


Abstract
Background/Aim: Malignant melanoma is a skin cancer originating from the oncogenic transformation of melanocytes located in the epidermal layers. Usually, the patient’s prognosis depends on timing of disease detection and molecular and genetic profiling, which may all significantly influence mortality rates. Genetic analyses often detect somatic BRAF, NRAS and cKIT mutations, germline substitutions in CDKN2A, and alterations of the PI3K-AKT-PTEN pathway. A peculiar molecular future of melanoma is its high immunogenicity, making this tumor targetable by programmed cell death protein 1-specific antibodies. Materials and Methods: Ten formalin-fixed paraffin embedded samples derived from melanoma patients were subjected to next-generation sequencing (NGS) analysis using the FDA-approved FoundationOne CDx™ test. The molecular features of each case were then analyzed employing several in silico prediction tools. Results: We analyzed the mutational landscape of patients with metastatic or relapsed cutaneous melanoma to define enriched pathways and protein-protein interactions. The analysis showed that both known genetic alterations and variants of unknown significance rely on redundant signaling converging on similar gene ontology biological processes. Complex informatics analyses of NGS-based genetic results identified pivotal signaling pathways that could provide additional targets for cancer treatment. Conclusion: Our data suggest an additional role for NGS in melanoma, as analysis of comprehensive genetic findings using innovative informatic tools may lengthen the list of druggable molecular targets that impact patient outcome.
Melanoma is the deadliest form of skin cancer due to its high invasion and metastatic potential. It is a heterogenous disease that originates from the pigment-generating cells known as melanocytes (1, 2). Although most melanocytes are usually located in the skin, their neural crest origin implies that they may be found in any location where neural crest cells migrate, such as the gastrointestinal tract and brain (3, 4). Family history, UV exposure over time, atypical mole syndrome and fair skin are among the major risk factors for developing melanoma.
The incidence of malignant melanoma is increasing worldwide, and is more common in Caucasians than Afro-American and Asian subjects. Melanoma cells have a high predisposition to invade adjacent tissues generating distant metastases even when the disease is in an early stage. Thus, while melanoma accounts for only 5% of skin cancers, it is responsible for more than 60% of the skin cancer-related deaths (1, 5).
During the past decade, several genetic alterations have been identified that are responsible for melanoma development in both sun-exposed and non-sun-exposed regions. In addition, as similar mutations and gene expression patterns have been detected in both primary and metastatic melanoma sites, a mechanism independent of clonal evolution is likely the major driver of disease progression (6).
The most frequent genetic alterations associated with melanoma involve constitutive signaling by the mitogen-activated protein kinase (MAPK) kinase and the phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT) pathway due to activating mutations in BRAF and NRAS (1, 7). In addition, a mechanism deregulating the cell cycle such as germline alterations of CDKN2A/B have also been reported (8).
Melanoma is considered the most immunogenic solid tumor as it is associated with documented recognition of melanoma antigens by tumor-infiltrating T lymphocytes making it targetable by anti-PD1 therapies (9).
In the last few years, the implementation of next generation sequencing (NGS) in clinical practice has shown important progress in the molecular profiling of several malignancies including skin tumors (10). Several NGS-based studies have identified additional genetic alterations that may improve our understanding of the biological features of melanoma (11-13). In this context, The Cancer Genome Atlas (TCGA) program performed a systematic multi-platform characterization of 333 cutaneous melanomas, analyzing DNA, RNA, and protein, to generate a list of somatic alterations found in this cancer, describing their biological, therapeutical and clinical significance (14).
Here, we describe the pathway enrichment and oncogenic protein-protein interactions associated with the mutational landscape of 10 melanoma patients.
Materials and Methods
Patient samples. Ten melanoma patients followed in the Division of Medical Oncology of the Azienda Ospedaliero-Universitaria Policlinico “G. Rodolico - San Marco” in Catania gave written informed consent to employ their formalin-fixed paraffin embedded (FFPE) tissue samples for these molecular analyses.
Next-generation sequencing. Genomic profiling was performed on FFPE tissue using the NGS platform provided by Foundation Medicine (15). Specifically, the panel employed was the FDA-approved FoundationOne CDx™ detecting 36 genetic rearrangements and interrogating 324 genes to reveal substitutions, insertions, deletions, and copy number alterations. FoundationOne CDx™ also identifies genomic signatures including microsatellite status (MS) and tumor mutational burden (TMB). For two patients, the genomic material was of insufficient quality and their samples were therefore excluded from the analysis.
In silico mutation prediction tools. To predict whether amino acid substitutions are tolerated or deleterious on the structure and function of the analyzed human variants of unknown significance (VUS), we chose in silico mutation prediction tools employing the following algorithms: POLYmorphism PHENotyping (Polyphen-2) v2 for single nucleotide variants (SNV) (16), PROtein variation effect analyzer (PROVEAN) v1.1 for insertion/deletion (Ins/Del) (17) and MutPredLOF for stop-codon and frame-shifts (18). We then classified VUS as pathogenetic or benign according to their impact on the structure and function of the proteins for which they encode.
Tile plot, gene pathway analysis and PPI Network generation. Tile plots were generated using Bioconductor R v3.13 applying the complex HeatMap section. The functional classification of both known genetic alterations and VUS was generated with the protein analysis through evolutionary relationships (PANTHER) v16.0 classification system (19), a functional annotation tool part of the Gene Ontology Reference Genome Project. Pathway enrichment analysis was performed by the Overrepresentation Test, applying Fisher’s exact test (p<0.01) corrected by the False discovery rate (FDR) setting a p<0.05 threshold value.
To construct a protein-protein interaction network (PPI) and cluster analysis, we employed the STRING database v11.5. We limited species to “Homo sapiens” and set the confidence score to >0.4 (20), while the Markow cluster alghorithm (MCL) was applied for cluster generation. Enrichr (21) was used to identify the Gene Ontology biological process of the PPI clusters constructed by STRING.
Kaplan-Meier survival estimates. Kaplan-Meier estimators for overall survival (OS), relapse-free survival (RFS), and progression-free survival (PFS) were generated using Prism Software v8.0.3.
Results
Study population. Ten melanoma patients were selected showing superficial spreading melanoma (n=4), nodular melanoma (n=5), and amelanotic melanoma (n=1). Two of 10 patients were in stage IV (metastatic stage) at diagnosis, while the remaining eight patients showed disease relapse after surgical therapy. Patient demographics and tumor clinical futures are summarized in Table I.
Patient demographics and tumor clinical futures.
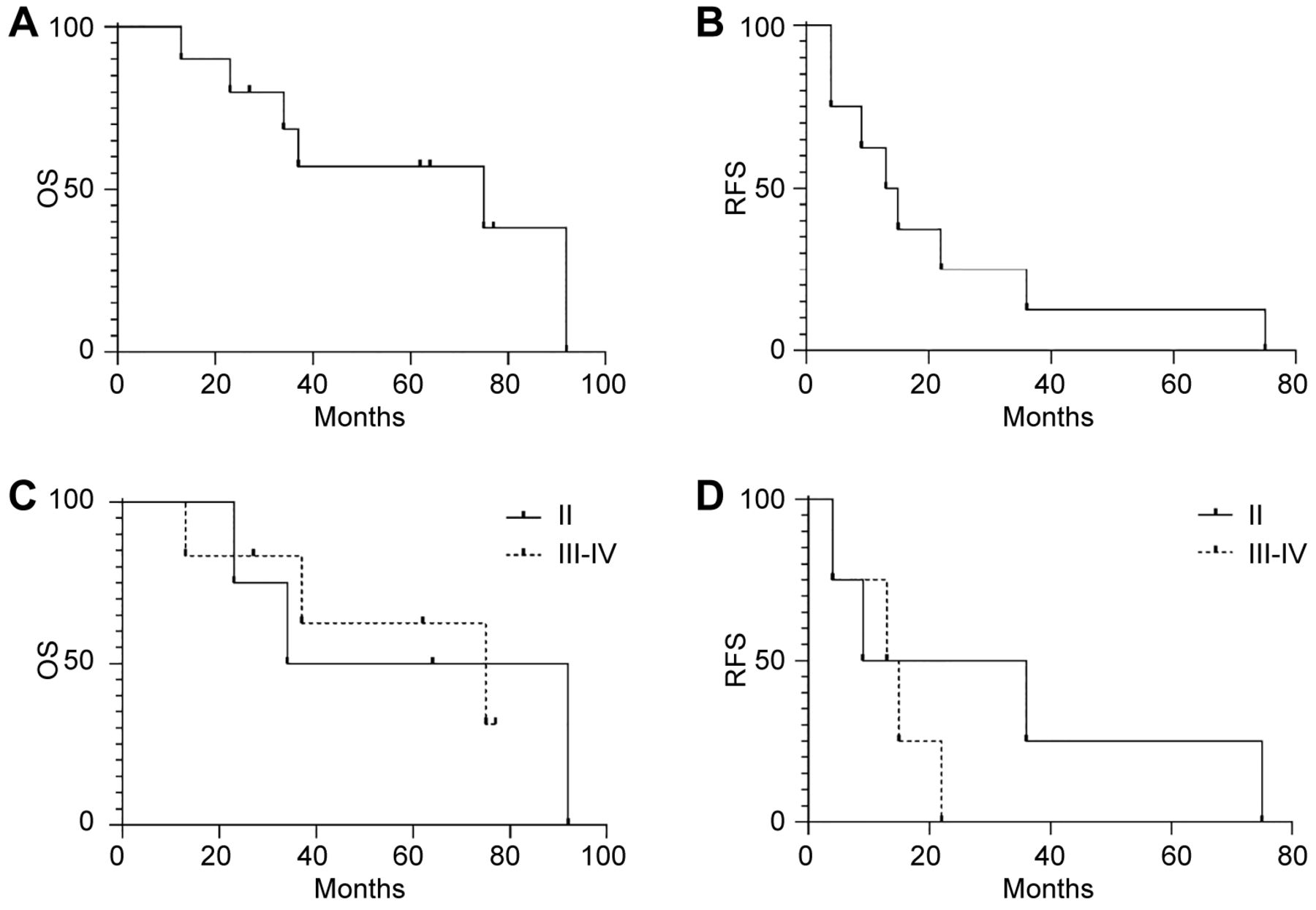
Using the Kaplan-Meier estimator we calculated median OS and RFS for the entire population or according to tumor stage (Figure 1). Our population showed median OS and RFS of 75 and 14 months, respectively (Figure 1A and B). Stratifying the population according to tumor stage at diagnosis we observed a median OS of 63 and 75 months for patients with stage II and III-IV disease (p=0.9). Although not significant, the subjects with stage III-IV displayed a median RFS of 14 months when compared with those in stage II, which was of 22.5 months (p=0.3) (Figure 1C and D).

Kaplan-Meier estimator of the study population. Kaplan-Meier curves showing the overall survival (OS) (A) and relapse-free survival (RFS) (B) of melanoma patients (n=10). Kaplan-Meier curves displaying the overall survival (C) (n=10) (p=0.9) and relapse-free survival (D) (n=8, M0:8) (p=0.3) in melanoma patients according to tumor stage. n: Number of patients, M0: number of non-metastatic patients at diagnosis.
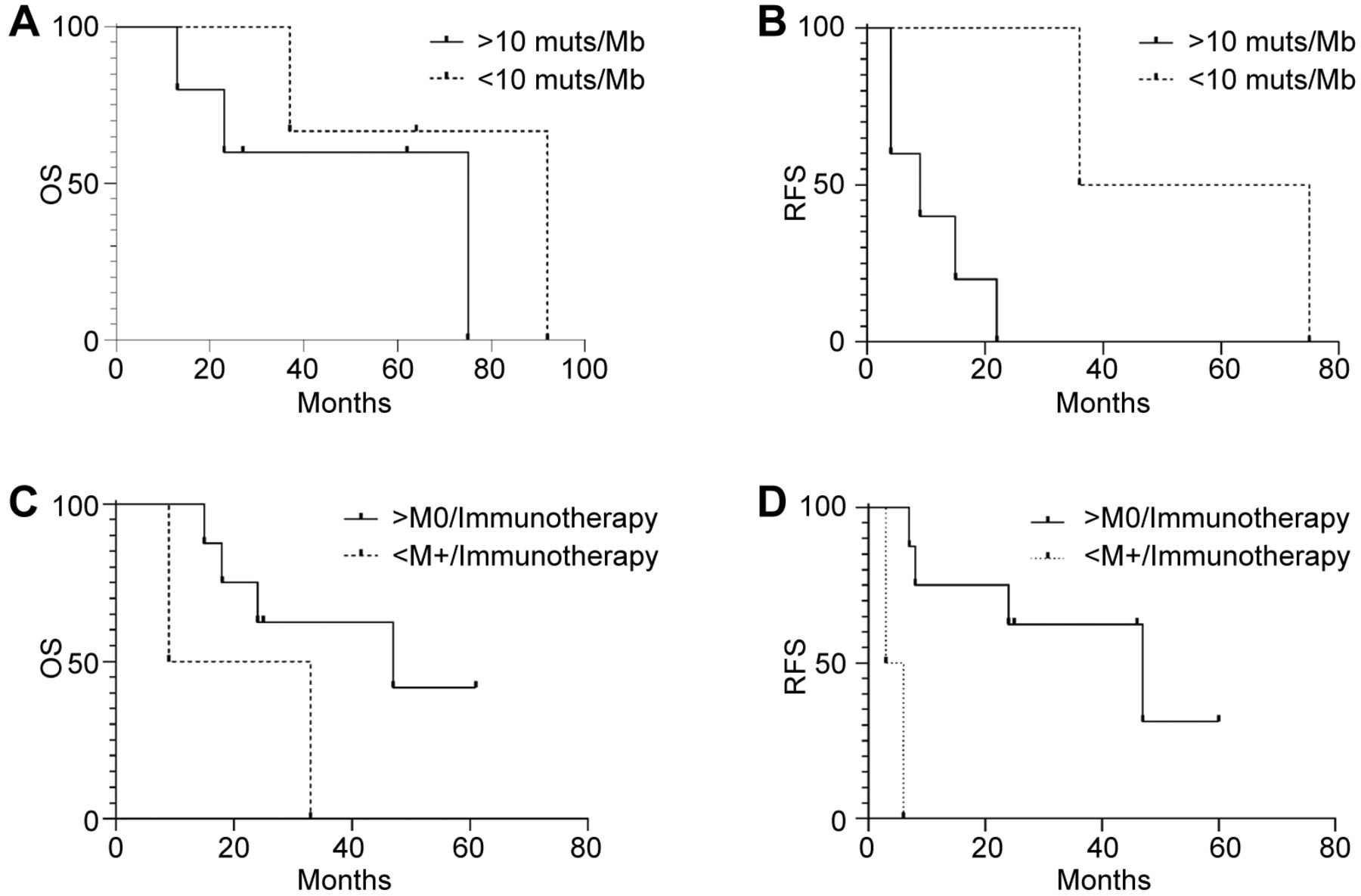
Impact of genomic signatures and anti-PD1 therapy on survival. While all patients lacked microsatellite instability, different data were obtained for TMB and anti-PD1 therapy. For TMB (defined as the number of somatic mutations per Mb harbored by tumor cells) the population was stratified using 10 mutations/Mb as threshold (TMB-High: >10 muts/Mb, TMB-Low: <10 muts/Mb), as previously reported (22). Kaplan-Meier estimates failed to show a significant effect on OS, which was 75 months for >10 muts/Mb and 92 months for <10 muts/Mb (p=0.3) (Figure 2A). However, differences in terms of recurrence were statistically significant, with a median RFS of 9 months for TMB-High versus 55.5 months for TMB-Low (p=0.03) (Figure 2B).
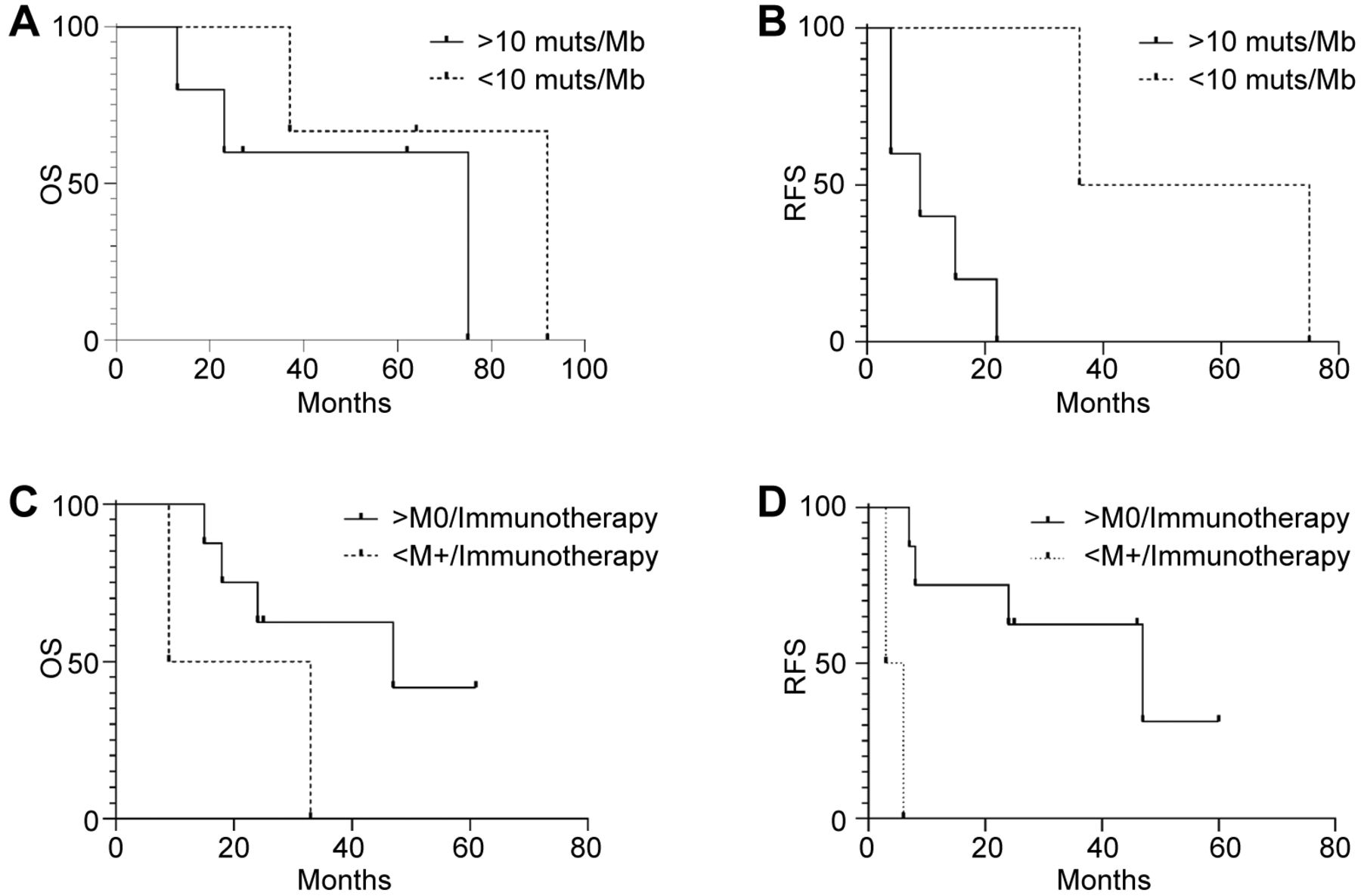
Kaplan-Meier estimator of overall, relapse-free and progression-free survival by tumor mutational burden (TMB) and immunotherapy treatment. Kaplan-Meier curves of overall survival (OS) (n=8, p=0.3) (A) and relapse-free survival (RFS) (n=7, M0=7) (p=0.03) (B) of melanoma patients according to TMB using 10muts/Kb as threshold. Kaplan-Meier curves of OS (C) (n=10, M0=8, M+=2) (p=0.4) and PFS(D) (n=10, M0=8, M+=2) (p=0.008) in melanoma patients (n=10) according to anti-PD-1 antibody therapy. n: Number of patients; M0: number of non-metastatic patients at diagnosis; M+: number of metastatic patients at diagnosis.
Next, we evaluated the efficacy of anti-PD1 therapy on OS and PFS. We compared metastatic patients (M+) at diagnosis with those relapsing after surgical therapy (M0). Although not statistically significant, we observed a median OS of 21 months for M+ individuals and 47 months for M0 patients (p=0.15) (Figure 2C). However, Kaplan-Meier estimates showed a significant correlation between efficacy of an anti-PD1 treatment and disease extent at diagnosis, with a median PFS of 4.5 (M+) versus 47 (M0) months, respectively (p=0.0009) (Figure 2D). These findings suggest that patients displaying metastatic disease at diagnosis may have limited benefit from anti-PD1 therapy compared to those relapsing after surgery.
Mutational landscape and VUS classification. We identified 45 known gene alterations represented by SNVs (65%), amplifications (19%), deletions (4%), losses (2%), rearrangements (2%), and frame-shifts (2%). No insertions were detected. In detail, patients affected by superficial spreading melanoma (37%) presented alterations in the TERT promoter, whereas individuals with nodular melanoma (50%) displayed mutations in both the TERT promoter and BRAF. Several additional genetic alterations were identified involving ARFRP1, ARID1A, ATRX, BRCA2, CDKN2A/B, CTNNB1, EP300, FLT3, GNAS, IDH1, IGF1R, KDR, Kit, MAP3K1, MEK1, MTAP, NF1, NRAS, PDGFRA, PTEN, ROS1, SMARCA4, SMARCB1, SNCAIP, SRC, TBX3, and TP53 (Figure 3A) (Table II).
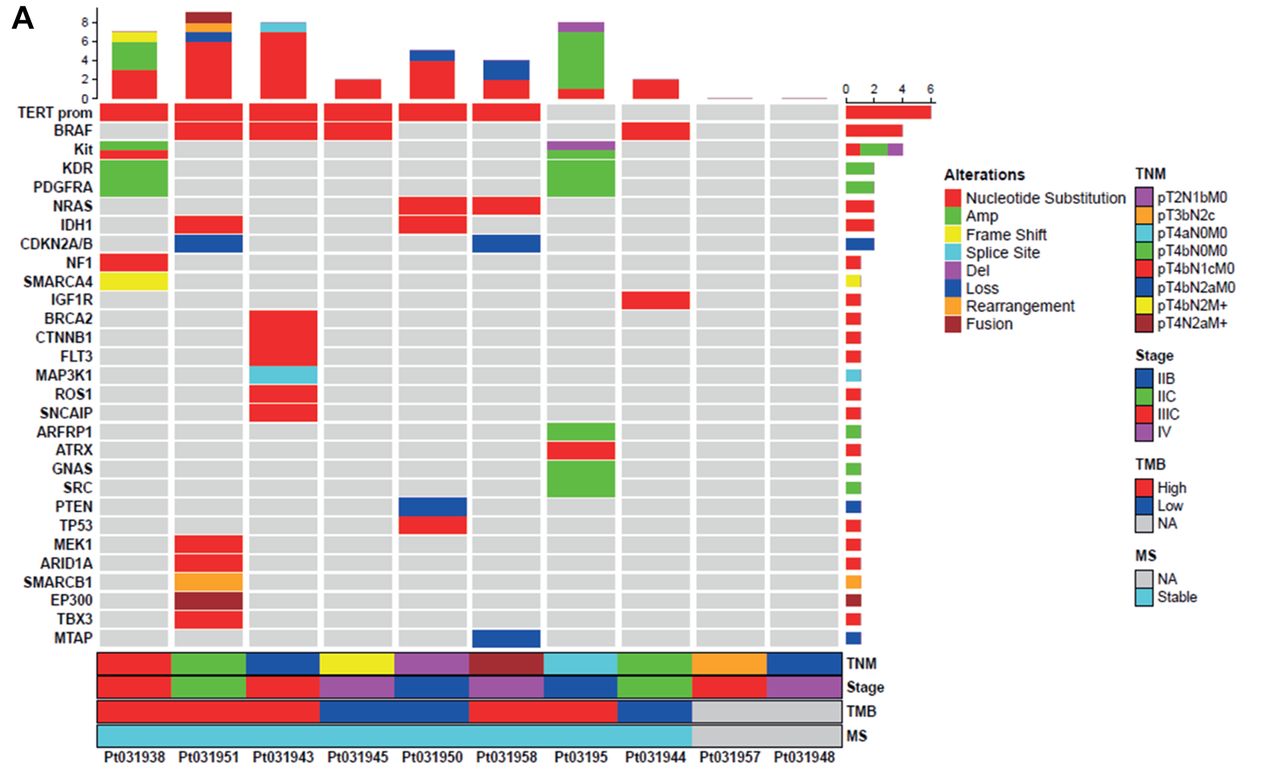
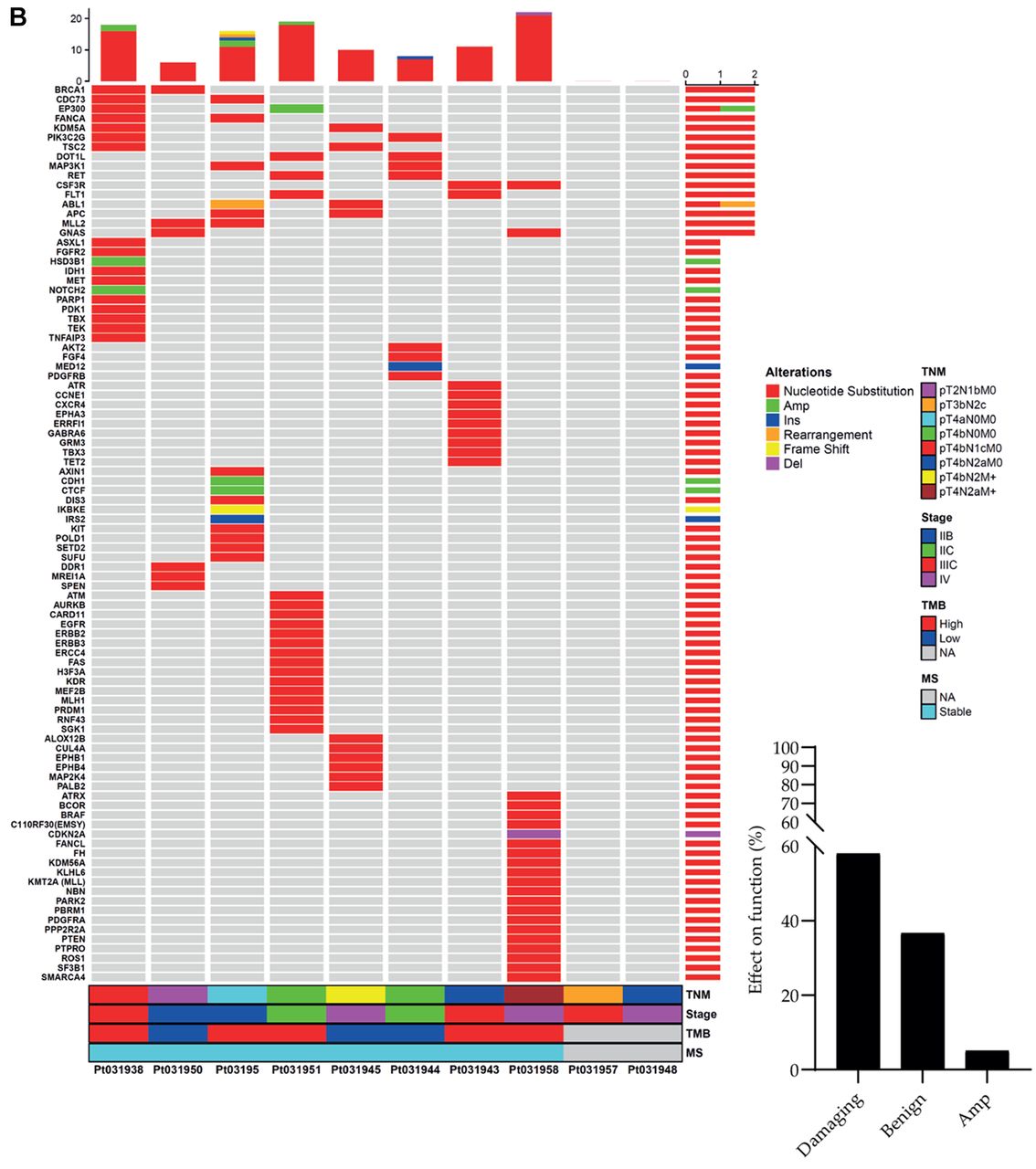
Tile plot and mutational landscape. Tile plots showing the mutational landscape of known gene alterations (A) and VUS (B) associated with the indicated clinical and molecular futures including TNM, stage, TMB, and MS of melanoma patients (n=8). The black columns report the percentage of distribution of pathogenic or benign VUS analyzed by Polyphen-2 v2 for single nucleotide variants (SNV), PROtein variation effect analyzer (PROVEAN) v1.1 for insertion/deletion (Ins/Del) and MutPredLOF for stop-codon and frame-shift. For gene amplifications, it was not possible to determine the impact on protein function and structure. Results derived from patients 957 and 948 are not applicable due to the poor quality of genomic material. n: Number of patients; TNM: tumor node metastasis; TMB: tumor mutational burden; MS: microsatellite status; VUS: variants of unknown significance.
Overview of clinical trial and targeted therapy approved for melanoma.
We then analyzed human 110 unknown genetic alterations represented by SNVs (91%), insertions (1.8%), deletions (0.9%), amplifications (4.5%), frame-shifts (0.9%), and rearrangements (0.9%). In order to evaluate the deleterious or tolerated impact of these genetic events on protein structure and function we used in silico prediction tools and generated the following categorization: 58% of VUS were classified as pathogenic and 36.7% as benign. A 5.3% of the genetic VUS could not be accurately categorized by our informatic predictors (Figure 3B, black columns).
Finally, patients displaying a TMB-High (62.5%) showed a superior mutation rate for both known genetic alterations and VUS when compared to those presenting a TMB-Low (37.5%) (Figure 3A and B).
Pathway enrichment. To translate the alterations found in our genetic analyses in information concerning improperly activated/suppressed intracellular pathways we performed a Panther analysis (Figure 4). Known genetic modifications altered the following pathways: angiogenesis and CCKR, EGFR, inflammation, insulin/IGF system, PDGF, PI3K, T cell, Toll like receptor, VEGF and Wnt (Figure 4A). Likewise, VUS altered the following pathways: angiogenesis and EGFR, FGF, Inflammation, Insulin/IGF system, p53 and PDGF signaling (Figure 4B). Interestingly, we observed a 71% overlap in the five signaling pathways (angiogenesis, EGFR, inflammation, insulin/IGF system and PDGF) affected by both known genetic alterations and VUS.

Panther classification system. Graphs report the percentage of significantly altered pathways obtained by Panther analysis using genes having known gene alterations (A) and pathogenic variants of unknown significance (B) as input.
PPI network and functional enrichment analysis of clusters. The search tool for retrieval of interacting genes (STRING) database, which integrates both known and predicted PPIs, was applied to predict functional interactions and cluster generation of the indicated proteins. To associate PPIs and clusters to a specific biological process we used the Enrichr tool (Figure 5).

STRING analysis of protein-protein interaction (PPI) network and clustering analysis by gene ontology (GO) biological process enrichment. Cartoon showing the PPI network and clustering analysis (colored circles) of known gene alterations (A) and variant of unknown significance (B). C indicates the clusters and GO the biological process. Red circles indicate the GO biological process shared between known gene alterations and VUS.
We identified one cluster for known genetic alterations (C1A) and seven clusters (from C1B to C7B) for VUS (Figure 5). Cluster C1A was enriched in kinase signaling (Figure 5A), whereas VUS-derived clusters showed the following enrichment: C1B, C4B and C7B: kinase signaling, C2B: DNA repair, C3B: DNA methylation, C5B: cell adhesion and migration, C6B: metabolic processes (Figure 5B). The association between clusters and the Gene Ontology biological process for known genetic alterations and VUS is reported in Table III and Table IV, respectively.
Top five gene ontology (GO) biological process of cluster derived from protein-protein interaction (PPI) of known gene alterations.
Top five gene ontology (GO) biological process of clusters derived from protein-protein interaction (PPI) of variant of unknown significance VUS.
Discussion
Most medical treatments are designed for the “average patient” as they can be successful for some but not all individuals. Personalized medicine is an innovative approach trying to distinguish patients who will benefit from a specific targeted treatment from those that should be considered for alternative therapeutic strategies. This approach has become increasingly successful in Oncology and, to date, a plethora of FDA-approved treatments are available according to the individual genetic profile of a patient with a specific form of cancer (23-28).
Here, we defined the molecular alterations of eight melanoma patients using the FDA-approved NGS-based platform FoundationOne CDx™. In agreement with previous evidence, we described a genetic landscape including alterations involving - among others - BRAF, CDKN2A, EGFR, IGF1R, NF1, PTEN, and the TERT promoter (29-32). Interestingly, we found co-existence of mutations on the TERT promoter and BRAF in patients with nodular melanoma, whereas those diagnosed with superficial spreading melanoma only presented TERT promoter alterations. In addition to known genetic modifications, we also identified a plethora of VUS that are of uncertain relevance as they cannot be used to inform clinical decision making in the absence of clear scientific evidence.
To gain further insight into the molecular mechanisms linking the above-described alterations to melanocyte neoplastic transformation, we employed the Panther enrichment tool. We found that both known genetic alterations and VUS mostly rely on the angiogenesis pathway, on EGFR, PDGF, and inflammation signaling, and on the insulin/IGF system (33-35). The 71% overlap in signal transduction between known genetic alterations and VUS supports the hypothesis that multiple genetic modifications - either well established or yet uncharacterized - share the same molecular mechanisms to drive the complex network required for melanoma tumorigenesis.
These data are consistent with those obtained after PPI network construction and cluster generation that identified kinase signaling as a common denominator linking most known genetic alterations. These findings were further strengthened by the observation that 42.8% of the VUS detected by our molecular analysis also converged on the kinase signaling cluster.
Since our cohort has a modest number of patients, further analyses are mandatory to extend the observed data in a larger number of patients. However, although this observation represents a limit of this study, our finding demonstrates that the combination of NGS analysis with complex bioinformatics tools generates accurate results as supported by previous published data (36).
In conclusion, we have analyzed the mutational landscape of patients with metastatic or relapsed cutaneous melanoma to define enriched pathways and protein-protein interactions contributing to melanocyte malignant transformation. We found that both known genetic alterations and VUS rely on redundant signaling converging on similar GO biological processes. Hence, complex informatic analyses of NGS-based genetic results identify pivotal signaling pathways that could provide additional druggable targets for cancer treatment.
Footnotes
Authors’ Contributions
Conceptualization, MM, and PV.; methodology, MM and SS; software, MM and GMi; validation, MM and SS; formal analysis MM and SS.; investigation, MM, SS and PV; resources, LM, GP, GMi, GM, HJSP, GB and EP; data curation, MM, SS, LM and PV; writing - original draft preparation, MM; writing - review and editing, PV, SS and LM, visualization, MM, SS, GMi, LM, GP, GB, EP, GM, HJSP, LM and PV; supervision, PV. All Authors have read and agreed to the published version of the manuscript.
Conflicts of Interest
The Authors declare no conflicts of interest in relation to this study.
- Received December 10, 2021.
- Revision received January 31, 2022.
- Accepted February 2, 2022.
- Copyright© 2022, International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved
This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY-NC-ND) 4.0 international license (https://creativecommons.org/licenses/by-nc-nd/4.0).
References
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.