP3A5 *3 variations in OSU11055 patients (N=108).
The genotype frequencies of these two SNPs in this cohort of MM patients were in Hardy-Weinberg equilibrium (p>0.1). Further allele association analyses showed that CYP3A4*1B and CYP2A5*3 SNPs were significantly associated with each other in this cohort of MM patients (p<0.001).
A total of 81 patients (75%) were CYP3A4*1B/*1B and CYP3A5*3/*3 carriers, and the rest of patients had genotypes different from CYP3A4*1B/*1B and CYP3A5*3/*3. Table II summarizes the demographic and clinical characteristics of these two groups of MM patients with different CYP3A4 and CYP3A5 genotypes (defined as CYP3A4*1B/*1B and CYP3A5*3/*3 carriers and Others). There are no statistically significant differences between these two groups of MM patients regarding age, sex, length of stay in hospital (LOS), melphalan dose, and risk (p-Values>0.1). Due to the known association between CYP3A5 *3 allele with Caucasian (white) people, there is a statistically significant difference in race between these two groups of MM patients (p<0.001) (38).
Demographic and clinical characteristics of patients in the current study.
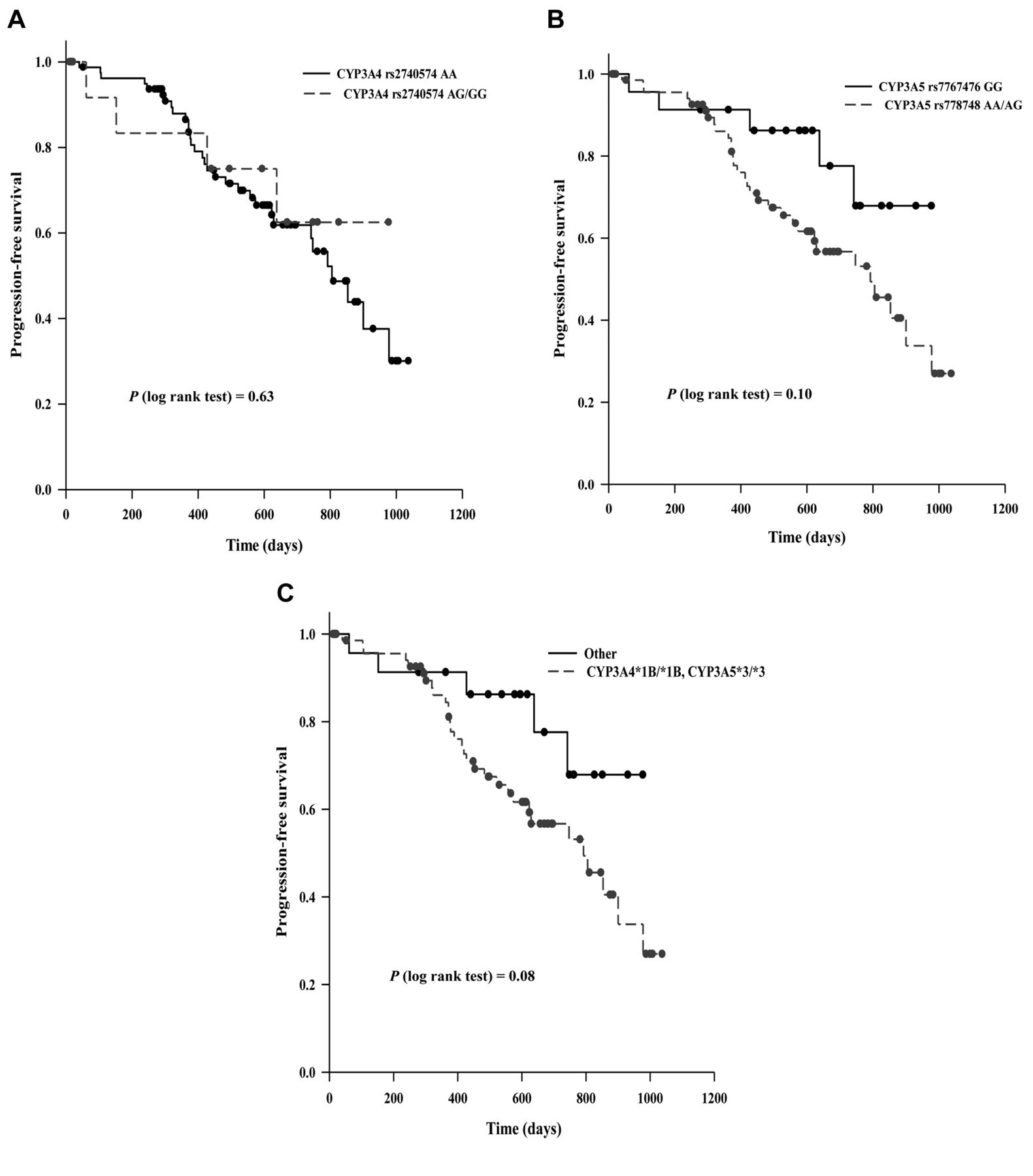
Association between CYP3A4*1B and CYP3A5*3 variations and clinical outcomes in MM patients. We then evaluated the potential association between CYP3A4*1B and CYP3A5*3 variations with progression-free survival after HSCT-HDM. Survival analyses using univariate models showed that CYP3A5*3 variation trended to be associated with PFS (p=0.10) (*3/*3 vs. *1/*1 and *1/*3, HR=2.20, 95% CI=0.85-5.84) (Table III). In comparison, there was no statistically significant association between CYP3A4*1B variation itself and PFS (p=0.63). Interestingly, the genotype of CYP3A4*1B/*1B and CYP3A5*3/*3 was associated with PFS with borderline significance (p=0.08) (CYP3A4*1B/*1B and CYP3A5*3/*3 vs. Others, HR=2.23, 95% CI=0.86-5.78). For patients carrying CYP3A4*1B/*1B and CYP3A5*3/*3, the median PFS time was 792 days (95% CI=573-978); for other patients, the median PFS time was longer than 950 days (Figure 1). Apparently, patients carrying CYP3A4*1B/*1B and CYP3A5*3/*3 had a shorter median PFS time. Since our previous studies have showed that sex, risk level, ANRIL rs2151280 SNP (6), and XRCC1 rs25487 SNP (40) appeared to be associated with PFS in univariate survival models, we continued to explore the association between the genotypes of CYP3A4*1B/*1B and CYP3A5*3/*3 and PFS using multivariate models. Of note, due to the relatively small sample size in our current study, we used p<0.1 and not p<0.05, in this multivariate analysis. Our results indicate that the genotype of CYP3A4*1B/*1B and CYP3A5*3/*3, ANRIL rs2151280 SNP, and XRCC1 rs25487 SNP appeared to be three independent factors associated with PFS in MM patients after ASCT-HDM. After adjustment for ANRIL rs2151280 and XRCC1 rs25487 statuses, the HR between patients carrying CYP3A4*1B/*1B and CYP3A5*3/*3 and other patients was 2.44 (p=0.072, 95% CI=0.92-6.57). Taken together, these results support further studies on the potential role of CYP3A4*1B and CYP3A5*3 variations as a prognostic factor for PFS in MM patients after ASCT-HDM.
Univariate and multivariate Cox Proportional Hazard regression analyses of progression-free survival in multiple myeloma patients with melphalan therapy.

Kaplan-Meier curves showing the progression-free survival (PFS) of 108 multiple myeloma (MM) patients with different CYP3A4*1B (rs2940574) and CYP3A5*3 (rs776746) genotypes. The differences in PFS between different groups were analyzed using log-rank tests.
Additionally, we investigated the potential associations between CYP3A4*1B and CYP3A5*3 variations and another two clinical outcomes, 90-day response and the severity of oral mucositis. Our results show that none of CYP3A4*1B variation, CYP3A5*3 variation, and the combination of CYP3A4*1B/*1B and CYP3A5*3/*3 variations was associated with 90-day response and the severity of oral mucositis (p-Values>0.2).
Association between CYP3A4*1B and CYP3A5*3 variations and PK properties of melphalan in MM patients. Considering the predominant roles of CYP3A4 and CYP3A5 in drug metabolism, we further explored the potential impacts of CYP3A4*1B and CYP3A5*3 variations on the PK properties of melphalan in these MM patients. As shown in Figure 2, there was no statistically significant association between the genotype of CYP3A4*1B/*1B and CYP3A5*3/*3 and any of AUCinf, CrCL, and Tmax in MM patients (p-Values>0.2). In contrast, the genotype of CYP3A4*1B/*1B and CYP3A5*3/*3 was statistically associated with Cmax, the maximum plasma concentration of melphalan (p=0.039). After stratification by the dose level (140 and 200 mg/m2), such association remained to be of borderline significance (p=0.07). Carriers of CYP3A4*1B/*1B and CYP3A5*3/*3 appeared to have higher Cmax values than their non-carrier counterparts. Apparently, such observation was consistent with the fact that carriers of CYP3A4*1B/*1B and CYP3A5*3/*3 had lower CYP3A5 enzymatic activities as mentioned earlier (38). Interestingly, even though carriers of CYP3A4*1B/*1B and CYP3A5*3/*3 had higher Cmax values, these patients had unfavored PFS (Figure 1). It has demonstrated in our previous study that Cmax was not associated with PFS in the same cohort of MM patients (6). Hence, we postulated that the impact of the genotype of CYP3A4*1B/*1B and CYP3A5*3/*3 on Cmax would not directly underscore the observed association between this genotype and PFS.

Comparison of pharmacokinetic (PK) parameters of melphalan between carriers of CYP3A4*1B/*1B and CYP3A5*3/*3 and the corresponding non-carriers. (A) AUCinf (the area under the plasma concentration-time curve extrapolated to infinity) (B) CrCL (creatine clearance); (C) Cmax (the maximum plasma concentration); (D) Tmax (time to reach the maximum concentration). Two-sided U rank sum tests were used to analyze the data.
Conclusion
In this study, we evaluated the impacts of the CYP3A4*1B and CYP3A5*3 variations on pharmacokinetic properties of melphalan and clinical outcomes in a cohort of 108 MM patients with ASCT-HDM therapy. Our results show that CYP3A4*1B and CYP3A5*3 variations led to unfavored PFS in MM patients. Interestingly, CYP3A4*1B and CYP3A5*3 carriers had higher maximum melphalan plasma concentrations than non-carriers. These results suggest that CYP3A4*1B and CYP3A5*3 variations might contribute to the inter-patient diversities of melphalan exposure and clinical outcomes of MM patients through yet-to-be-elucidated mechanisms. It is known that most of gene polymorphisms exert a small or moderate effect on drug metabolisms and clinical responses, and the impact of a gene polymorphism could be cofounded with other polymorphisms in the same gene or in other genes (27). Apparently, in the era of precision medicine, controlled clinical trials in large cohorts of patients are essential to extensively evaluate the effects of functional polymorphisms of drug-metabolizing enzymes, thus elucidating the interpatient heterogeneity of drug response. Nonetheless, our findings, from a relatively small cohort of MM patients, provide insights in support of continuing investigation on the potential roles of CYP3A4*1B and CYP3A5*3 variations in large cohorts of MM patients.
Acknowledgements
This work was partially supported by a Pelotonia IDEA award (46050-502048) (MAP, CCH, MJP) and a startup research grant from the College of Pharmacy, The Ohio State University (MJP).
Footnotes
Authors’ Contributions
DS, MAP and CH initiated the original clinical trial. JL and MJP developed the concept and designed the current study. JL and YKC conducted the experiments and collected the data. JL and MJP analyzed the data and drafted the manuscript. All Authors critically revised the manuscript and approved the final submitted version.
Conflicts of Interest
The Authors have no conflicts of interest to disclose.
- Received October 29, 2022.
- Revision received November 10, 2022.
- Accepted November 14, 2022.
- Copyright © 2023, International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved
This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY-NC-ND) 4.0 international license (https://creativecommons.org/licenses/by-nc-nd/4.0).
References
In this issue





{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.