


Abstract
Background/Aim: Opioid-binding protein/cell adhesion molecule-like (OPCML) plays a crucial role in the suppression of tumor progression in several cancer types. Nevertheless, the association between OPCML functions and cholangiocarcinoma (CCA) progression remains unknown. We aimed to investigate biological functions of OPCML and related signaling pathways in CCA cell lines. Materials and Methods: Methylation status and ectopic expression of OPCML were determined in CCA cell lines using methylation-specific polymerase chain reaction and pcDNA3.1+/C-(K)DYK-OPCML, respectively. Cell proliferation, migration and invasion were investigated. Results: OPCML was found to be epigenetically silenced by DNA methylation. Ectopic expression of OPCML inhibited CCA proliferation by inducing apoptosis via AXL receptor tyrosine kinase/signal transducer and activator of transcription 3 (AXL/STAT3) inactivation. It also suppressed cell migration and invasion via down-regulation of Rho GTPases, ras homolog family member A (RHOA), Rac family small GTPase 1 (RAC1) and cell division cycle 42 (CDC42). Conclusion: We are the first to unravel the antitumor effects and the related signaling pathways of OPCML in CCA. The loss of OPCML expression due to promoter hypermethylation can cause a decrease in cell death but increase in cell migration and invasion, which may at least in part contribute to CCA progression.
Cholangiocarcinoma (CCA) is an epithelial cancer of bile ducts (1), which is a rare cancer worldwide (2). However, it is the most common in the Northeastern region of Thailand, with the highest incidence approximately 85 per 100,000 (3). CCA is characterized by poor prognosis, and usually diagnosed at a late stage (4). Hence, the study of new molecules to be used as biomarkers is vital for early diagnosis. A previous study by Sriraksa et al. reported the methylation frequency of 26 CpG-island loci in CCA, of which opioid-binding protein/cell adhesion molecule like (OPCML) was the most frequently methylated locus along with low or no expression (5), suggesting the effect of DNA methylation on gene expression. In addition, Amonpisutt et al. also reported the significantly high level of methylation of OPCML in CCA when compared to adjacent normal tissues. They suggested that DNA methylation level of OPCML might be used as a potential diagnostic biomarker for CCA (6).
OPCML is a glycosyl phosphatidylinositol-anchored cell adhesion-like molecule. It normally acts as a cell adhesion molecule and negative regulator, and is associated with signal transduction by regulating a set of receptor tyrosine kinases including EPH receptor A2 (EPHA2), fibroblast growth factor receptor 1 (FGFR1), FGFR3, erb-b2 receptor tyrosine kinase 2 (HER2) and HER4 in normal ovarian epithelial cells (7). Moreover, OPCML acts as a tumor suppressor and is frequently inactivated by promoter hypermethylation, resulting in gene silencing in multiple cancer types, including invasive cervical carcinoma (8), bladder carcinoma (9), colorectal cancer (10), epithelial ovarian cancer (11), and CCA (5). Many studies have demonstrated the important roles of OPCML in the suppression of tumor progression in various types of human cancer (7, 10, 12–14). Nonetheless, the relationship between its biological roles and CCA progression remains unknown. Herein, we explored the involvement of OPCML methylation status and its endogenous expression in CCA cell lines. We also investigated the biological functions, as well as the related signaling pathways, when OPCML was ectopically expressed.
Materials and Methods
Cell lines and culture. The CCA cell lines (KKU-M213A, KKU-M055, and KKU-100) were established and kindly provided by the Cholangiocarcinoma Research Institute, Khon Kaen University, Thailand. KKU-M055 and KKU-M213A were cultured in Dulbecco’s modified Eagle’s medium, whereas KKU-100 was cultured in Ham’s F-12 Nutrient Mixture medium containing 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin (all from Thermo Fisher Scientific, Waltham, MA, USA). Cell culture was maintained at 37˚C with 5% CO2.
Vectors and transient transfection. pcDNA3.1+/C-(K)DYK-OPCML (GeneScript, Nanjing, PR China) and control vectors were included in this study. In brief, KKU-M213A (7.5×105 cells/well) and KKU-100 (4×105 cells/well) were seeded in six-well plates and incubated at 37˚C with 5% CO2 overnight. At the time of transfection, 70–90% confluent cells were then transfected with OPCML and control vectors using Lipofectamine 3000 reagent (Thermo Fisher Scientific) according to the manufacturer’s protocols. After 6-h incubation, medium was replaced before being incubated at 37˚C with 5% CO2 for 48 h, after which cells were harvested and used for further experiments.
Genomic DNA extraction, bisulfite treatment and methylation-specific polymerase chain reaction (MSP). Genomic DNA was extracted from CCA cell lines using QIAamp DNA blood mini kit (QIAGEN, Hilden, Germany) according to the manufacturer’s instructions, and subsequently modified with bisulfite using EZ DNA Methylation-GoldTM kit (Zymo Research, Irvine, CA, USA) following the manufacturer’s instructions. Afterward, bisulfite-modified DNA samples were amplified by MSP with methylation-specific primer set for OPCML promoter (Table I). The hot-start PCR reaction steps were carried out as follows: An initial denaturation at 95˚C for 5 min, followed by 35 cycles of the following steps: 95˚C for 30 s, 61˚C for 30 s, and 72˚C for 30 s, with a final elongation at 72˚C for 5 min. The amplified products were evaluated by 2% agarose gel electrophoresis.
List of primer sequences.
Total RNA extraction, cDNA synthesis and reverse transcription polymerase chain reaction (RT-PCR). Total RNA was extracted from CCA cell lines using TRIzol reagent (Thermo Fisher Scientific) and was then converted to cDNA using oligo-(dT)15 by ImProm-II™ Reverse Transcription System kit (Promega, Madison, WI, USA) according to the manufacturer’s instructions. PCR primers for human OPCML and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as an internal control are shown in Table I. RT-PCR was carried out as follows: An initial denaturation at 95˚C for 5 min, followed by 35 cycles of the following steps: 95˚C for 30 s, 58˚C for 30 s, and 72˚C for 30 s, with a final elongation at 72˚C for 10 min. The amplified products were evaluated by 2% agarose gel electrophoresis.
Protein extraction, concentration determination, and western blot analysis. Cells were lysed on ice for 15 min in RIPA buffer (Cell Signaling Technology, Danvers, MA, USA) containing protease and phosphatase inhibitors (Roche, Basel, Switzerland), then subjected to centrifugation at 16,000 × g for 20 min at 4˚C. After centrifugation, supernatants were collected and protein concentrations were determined by Coomassie Plus-The Better Bradford assayTM kit (Thermo Fisher Scientific). Protein lysates (20 μg) were separated on 8% or 12.5% sodium dodecyl sulphate-polyacrylamide gel electrophoresis and transferred onto a polyvinylidene difluoride membrane. The membrane was then blocked with 5% skim milk in Tris-buffered saline with 0.1% Tween 20 for 1 h, and incubated with primary antibodies overnight at 4˚C. The antibodies used were as follows: 1:1,000 rabbit monoclonal anti-OPCML (Abcam, Cambridge, UK), 1:1,000 rabbit monoclonal anti-AXL receptor tyrosine kinase (AXL) (C89E7), 1:500 rabbit monoclonal anti-phospho-AXL (Y779) (R&D systems, Minneapolis, MN, USA), 1:2,000 rabbit monoclonal anti-signal transducer and activator of transcription 3 (STAT3) (79D7) (Cell Signaling Technology), 1:1,000 rabbit monoclonal anti-phospho-STAT3 (Ser727), 1:500 mouse monoclonal anti-RAS homolog family member A (RHOA), 1:500 mouse monoclonal anti-RAC family small GTPase 1 (RAC1), 1:500 mouse monoclonal anti-cell division cycle 42 (CDC42) (Cell Biolabs, San Diego, CA, USA), and 1:10,000 rabbit polyclonal anti-actin (Abcam). The membrane was washed three times, then incubated with horseradish peroxidase-conjugated secondary antibodies: 1:2,000 donkey anti-rabbit IgG or goat anti-mouse IgG (Abcam) for 1 h. After washing three times, proteins were detected by ECL™ reagents (GE Healthcare, Chicago, IL, USA). The protein images were analyzed by Amersham Imager 600 (GE Healthcare), and band intensities were quantified by Image J software (U. S. National Institutes of Health, Bethesda, MD, USA).
Proliferation assay. Cell proliferation was performed using CellTiter 96® AQueous One Solution Cell Proliferation Assay (Promega). Transfected KKU-M213A (5×103 cells) and KKU-100 (2×103 cells) in 100 μl of complete medium were seeded in a 96-well plate in triplicates, and baseline readings were taken on the day of seeding as soon as cells were attached (15). At baseline and each time-point (day 0–4 for KKU-M213A and day 0–6 for KKU-100), 20 μl of 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium solution (MTS) was added to each well and plates were incubated for 3 h before absorbance was measured at 490 nm using a RT-2100C Microplate Reader (Rayto Life and Analytical Sciences, Shenzhen, PR China).
Migration and invasion assays. Cell migration and invasion assays were performed using Transwell® with an 8.0 μm Pore PET Membrane Insert with and without Matrigel (Corning Incorporated, Corning, NY, USA). Briefly, transfected KKU-M213A (4×104 cells) and KKU-100 (3×104 cells) in 300 μl of serum-free medium were seeded into the upper chamber in duplicate and 750 μl of complete medium was placed in the lower chamber. After 18 h incubation for KKU-M213A and 48 h for KKU-100, non-migrating or non-invading cells residing at the upper filter were removed. Migrating or invading cells that were attached to the underside of the filter were fixed with 100% ice-cold methanol and stained by 2% crystal violet in 2% ethanol at room temperature for 15 min. The number of migrating or invading cells was quantified by counting under light microscope. The mean value of five fields at ×100 magnification was calculated.
Apoptosis assay. Cell apoptosis was executed by Alexa Fluor® 488 annexin V/Dead Cell Apoptosis kit (Thermo Fisher Scientific). In brief, transfected cells were harvested and washed twice with ice-cold phosphate-buffered saline. Cells were then resuspended in 1x annexin binding buffer, and stained with 5 μl of annexin V and 1 μl of 100 μg/ml propidium iodide for 15 min at room temperature in the dark. Samples prepared in duplicate were measured by Becton Dickinson flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA). The data were analyzed by BD FACSDiva software (BD Biosciences).
Statistical analysis. SPSS version 25 software (IBM, Armonk, NY, USA) was used for statistical analyses. Values are presented as the mean±standard deviation (SD) and significance was analyzed by independent-samples t-test. A value of p<0.05 was considered statistically significant.
Results
OPCML is epigenetically silent by promoter hypermethylation in all CCA cell lines. We initially determined the expression of mRNA and protein of OPCML in CCA cell lines using RT-PCR and western blot analysis, respectively. Neither mRNA nor protein expression of OPCML was observed in any of the three CCA cell lines (Figure 1A and B). The methylation status of OPCML promoter was further investigated in CCA cell lines using MSP. As shown in Figure 1C, all three CCA cell lines were methylated at the OPCML promoter, indicating that DNA methylation regulated OPCML expression in CCA.
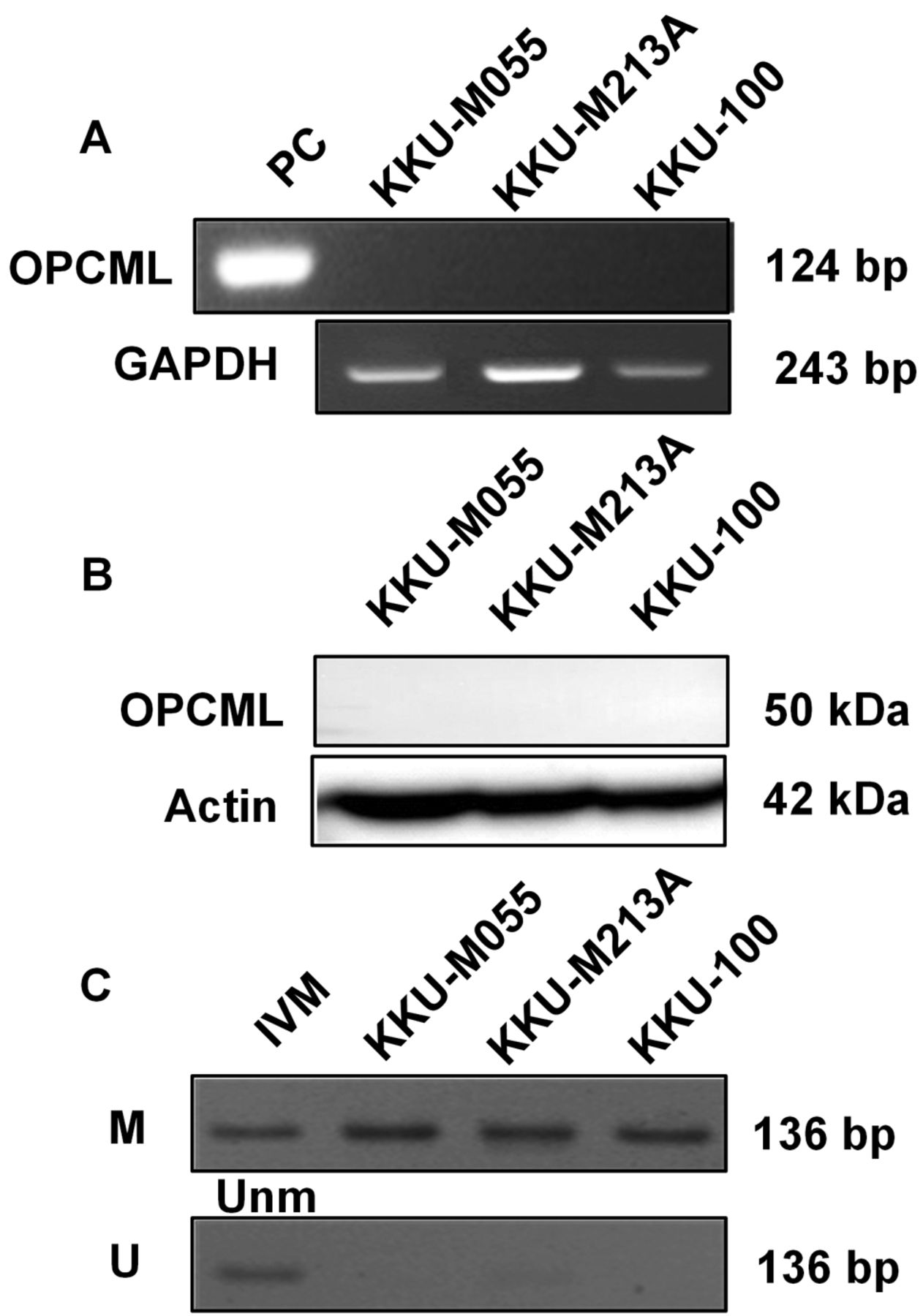
Loss of opioid binding protein/cell adhesion molecule like (OPCML) expression is regulated by promoter methylation in all cholangiocarcinoma (CCA) cell lines. Neither the expression of mRNA (A) nor protein (B) was detected in CCA cell lines while DNA methylation of the OPCML promoter was observed in all (C). IVM: In vitro methylation; M: methylated; PC: positive control; U/Unm: unmethylated.
Overexpression of OPCML in CCA cell lines after transient transfection. To investigate the biological functions of OPCML in CCA, pcDNA3.1+/C-(K)DYK-OPCML was transiently transfected into KKU-M213A and KKU-100 cells. As shown by RT-PCR and western blot, OPCML was significantly overexpressed in KKU-M213A and KKU-100 cells after transient transfection with OPCML plasmid compared to control vector (Figure 2).

Opioid binding protein/cell adhesion molecule like (OPCML) was significantly overexpressed in representative cholangiocarcinoma cell lines after transient transfection. Expression of OPCML mRNA (A) and protein (B) in KKU-M213A and KKU-100 cells after transfection with OPCML plasmid for 48 h.
Ectopic OPCML expression suppresses cell proliferation by inducing apoptosis of CCA cells. We assessed the effect of ectopic OPCML expression on the proliferation of CCA cells by carrying out MTS proliferation assay. We found that ectopic OPCML expression significantly reduced the viability of KKU-M213A on days 1–4 (Figure 3A, left panel) and of KKU-100 on days 1–6 (Figure 3A, right panel), indicating the anti-proliferative effect of OPCML on CCA cells. Furthermore, we investigated whether OPCML inhibited CCA cell proliferation by inducing apoptosis. Annexin V-fluorescein isothiocyanate/propidium iodide flow cytometry was used to detect the effect of ectopic OPCML expression on apoptosis of KKU-M213A and KKU-100 cells. We found that OPCML increased the populations of both early and late apoptotic cells from 8.4% to 14.2% (p<0.05) in KKU-M213A cells transfected with OPCML, when compared to control vector. Similar results were observed in OPCML-transfected KKU-100 cells, with a significant induction of the apoptotic cell population from 26.3% to 33.8% (p<0.01), when compared to cells transfected with control vector (Figure 3B). These data indicated that OPCML inhibited proliferation of CCA cells by inducing cell apoptosis.

Opioid binding protein/cell adhesion molecule like (OPCML) diminished cholangiocarcinoma cell proliferation by inducing apoptosis. A: The effect of forced expression of OPCML on cell proliferation as shown by 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay in KKU-M213A and KKU-100 cells. B: Representative cytograms and quantification of KKU-M213A and KKU-100 cells undergoing apoptosis after transfection with OPCML compared to control vectors. Values are expressed as mean±SD from independent experiments running in triplicate (MTS assay) or duplicate (flow cytometry). Statistically significantly different compared to control vector at: *p<0.05, **p<0.01 and ***p<0.001.
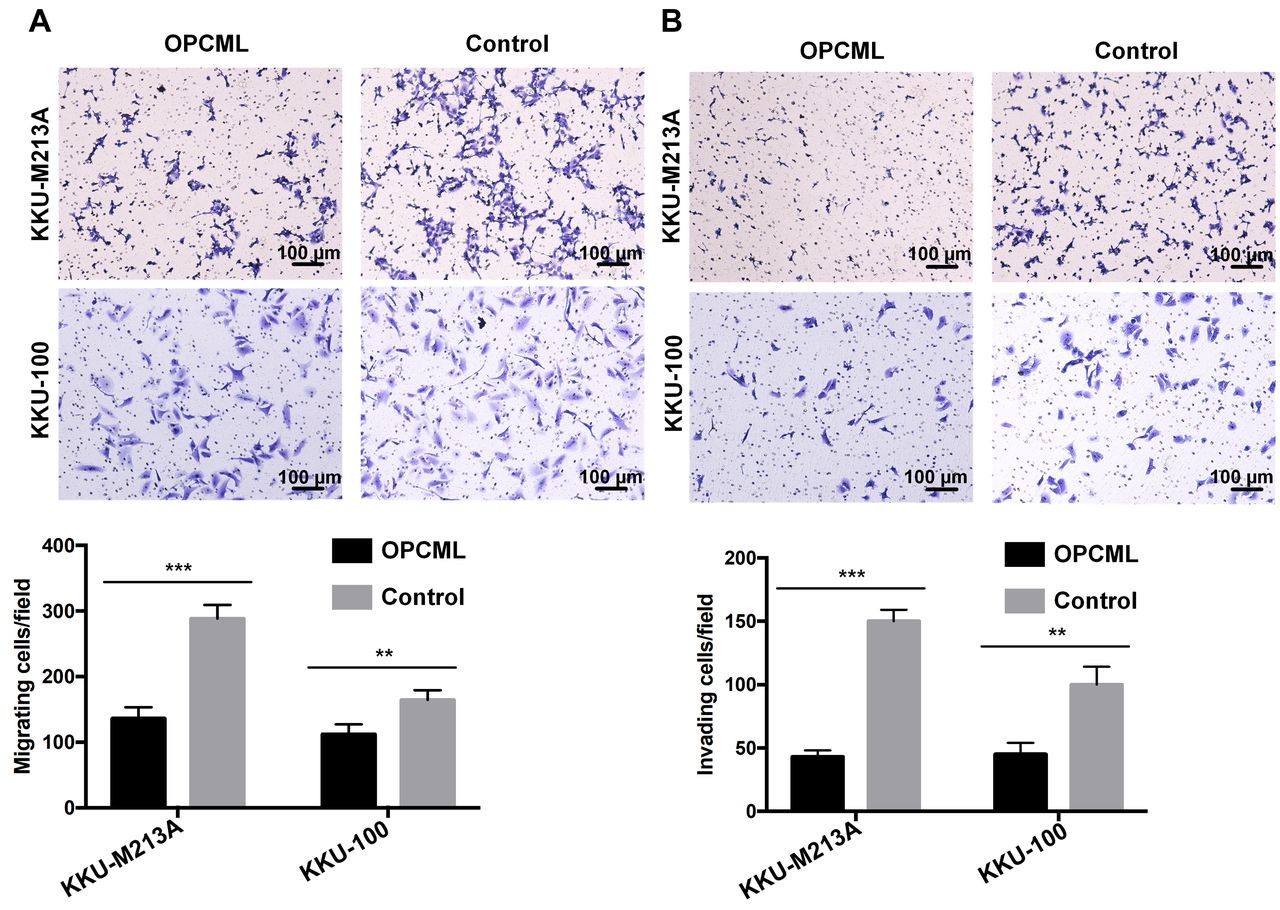
OPCML inhibits migration and invasion of CCA cells. To further explore the effect of OPCML on the migration and invasion of CCA cells, we performed transwell assays with and without Matrigel-coated chambers. The results showed that fewer OPCML-transfected KKU-M213A and KKU-100 cells migrated to the lower chamber than those transfected with control vector (p<0.01, Figure 4A). In addition, a similar result was also observed in the cell invasion assay (p<0.01, Figure 4B). Our findings indicated that OPCML suppressed migration and invasion by CCA cells.

Opioid binding protein/cell adhesion molecule like (OPCML) suppressed migration and invasion of cholangiocarcinoma cell lines. Representative results showing the effect of forced expression of OPCML on cell migration (A) and invasion (B) as shown by transwell assay. The migrating and invading cells were counted under light microscopy with an objective lens of 10×. Five randomly selected fields for each membrane filter were counted. Values are expressed as the mean±SD from independent experiments running in duplicate. Statistically significantly different compared to control vector at: **p<0.01 and ***p<0.001.
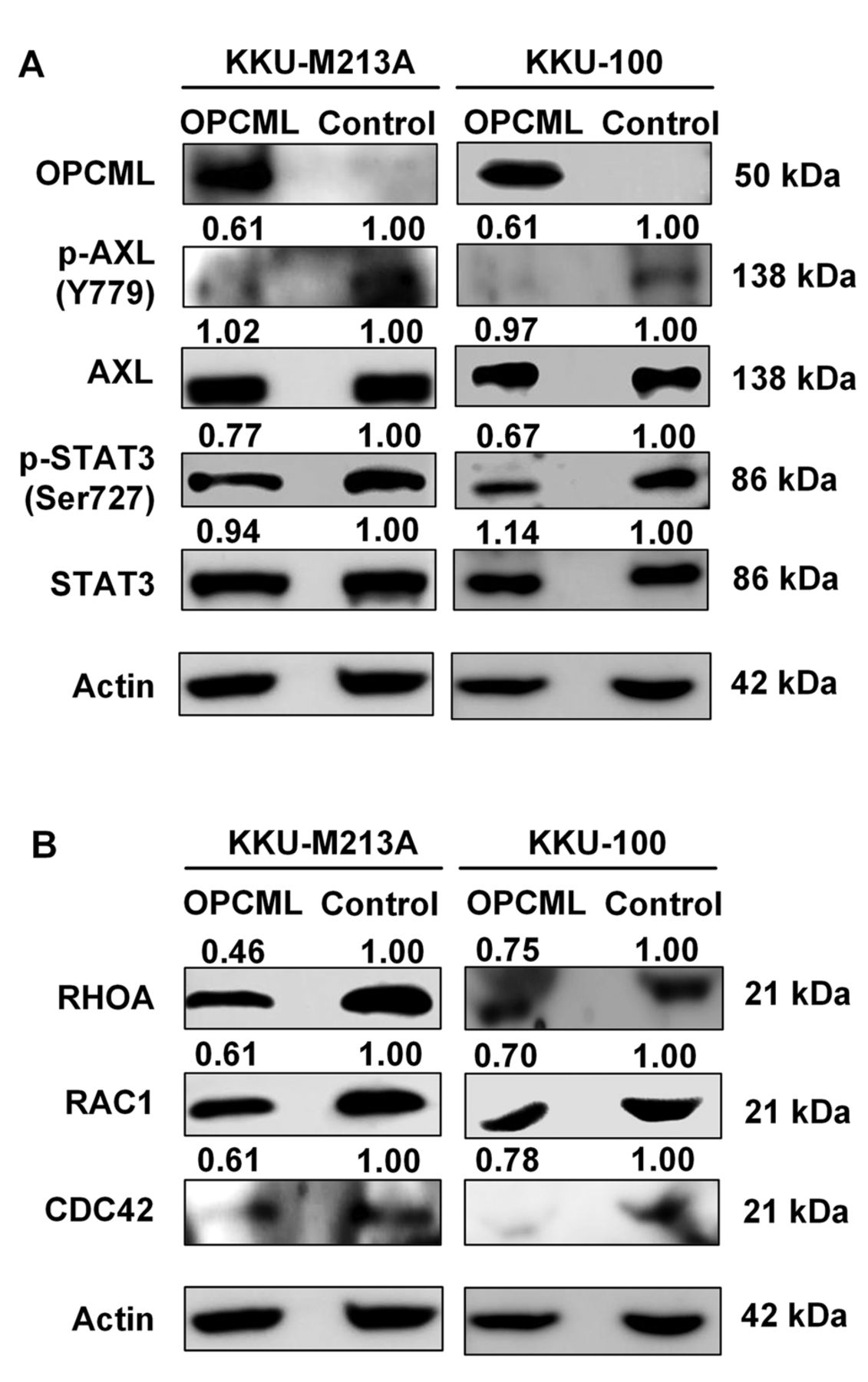
OPCML expression is associated with AXL/STAT3 inactivation and down-regulation of Rho GTPases in CCA cells. We further sought the OPCML-mediated signaling pathways. To address whether OPCML interacted with pro-metastatic receptor tyrosine kinase, AXL and inhibited proliferation of CCA, we assessed the phosphorylation status of AXL and its downstream protein, STAT3 after OPCML overexpression. In the absence of OPCML, AXL and STAT3 were activated in both KKU-M213A and KKU-100 cells, whereas the levels of phosphorylated AXL and STAT3 markedly declined after ectopic OPCML expression (Figure 5A). We next investigated whether the effects of OPCML on migration and invasion were related to Rho GTPase expression. After 48-h transfection with OPCML plasmid vector, expression of Rho GTPases RHOA, RAC1, and CDC42 was significantly reduced in both KKU-M213A and KKU-100 cells, compared to the controls (Figure 5B). Activation of Rho GTPases was also determined in CCA cell lysates using pull-down assay. However, Rho GTPase activation was not observed in CCA cell lines, neither in the absence nor presence of OPCML. Overall, our findings suggest that OPCML may regulate CCA cell proliferation through the inhibition of AXL/STAT3 activation, and perhaps control the migration and invasion of CCA cells, at least in part, via down-regulation of Rho GTPases (Figure 6).

Opioid binding protein/cell adhesion molecule like (OPCML)-related signaling component proteins as revealed by western blot analysis in cholangiocarcinoma cells. A: The levels of phosphorylated AXL receptor tyrosine kinase (AXL)/signal transducer and activator of transcription 3(STAT3) signaling pathway proteins under forced expression of OPCML. B: Expression of Rho GTPases; ras homolog family member A (RHOA), Rac family small GTPase 1 (RAC1) and cell division cycle 42 (CDC42). The band intensity of each protein was normalized to that of the internal control, actin. The level of the phosphorylated form was then normalized to its total form. The numbers represented the fold change from the respective control group.

Schematic representation of opioid binding protein/cell adhesion molecule like (OPCML)-mediated signaling pathways in cholangiocarcinoma (CCA) cells. OPCML interferes with AXL receptor tyrosine kinase (AXL) homodimerization and phosphorylation, resulting in reduced phosphorylation of signal transducer and activator of transcription 3 (STAT3), which is an important transcription factor in activating proliferation-related genes. Moreover, OPCML can also reduce expression of Rho GTPases, leading to the suppression of CCA cell migration and invasion.
Discussion
OPCML gene is located on chromosome 11q25 and was initially identified as a candidate tumor-suppressor gene in epithelial ovarian cancer, in which it is frequently epigenetically inactivated (11). Further studies later illustrated the down-regulation or silencing of OPCML due to its promoter methylation in various primary tumor types and cancer cell lines but not in non-cancer cell lines, suggesting its broad tumor suppressor role for multiple tumor types (16). In addition, previous studies demonstrated low expression of OPCML to be associated with the carcinogenesis of many cancer types (6, 8, 9, 12, 17–19). In the current study, we showed the loss of mRNA and protein expression, and promoter hypermethylation of OPCML in CCA cell lines. Sriraksa et al. demonstrated the high methylation frequency (72.5%) for OPCML in primary CCA, in which low protein expression was found in 88% of OPCML-hypermethylated CCA (5). The data from primary CCA and cell lines indicate the expression of OPCML is epigenetically regulated by DNA methylation. Moreover, methylation of OPCML was found more frequently in less differentiated than in well-differentiated CCA, supporting its role in tumor progression (5). A previous study in CCA illustrated that less differentiated tumors conferred a poorer outcome and high incidence of metastases than well differentiated ones (20).
To address the roles of OPCML in CCA progression, CCA cell lines KKU-M213A and KKU-100 were transfected with OPCML plasmid vector. As a result, overexpression of OPCML inhibited proliferation of CCA via induction of apoptosis. Previous studies also showed that ectopic OPCML expression inhibited the proliferation of gastric and breast cancer cells (12, 13), in which OPCML also activated apoptosis of gastric cancer cells (12). Metastasis is a key driver of tumor progression that involves cell migration and invasion. We found that ectopic expression of OPCML reduced migration and invasion of CCA cells. The effect of OPCML on the suppression of cell migration and invasion was also reported in breast and ovarian cancer cells (13, 14). Moreover, Li et al. found that OPCML induced the reversal of epithelial-mesenchymal transition, which was associated with migration and invasion of colorectal cancer cells (10). The data from our and previous studies indicate a tumor-suppressive role of OPCML whose loss of its expression can lead to cancer progression.
The molecular mechanism underlying OPCML repression of cell proliferation by inducing apoptosis was investigated. Our study demonstrated that the activation of receptor tyrosine kinase, phospho-AXL was reduced in CCA cell lines after ectopic expression of OPCML. Similarly, the activation of its downstream target, phospho-STAT3 was also reduced. AXL is one of the three members of the TYRO3 protein tyrosine kinase, AXL and MER proto-oncogene tyrosine kinase (TAM) receptor tyrosine kinase family which play critical roles in regulating cell proliferation, adhesion, and migration (21). Activation of AXL has been linked to tumor growth, high invasiveness and metastasis of many cancer types (22). Additionally, AXL was recently reported to be a crucial target of OPCML, resulting in the inhibition of motility and invasion of ovarian cancer cells (14). Many studies have elucidated that AXL may potentially drive cell proliferation through effector molecules in the Janus kinase/STAT signaling pathway (23-26). STAT3 plays important roles in proliferation, survival, apoptosis, angiogenesis, and metastasis (27). Furthermore, the activation of STAT3 is also vital in CCA for cell growth and metastasis (28). In this study, the effect of OPCML on the expression but not activation of Rho GTPases (RHOA, RAC1 and CDC42) resulting in reduced cell migration and invasion was observed in CCA cell lines. The Rho GTPases belong to a subgroup of the Ras superfamily of 20–30 kDa GTP-binding proteins (29). They promote many cellular processes including actin organization, and regulation of gene expression and cell migration (30). Alterations of expression levels or activity of Rho GTPases have been reported in many cancer types. Rho GTPases are often up-regulated in human cancer, RHOA has particularly been found to be overexpressed in testicular cancer and esophageal squamous cell carcinoma (31, 32). In addition, RAC1 has been shown to be up-regulated in testicular, breast, prostate, gastric and lung tumors (31, 33-36). CDC42 is also overexpressed in testicular and breast cancer (31, 33), non-small cell lung carcinoma (37) and colorectal adenocarcinoma (38). Moreover, many studies illustrated a crucial role of Rho GTPases in the progression and metastasis of various human cancer types (29, 30, 38–40). The mechanism by which OPCML regulates the expression of Rho GTPases in CCA requires further investigation.
Taken together, our findings suggest that OPCML plays an important role as a tumor suppressor by regulating AXL, in which its inactivation by promoter hypermethylation results in the activation of STAT3 and up-regulation of Rho GTPase signaling pathways, leading to the progression of CCA. In conclusion, our study confirmed that OPCML was epigenetically silenced in CCA. Overexpression of OPCML suppressed CCA cell proliferation, migration, and invasion, suggesting that OPCML exerts its suppressor activity at least in part through AXL/STAT3 and Rho GTPase signaling pathways. To our knowledge, this is the first time that the mechanisms related to antitumor effects of OPCML in CCA were unraveled.
Acknowledgements
This work was financially supported by the Thailand Science Research and Innovation (TSRI) through the Royal Golden Jubilee Ph.D. Program (grant no. PHD/0015/2557 to R. Khamko) and Grant-in-aid from KKU Research Funds and the Centre for Research and Development of Medical Diagnostic Laboratories, Faculty of Associated Medical Sciences, Khon Kaen University, Thailand. We express our appreciation to the Cholangiocarcinoma Research Institute, Khon Kaen University, Khon Kaen, Thailand for kindly providing CCA cell lines.
Footnotes
Authors’ Contributions
R. Khamko: Conceptualization, methodology, formal analysis, investigation, data curation, writing – original draft, visualization and project administration. J. Daduang: Conceptualization, methodology and validation. C. Settasatian: Conceptualization, methodology and validation. T. Limpaiboon: Conceptualization, methodology, validation, resources, writing – review and editing, supervision and funding acquisition.
This article is freely accessible online.
Conflicts of Interest
The Authors declare no conflicts of interest.
- Received August 11, 2021.
- Revision received October 9, 2021.
- Accepted October 12, 2021.
- Copyright© 2021, International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}