


Abstract
Background/Aim: Cervical cancer is the fourth most common type of cancer in women worldwide and it is a major cause of cancer-related deaths in developing countries. Despite the marked reduction observed in the rates of the disease as a result of screening programs, it is necessary to develop robust biomarkers that can detect the neoplastic progression early in HPV-related cervical lesions. Materials and Methods: We performed comparative mRNA sequencing from exfoliative cervical cytology samples from nine Korean women using the Illumina NovaSeq6000 platform. Each pathological tissue was matched to the corresponding cytological sample. The pathologic diagnosis was scrutinized with ancillary immunohistochemistry and was considered a confirmative (endpoint) diagnosis. The pathological diagnoses consisted of three cases of chronic cervicitis, 2 high-grade squamous intraepithelial lesions (HSILs), 2 squamous cell carcinomas in situ (CIS), and 2 invasive squamous cell carcinomas (SQCCs), respectively. Using bioinformatic analyses, differentially expressed genes (DEGs; fold change ≥1.5; p<0.05) were applied for Gene Ontology (GO), Gene Set Enrichment Analysis (GSEA), and protein-protein interaction (PPI) networks. Results: From a total of 55,882 genes, 438 DEGs were pinpointed; 282 genes were up-regulated and 156 genes down-regulated. These transcriptomic profiles were clearly divided into neoplastic (HSIL, CIS, and SQCC; ≥HSILs) and non-neoplastic lesions. The up-regulated DEGs were HIF-1a, EDN1, PIK3R3, PPP1CA and AKR1C1. GO, GSEA, and PPI network analyses showed marked associations with metabolism, proteolysis, or proteoglycan process pathways in cervical carcinogenesis. Conclusion: The transcriptomic analysis using exfoliative cervical cells was more likely representative of its corresponding histopathological diagnosis, thus emphasizing its potential utility in clinical practice. This study provides comprehensive transcriptomic network analyses for robust biomarkers that might present a high potential risk of progression to cancer in the exfoliative cervical cytology; our findings support their clinical utility for improved cervical cancer screening.
Human papillomavirus (HPV) is the etiological agent responsible for more than 90% of all cervical cancer cases worldwide. During cervical carcinogenesis, the infective pattern of HPV is associated with a shift from productive infection–which in most cases would be cleared by the immune system–toward nonproductive persistent infection in a minority of cases (1). High-risk HPV infection contributes to the majority (80-90%) of low-grade squamous intraepithelial lesions (LSIL) (2), with a high rate of spontaneous regression (1). High-risk persistent HPV infection is responsible for high-grade squamous intraepithelial lesions (HSIL), which could progress into invasive cancer in at least 30% of cases without treatment (3).
High-risk persistent HPV infection is associated with dysregulation of epithelial cell proliferation, cellular differentiation, and apoptosis, accompanied with increased genetic instability (4). Genetically altered epithelial cells interact with the tumor microenvironments by changing intercellular contacts and releasing biochemical factors that can promote invasiveness to adjacent tissues, stimulate neoangiogenesis, and deregulate the balance between tumor cells and immune cells (5). The exfoliative epithelial cells from the cervix or vagina contain a variety of genetic information of human somatic cells and HPV. Moreover, the exfoliative dysplastic cells are easily obtained and are accessible through either visual inspection or other techniques including colposcopy.
Cervical cytology screening has been the most effective exfoliative cytology test for cancer detection and treatment of precancerous lesions before the development of cervical cancer. However, the practice of cervical cytology has several critical issues: 1) false-negative diagnoses for either precancerous lesions or invasive cancer due to sampling errors, screening errors, and interpretation errors; 2) low specificity in cases diagnosed as atypical squamous cells of undetermined significance (ASCUS) or LSIL; and 3) variability of inter- and intra-observer interpretation of cytological diagnoses (5). HPV DNA tests are currently performed to overcome these shortcomings of cervical cytology. However, HPV DNA detection only shows the state of HPV infection regardless of being transient or asymptomatic and does not provide any information regarding disease progressiveness (6). As a result, HPV DNA tests have been used as triage tests to confirm the cytological diagnosis. Therefore, it is necessary to identify novel biomarkers that could serve to better identify clinically significant lesions which can facilitate disease progression.
Recent advances in genomic analysis, such as next-generation sequencing and mRNA sequencing, aided by bioinformatics have become a powerful tool for functional genomics studies analyzing transcriptome profiles towards detection of alternative splicing, isoform variants, fusion transcripts etc., and for noting novel transcripts as efficient and reliable biomarkers for cancer diagnosis and therapeutic targets in various cancers (7, 8). Currently, transcriptome analyses of cervical specimens, including normal tissues, cervical intraepithelial neoplasia (CIN) and/or cancer have shown that functional transcriptomic and genomic pathway analyses could provide an improved understanding of the genetic regulation in cervical carcinogenesis (8). However, to date there are no studies reporting on transcriptome analysis using mRNA sequencing in cervicovaginal exfoliative cells.
In this study, we aimed to evaluate differential transcriptome profiles of exfoliative cervicovaginal cells for identification of lesions with a high potential risk of progression to cancer. For this study, we performed comparative mRNA sequencing (mRNAseq) analyses between non-neoplastic and neoplastic lesions from cervicovaginal swab samples of the patients, and assessed the significant biological processes and pathways involved in the progress of cervical carcinogenesis. This study provides the first transcriptome and network analysis for novel cervical progressive biomarkers in a setting of exfoliative cytology for cervical cancer screening.
Materials and Methods
Patients and sample collection. The study was approved by the Institutional Review Board of Kyungpook National University Chilgok Hospital (KNUCH 2017-08-004-001). This study included patients with tissue-proven uterine cervical lesions between July 2019 and February 2020 at the Kyungpook National University Chilgok Hospital, Daegu, Republic of Korea. Among the patients, nine available cervicovaginal swab samples for each mRNA sequencing and cytologic diagnosis and matched biopsy tissue samples were selected for the current pilot study.
For mRNA sequencing, cervicovaginal smears were performed using pap brush lines (Bion, Guri, Republic of Korea). The brush was placed into the cervical canal and was rotated 360 degrees in a clockwise direction. The samples were transported in DNase, RNase, and pyro-genic free tubes. At the same time, for a cytologic diagnosis, liquid-based cytology (LBC) samples were collected using broom-like devices with detachable heads (BD SurePathTM collection vial, Becton, Dickinson, BD Life Sciences-Integrated Diagnostic Solutions, Sparks, MD, USA) according to the manufacturer’s instructions. The cervical broom was placed into the cervical canal and was rotated 360 degrees around the entire cervical canal. The detachable head was placed in a vial with preservative fluid.
The cytological diagnoses consisted of two cases negative for malignancy, three ASCUS, one atypical squamous cells-cannot exclude HSIL (ASC-H), and three SIL (one low-grade and two high-grade, respectively). The pathological diagnoses were confirmed on the corresponding cervical tissue biopsies, and these were considered to be the end-point of the study. They consisted of three cases of chronic cervicitis, two HSILs (cervical intraepithelial neoplasia, CIN2), two squamous cell carcinoma in situ (CIS; CIN3), and two invasive squamous cell carcinomas (SQCCs) (Figure 1). Other clinicopathological features were obtained from the medical records database, including age, HPV status, parity, procedure, and FIGO stage etc. The information of each patient is summarized in Table I.
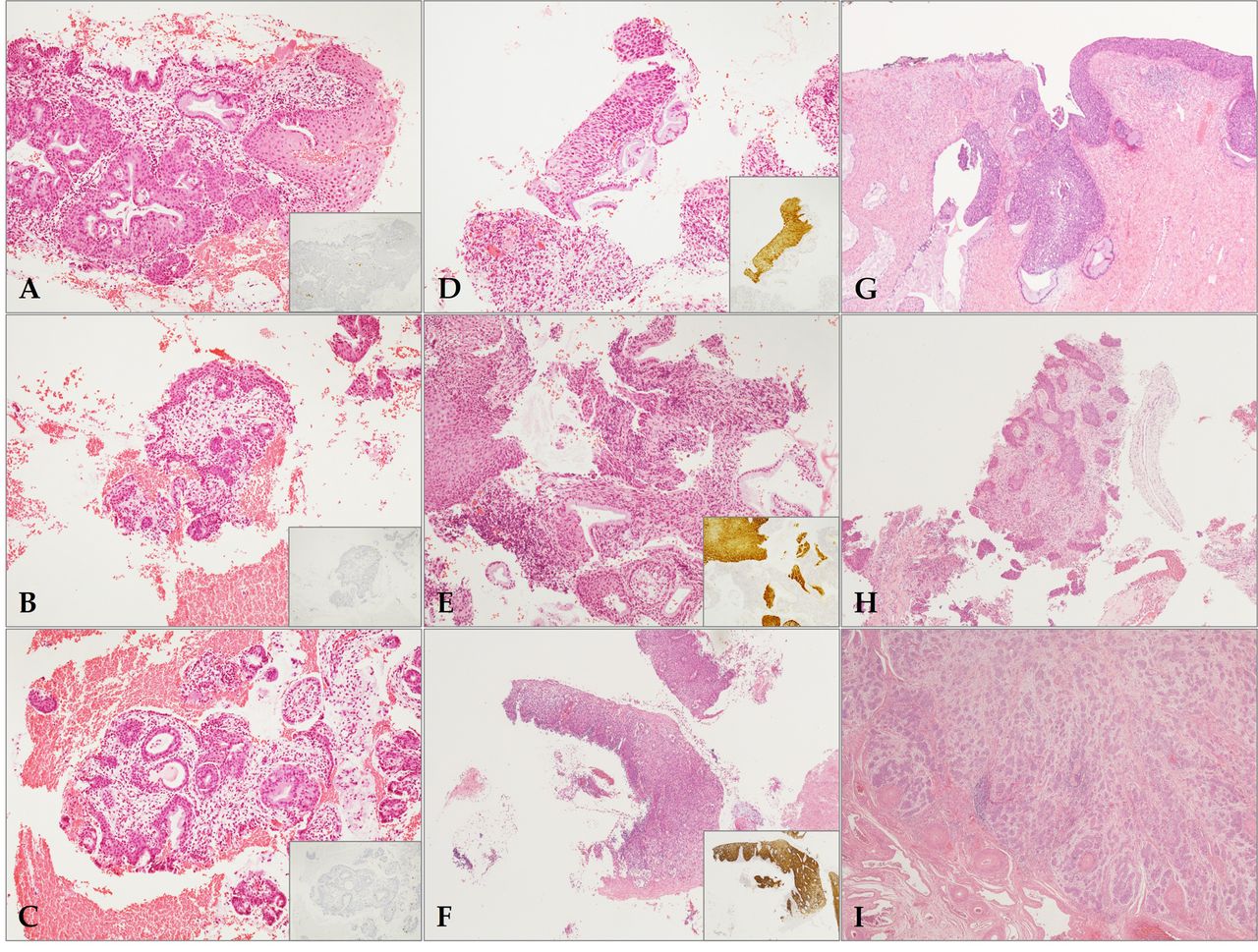
Histopathological findings of pathologic tissues matched with corresponding cytologic samples. The pathologic (endpoint) diagnosis was confirmed with ancillary P16 immunohistochemistry (A-I). Three cases (A-C) of chronic cervicitis showed no immunoreactivity to P16 (inset). In contrast, two high-grade squamous intraepithelial lesions (HSILs) (D and E) and one squamous cell carcinomas in situ (CIS) (F) were compatible with the diagnosis with P16 block positivity (inset). The single case of CIS (G) showed endocervical extension by CIS, but no stromal invasion. Two invasive squamous cell carcinomas (SQCCs) (H and I) demonstrated highly infiltrative features of tumor cells. (A-I, hematoxylin and eosin stain; original magnification, A-E, 100×; F-I, 40×; inset, P16 immunohistochemistry).
Clinicopathological features of patients with uterine cervical cytology.
HPV genotyping. HPV genotyping was performed using cervicovaginal swab specimens with the Anyplex II HPV 28 detection kit (Cat No. HP7S00X, Seegene, Seoul, Republic of Korea). The Anyplex II HPV 28 assay was performed according to the manufacturer’s instructions. Briefly, 5 μl of DNA was used in both 20-μl reactions, one with primer set A and the other with B. The Anyplex II HPV 28 assay uses HPV-specific dual priming oligonucleotides for multiplex (real-time) polymerase chain reaction (PCR). A total of 28 HPV types could be simultaneously detected, including 18 high-risk types (HPV 16, 18, 26, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59, 66, 68, 69, 73, and 82) and 8 low-risk types (HPV 6, 11, 40, 42, 44, 53, 54, and 70).
mRNA sequencing. The libraries were prepared for 151bp paired-end sequencing using the TruSeq stranded mRNA Sample Preparation Kit (Illumina, CA, USA). Namely, mRNA molecules were purified and fragmented from 1 μg of total RNA using oligo (dT) magnetic beads. The fragmented mRNAs were synthesized as single-stranded cDNAs through random hexamer priming. The application of this as a template for second strand synthesis allowed for the preparation of double-stranded cDNA. After sequential process of end repair, A-tailing and adapter ligation, cDNA libraries were amplified with PCR. The quality of these cDNA libraries was evaluated with the Agilent 2100 BioAnalyzer (Agilent, Palo Alto, CA, USA), and their quantification was performed with the KAPA library quantification kit (Kapa Biosystems, MA, USA) according to the manufacturer’s library quantification protocol. Following cluster amplification of denatured templates, sequencing was progressed as paired-end (2×151 bp) using Illumina NovaSeq6000 (Illumina, San Diego, CA, USA).
Transcriptome data analysis. The adapter sequences and the ends of the reads that had a Phred quality score less than 20 were trimmed. At the same time, reads shorter than 50 bp were removed by using cutadapt v.2.8 (9). Filtered reads were mapped to the reference genome related to the species using the STAR v.2.7.1a aligner (10), following ENCODE standard options with the “quantMode TranscriptomeSAM” option for estimation of the transcriptome expression level. Gene expression estimation was performed by RSEM v.1.3.1 (11) considering the direction of the reads, which are corresponding to the library protocol using the option—strandedness. To improve the accuracy of the measurement, the “—estimate-rspd” option was also applied. All other options were set to default values. To normalize sequencing depth among samples, the FPKM and TPM values were calculated.
Differentially expressed gene (DEG) analysis. Based on the estimated read counts in the previous step, DEGs were identified using the R package called TCC v.1.26.0 (12). In general, the TCC package applies robust normalization strategies to compare tag count data. Normalization factors were calculated using the iterative DESeq2 (13) edgeR (14) method. q-Values were calculated based on the p-Values using the p.adjust function of the R package with default parameter settings. The DEGs were identified based on a q-Value threshold less than 0.05, correcting for errors caused by multiple-testing (15). A fold change (FC) ≥1.5 was chosen as the cut-off criterion.
Gene ontology (GO) analysis. GO database provides a set of hierarchical controlled vocabulary classified in three categories: Biological process (BP), cellular component (CC) and molecular function (MF). For functional characterization of the DEGs, a GO-based trend test was carried out using the R package, which is called GOseq (16) through the Wallenius non-central hypergeometric distribution (17). Selected genes of p<0.05 following the test were regarded as statistically significant.
Gene set enrichment analysis (GSEA). Functional annotations for DEGs were performed by the database for annotation, visualization, and integrated discovery (DAVID, https://david.ncifcrf.gov). GSEA was performed to examine the critical GO and the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways of the interesting transcripts with GSEA software 3.0 from the Broad Institute. A p-Value <0.05 was considered a significant enrichment. The estimated expression levels of all genes were applied in GSEA, and the enrichment scores were then calculated according to the ranked-ordered gene list. The enrichment map of annotation analysis was also conducted with the use of Cytoscape (https://cytoscape.org, version 3.3.1.) (18).
Protein-protein interaction (PPI) networks. The Search Tool for Retrieval of Interacting Genes/Proteins (STRING) [https://string-db.org/, version 11.0 accessed on 20 September, 2020 online database was applied to construct the PPI network (19)].
Open datasets of cervical cancer. The microarray datasets GSE7803, GSE9750, and GSE63514 were downloaded from the NCBI gene expression omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo). The dataset GSE7803 was based on the GPL96 platform (Affymetrix Human Genome U133A Array; Thermo Fisher Scientific, Inc., Waltham, MA, USA), which included 10 normal cervical tissues and 28 cancerous samples (7 HSILs, and 21 invasive SQCCs, respectively). The dataset GSE9750 was also based on the GPL96, including 24 normal cervical tissues and 33 cervical cancer tissues. The dataset GSE63514 was based on the GPL570 platform (Affymetrix Human Genome U133 Plus 2.0 Array) and included gene expression profiles for cervical cancer progression. The GSE64514 consisted of 24 normal cervical tissues, 14 CIN1, 22 CIN2, 40 CIN3, and 28 cervical cancer samples. A lesion of ≥CIN2/HSIL was considered to be neoplastic in this study. To identify and compare DEGs of each group of our samples and GEO gene sets, GEO2R with the limma package was processed and normalized with an FC ≥1.5, and a p-Value <0.05 was considered to be statistically significant.
Survival analysis using public mRNAseq data. We also conducted the web-based survival analysis using Kaplan-Meier Plotter (19) to identify the prognostic significances for the selective genes in the public mRNAseq data repositories of cervical squamous cell carcinoma. A p<0.05 was considered statistically significant.
Results
Identification of DEGs. We identified 438 DEGs from a total of 55,882 genes in our transcriptomic data. Among 438 genes, 282 were found to be up-regulated DEGs including EDN1, ANG, PP1R15A, and CTSL, and 156 were down-regulated DEGs including PAF1, CTNS, TTC7A, and ITPKC. To further assess the predictive significance of progressiveness in cervical cancer, we statistically analyzed 282 overexpressed DEGs. A heatmap and a volcano plot were constructed using the top 30 of the up-regulated and down-regulated DEGs, respectively (Figure 2A and B). The heatmap visually provided clear clustering between six neoplastic cases (HSIL, CIS, and SQCC; ≥HSILs) and three non-neoplastic cases.

Heatmap of the top 30 up-regulated and top 30 down-regulated differentially expressed genes (DEGs) (A). Volcano plot of the top 30 up-regulated and down-regulated DEGs (B). The volcano plot depicts statistical significance (−log10 of p-Value) on the y-axis versus fold-change (FC) on the x-axis. Red marks indicate points of interest that display both large magnitudes, up-regulated and down-regulated FCs (left or right of center), and high statistical significance (hence being toward the top). The dashed horizontal gray line shows where p=0.05 with points above the line having p<0.05 (red and blue marks) and points below the line having p>0.05 (green and gray marks). The dashed vertical gray line shows where FC=1.5. The transcriptome profiles of patients were distinctively classified into six neoplastic [high-grade squamous intraepithelial lesions (HSILs), squamous cell carcinomas in situ, and invasive squamous cell carcinomas; ≥HSILs] and three non-neoplastic lesions.
Function and pathway enrichment analysis. To analyze the functional annotations in overexpressed DEGs, all 282 up-regulated DEGs were analyzed on the DAVID database (https://david.ncifcrf.gov/ accessed on 15 September 2020) (20). In GO analysis, we specifically focused on the BP analysis. A total of 21 BP terms were identified, of which eight BP terms were considered statistically significant: positive regulation of heart rate; regulation of translational initiation by eIF2 alpha dephosphorylation; positive regulation of chemokine-mediated signaling pathway; negative regulation of RNA polymerase II transcriptional preinitiation complex assembly; proximal/distal pattern formation; lung development; lactate metabolic process, and negative regulation of urine volume. The genes involved in these BP terms were as follows: ALDH1A2, ATP2A2, AVPR2, EDN1, GLI1, HIF1A, HMGB1, HOXA10, LDHA, PPP1CA, PPP1R15A, THRA, and UTS2.
GSEA analyses also provided pathways identified from the KEGG database of 282 overexpressed DEGs. Taking p<0.05 as the cut-off criterion, three out of a total of seven up-regulated pathways were clarified as significant: proteoglycans in cancer; lysosome, and regulation of actin cytoskeleton. Genes involved in these three pathways were as follows: AP3M1, ASAH1, BAIAP2, CLN3, CLN5, CTSL, GNS, HIF1A, PIK3R3, PIP4K2B, PLAUR, PPP1CA, PPP1R12B, RAC1, and WNT4. Furthermore, genes present in both GO and GSEA pathways were HIF1-a and PPP1CA. Identification of these genes provides a correlation between the roles of those genes and cervical carcinogenesis, but it also proposes the possibility to use these genes as a concrete biomarker for screening HPV-related neoplastic lesions of uterine cervix and their potential progressiveness.
PPI analysis. To visualize the functional protein association network between the 282 overexpressed DEGs, the PPI was constructed using the STRING database (Version 11.0). A total of 195 nodes were mapped with 205 edges in PPI enrichment with a statistical significance (p<0.0001). From 14 statistically significant pathways, we selected six which were closely related to our interests: HIF-1 signaling pathway (hsa04066; p=0.049); lysosome (hsa04142; p=0.026); proteoglycans in cancer (hsa05205; p=0.016); metabolic pathways (hsa01100; p=0.007); mRNA surveillance pathway (hsa03015; p=0.001), and oxidative phosphorylation (has00190; p<0.0001). The colored PPI network illustrated noteworthy results as follows. First, genes involved in the HIF-1 signaling pathway appeared repeatedly in the DAVID database, such as HIF1-a, PIK3R3, EDN1, and LDHA. Moreover, PIK3R3 and HIF1-a were also involved in proteoglycan in the cancer pathway. Sharing overlapped genes, two pathways were closely mapped in the PPI network. Second, the metabolic pathway had the most genes involved, but they were located throughout the overall PPI network. However, certain genes were correlated with genes from the oxidative phosphorylation pathway, and together they composed a cluster of their own. Lastly, genes involved in the lysosome and the mRNA surveillance pathways did not reveal any significant tendencies and were sporadically mapped within the PPI network. The PPI network with each pathway colored is presented in Figure 3. The enriched sets of genes for interested PPI networks are described in Table II.

Protein-protein interaction network analysis of the current transcriptomic profiles using the STRING database (Version 11.0). Of the 14 statistically significant pathways, six were highly significant: the HIF-1 signaling pathway (hsa04066); lysosome (hsa04142); proteoglycans in cancer (hsa05205); metabolic pathways (hsa01100); mRNA surveillance pathway (hsa03015), and oxidative phosphorylation (has00190). Genes involved in each pathway were highlighted in different colors for a more accessible visual representation.
Enriched sets of genes for interested protein-protein interaction (PPI) networks. The statistically significant PPIs were listed by the functional protein association network analyses using the STRING database (Version 11.0) between the 282 overexpressed differentially expressed genes (DEGs).
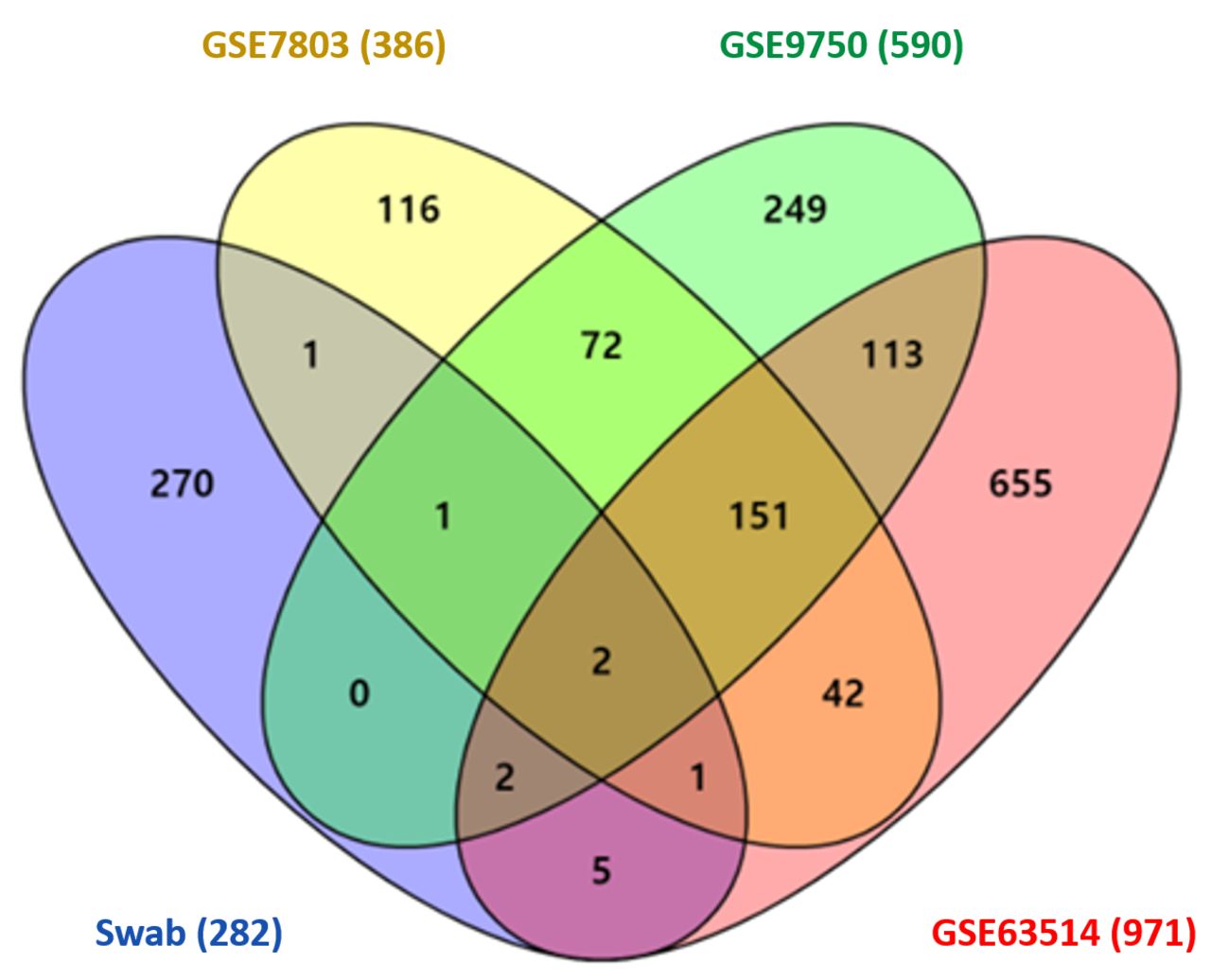
Public database validation. The consistency in our results from the analyses of different databases can validate the significance of our findings regarding the diversity of genes and pathways related to cervical cancer, even from cervical swab specimens. To validate our data, we selected three different gene sets from the GEO database, whose profile includes both normal and neoplastic cells: GSE7803, GSE9750, and GSE63514. Consequently, we compared the genes from our swab sample with each gene in the gene sets, as well as all four data combined.
When each dataset was normalized with a p<0.05 and an absolute FC ≥1.5, each GSE7803, GSE9750 and GSE63514 provided 386, 590 and 971 DEGs, respectively. DEGs from each gene set were compared against the 282 overexpressed DEGs from our data, and they were also analyzed using a Venn diagram (Venny 2.0.2). The overlapping genes in each of the three sets were: 1) Swab+GSE7803+GSE9750 (3 genes), PIK3R3, AKR1C1, and CTSL; 2) Swab+GSE7803+GSE63514 (3 genes), PIK3R3, AKR1C1, and ECI1; 3) Swab+GSE9750+ GSE63514 (4 genes): PIK3R3, AKR1C1, PLAUR, and SLC45A4. The overlapping genes in all four sets were PIK3R3 and AKR1C1. The comparison performed in the Venn diagram analysis is described in Figure 4.

Venn diagrams illustrating the number of up-regulated differentially expressed genes (DEGs) from four different gene sets, including our exfoliative cervical cytology samples (swab), GSE 803, GSE9750, and GSE63514 (The number of DEGs is included in parentheses next to the name of each dataset).
Web-based survival analysis. Kaplan-Meier survival curves showed that the expressions for HIF-1a, EDN1, PIK3R3, PPP1CA, and AKR1C1 were associated with distinct overall survival (OS) and recurrence-free survival (RFS) outcomes, respectively (Figure 5).

Prognostic significances HIF-1a, EDN1, PIK3R3, PPP1CA, and AKR1C1 expression in public mRNAseq data repositories (www.kmplot.com). Overall survival (A-E) and recurrence-free survival (F-J) for each gene were explored.
Discussion
Many countries have reduced the incidence of cervical cancers by implementing organized screening programs and efficient diagnostic processes. However, it is still necessary to identify clinically robust biomarkers that can detect the disease and screen patients at higher risk of progressiveness in cervical cancers (20).
To identify significant biomarkers for the discrimination of the potentially progressive cervical cancer, we performed mRNAseq using matched cervicovaginal swab samples collected from patients who had a tissue-proven diagnosis. We especially focused on up-regulated DEGs profiles and their relevant pathways to provide an interpretation for the discriminative intrinsic changes from the non-neoplastic lesion and the significantly neoplastic lesion. The transcriptomic profiles of patients were distinctively classified into the neoplastic (HSIL, CIS, and SQCC; ≥HSILs) and the non-neoplastic lesions; most notable the up-regulated DEGs were HIF-1a, EDN1, PIK3R3, PPP1CA, and AKR1C1 in the neoplastic lesions. Further PPI and network analyses revealed that the swab samples in the neoplastic lesions were significantly associated with the metabolic process, proteolysis, or proteoglycan process pathways, which might play a role in cervical carcinogenesis.
In our study, we found that HIF-1a was a significant gene that could be involved in cervical carcinogenesis. Considering the characteristics of cervicovaginal swab samples, however, there was insufficient evidence to interpret that exfoliative epithelial cells could directly represent the properties of hypoxia-dependent mechanisms in the tumor microenvironment including angiogenesis. Therefore, we concentrated on the comprehensive understanding of both hypoxia-dependent and hypoxia-independent mechanisms of the HIF-1a gene, which could be associated with the metabolic process, proteolysis, or proteoglycan process based on the PPI network analysis.
Hypoxia is a common condition in malignant tumors that induces hypoxia-inducible factor-1a (HIF-1a), which in turn activates the expression of various relevant genes that regulate angiogenesis, apoptosis, epithelial-mesenchymal transition, inflammation, metastasis, and resistance to therapies (21-26). In cervical cancers, HIF-1a has been known to play a potent dual function in the early phases of carcinogenesis (27), by stimulating angiogenesis through activation of the vascular endothelial growth factor (VEGF) gene and regulating hypoxia-mediated apoptosis through stabilization of p53 (20). In particular, inactivation of p53 through the viral protein E6 is essential for the stability of HIF-1a (20), and HIF-1a dependent VEGF expression in hypoxia (28).
Moreover, recent studies have revealed a variety of hypoxia-independent mechanisms for HIF-1a signaling activation (29, 30). Studies showed that HIF-1a played a critical role in both glucose metabolism and the glycolytic pathway. HIF-1a signaling activation leads to the accumulation of pyruvate and lactate (31), which can be used as an energy source for cancer cells, resulting in the growth advantage of cancers (32). In addition, accumulation of lactate can also stabilize HIF-1a, which is therefore involved in cell migration, invasion, immune escape, and radio-resistance of cancer cells (33). Similar to HIF-1a, the phosphatidylinositol 3-kinase (PI3K) signaling pathways are involved in various cellular processes, such as tumor metabolism, inflammation, survival, and progression (34). Notably, the PI3K/AKT signal can inactivate glycogen synthase kinase-3β (GSK3B), and thus result in HIF-1a stabilization (35) and glycolytic pathway activation. Consequently, both HIF-1a and PI3K/AKT/mTOR pathways may have an important role in cancer metabolism in carcinogenesis (36).
Interestingly, overexpression of HIF-1a has been proposed to be a biomarker for tumor progression in cervical cancer (20). More specifically, studies have shown that HIF-1a overexpression has been identified in the early stages of invasive cervical carcinomas and HSILs (81.3% and 80%, respectively), but it is not found in normal cervical epithelial cells (27). Therefore, considering our results and other previous studies, activation of HIF-1a signaling pathways, regardless of the oxygen situation, is speculated to play an important role in cervical cancer development and progression (20).
The endothelins 1 (EDN1) is one of EDN families that acts on cellular proliferation and transformation directly or synergistically with other growth factors (37). Studies reported that EDN1 up-regulated invasiveness through the activation of matrix metalloproteinases 2, 9, 3, 7, and 13 (38).
Furthermore, the protein phosphatases (PPs) regulate the diverse cellular processes. Protein phosphorylation acts as a molecular switch and it is controlled by the opposing actions of protein kinases and PPs (39). There are at least six families of serine/threonine PP with PP1, PP2A, and calcineurin (PPP3 formerly PP2B) representing the majority of protein phosphatase activity (40, 41). Protein phosphatase 1 catalytic subunit alpha (PPP1CA) is one of the three catalytic subunits of PP1 (42). PP1 is a serine/threonine specific PP that is known to be involved in the regulation of a variety of cellular processes, such as cell division, glycogen metabolism, muscle contractility, protein synthesis, and HIV-1 viral transcription (42). Studies showed that PP1 deregulation could be implicated in diabetes and multiple types of cancer (provided by RefSeq, Jul 2020), however, further specialized studies on the role of PPP1CA in cervical cancer are needed.
Another noteworthy feature was that the evaluation of the exfoliative cervical cell transcriptome profile showed that it could be more likely to represent the histological diagnosis than the cytological interpretation. The transcriptome analyses of four patients diagnosed with atypical squamous cells (three ASCUS and one ASC-H) on the cytological interpretation were classified into one non-neoplastic and three neoplastic lesions. These transcriptomic results were consistent with the confirmative histopathological diagnoses of one case of chronic inflammation and three cases of neoplastic lesion (≥HSIL) in those four patients (Table I). Therefore, in cases where it is difficult to obtain cervical tissues from patients in clinical practice, mRNA sequencing from the swab samples can provide us with an alternative and efficient testing method to accurately predict the disease status.
Furthermore, we analyzed the transcriptomic DEGs from our swab data and three GEO datasets. The overlapping genes in ≥HSIL lesions in all four datasets were PIK3R3 and AKR1C1. Added to the significance mentioned above of PIK3R3, AKR1C1 also showed potential clinical relevance. The level of AKR1C1 was up-regulated in neoplastic lesions, and it was frequently overlapped on the integrative analyses using the GEO databases of cervical cancers. AKR1C (AKR1C1–AKR1C4) are the members of the Aldo-keto reductase superfamily and involved in the metabolic processes of steroid hormones, prostaglandins, and lipid aldehydes (43). Previous studies have suggested that the overexpression of AKR1C is associated with the development of cancer in the endometrium (44), prostate (45) and breast (46). In addition, it has been suggested that they are closely related to the development of platinum drug resistance (47, 48) in colon (49), ovaries (50), lung (51), and uterine cervix (48). Notably, Badaracco et al. reported that cervical cancers harboring HPV showed a poor response to chemotherapy and an impaired chemotherapy-induced apoptosis (52). Further, cervical cancers with HPV-positive infection exhibited increased levels of AKR1C compared with HPV-negative tumors, a finding suggesting that HPV16 oncogenes are involved in the up-regulation of AKR1C1 and AKR1C3, while promoting drug resistance (53). These findings imply that AKR1C regulation can lead to implementation of more effective therapeutic modalities that focus on inhibiting drug resistance in HPV-related cervical cancers.
The main limitations of this study are the small sample size, the lack of true normal/benign controls, and the retrospective study design in the setting of a single time point. Moreover, the technical issues for the exfoliative cytology, including sample acquisition, preprocessing, wet- and dry-laboratory processes during mRNA sequencing, and data analysis and interpretation should be more standardized and generalized.
In particular, this study was designed as a pilot study to analyze transcriptomic DEGs. Therefore, it should be substantiated by further studies involving protein expression. In this regard, A. Ramirez-Torres et al. (54) recently investigated that cervical SQCC and adenocarcinomas have different protein profiles and suggested that RAB14 and RCN3 could represent valuable biomarkers related to disease prognosis and potential novel therapeutic targets for the treatment of CC. Nevertheless, our study provides several significant findings: first, the transcriptome profiles from the exfoliative cervical cells of patients could be distinctively classified into neoplastic versus non-neoplastic lesions, which could, in turn, be a significant biomarker to enable lesion differentiation according to its potential progressiveness in cervical cancers; second, the transcriptomic analysis revealed significant profiles including tumor metabolism, proteolysis or proteoglycan process, which could be involved in the early phases of cervical carcinogenesis; lastly, the transcriptomics using exfoliative cervical cells were more likely representative of the corresponding histological diagnosis, thus suggesting their potential utility in clinical practices.
Conclusion
This study examined the comprehensive transcriptomic network/pathway analyses using exfoliative cervical cytology. Most notably, the transcriptome profiles from the exfoliative cervical cells of patients could be distinctively classified into neoplastic (≥HSILs) versus non-neoplastic lesions. HIF-1a, EDN1, PIK3R3, PPP1CA, and AKR1C1 were found to be up-regulated and these genes are significantly involved in metabolism, proteolysis, or proteoglycan process pathways in cervical carcinogenesis. This study enhanced the transcriptomic network analysis that could support their clinical utility for improved cervical cancer screening in the exfoliative cytology sample by identifying potential biomarkers that may indicate a high risk of progression to cancer.
Acknowledgements
This work was supported by the Biomedical Research Institute grant, Kyungpook National University Hospital (2019).
Footnotes
↵*† These Authors contributed equally to this study.
Authors’ Contributions
The contributions by each author (as indicated by initials) are as follows: GOC and HSH conceptualized the research. GOC earned the funding resource. JMK, YHL, DGH, and GOC curated data. NJP, YC, and GOC conceived and designed the experiments. NJP, YC and JYP performed the experiments and analyzed the data. DL validated the data after the requirement of revision. NJP and YC wrote the original manuscript. GOC and HSH supervised the project and edited the manuscript.
Availability of Data and Materials
The mRNAseq data were assigned to the GEO database with an accession number of GSE215744.
Conflicts of Interest
The Authors declare that there are no conflicts of interest.
- Received August 19, 2022.
- Revision received November 1, 2022.
- Accepted November 22, 2022.
- Copyright © 2023, International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved
This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY-NC-ND) 4.0 international license (https://creativecommons.org/licenses/by-nc-nd/4.0).
References
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.