


Abstract
Background/Aim: Benign smooth-muscle tumors, leiomyomas, occur in nearly every organ but are most common in the uterus. Whereas much is known about the genetics of uterine leiomyomas, little genetic information exists about leiomyomas of other organs. Here, we report and discuss the genetic findings in a para-testicular leiomyoma. Materials and Methods: Cytogenetic, array comparative genomic hybridization (aCGH) RNA sequencing, reverse-transcription polymerase chain reaction (RT- PCR), and Sanger sequencing analyses were performed on a leiomyoma of the spermatic cord removed from a 61-year-old man. Results: The karyotype was 48~50,XY,add(3) (p21),+4,+7,+8,+9,add(21)(q22)[cp9]/46,XY[2]. aCGH confirmed the trisomies and also detected multiple gains and losses from 3p and 21q. RNA sequencing detected the chimeras ARHGEF3–CACNA2D2, TRAK1–TIMP4, ITPR1– DT-NR2C2, CLASP2–IL17RD, ZNF621–LARS2, CNTN4– RHOA, and NR2C2–CFAP410. All chimeras were confirmed by RT-PCR and Sanger sequencing. Conclusion: Our data, together with those previously published, indicate that a group of leiomyomas may be cytogenetically characterized by aberrations of 3p and the formation of fusion genes.
Leiomyomas are benign smooth-muscle tumors. They have been described in nearly every organ but are most common in the uterus (fibroids) (1-5).
Much is known about the genetics, and hence the pathogenesis, of uterine leiomyomas (6-9). In brief, most uterine leiomyomas are cytogenetically characterized by the presence of one or more of the following cytogenetic aberrations: t(12;14)(q15;q23–24); del(7)(q21.2q31.2); rearrangements involving 6p21, 10q22, and 1p; trisomy 12; deletions of 3q; and changes of the X chromosome (10, 11). These chromosomal aberrations rearrange and deregulate the following genes: High mobility group AT-hook 2 (HMGA2) on 12q14, RAD51 paralog B (RAD51B) on 14q24, cut-like homeobox 1 (CUX1) on 7q22, procollagen C-endopeptidase enhancer (PCOLCE) on 7q22, HMGA1 (on 6p21), lysine acetyltransferase 6B (KAT6B) on 10q22, and collagen type IV alpha 5 chain/collagen type IV alpha 6 chain (COL4A5/ COL4A6) on Xq22 (6-9). Additionally, point mutations of the mediator complex subunit 12 (MED12) gene (on Xq13) have also been reported to be frequent in uterine leiomyomas (12-15). Very little genetic information exists on leiomyomas of other organs, such as gastrointestinal (16-20) and retroperitoneal leiomyomas (21-23). A single para-testicular leiomyoma has also been genetically examined (24).
In the present study, we report rearrangements of the p arm of chromosome 3 in a leiomyoma of the spermatic cord leading to multiple fusion genes. We review the literature and conclude that such 3p aberrations may be characteristic of a leiomyoma subgroup.
Materials and Methods
Ethics statement. The study was approved by the regional Ethics Committee (Regional komité for medisinsk forskningsetikk Sør-Øst, Norge, http://helseforskning.etikkom.no) and written informed consent was obtained from the patient. The Ethics Committee’s approval included a review of the consent procedure. All patient information has been de-identified.
Case description. A small lump was detected in the groin of a 60-year-old man. Core-needle biopsy showed a leiomyomatous tumor with uncertain malignant potential. The tumor was resected. The tumor consisted of bundles of smooth muscle cells without atypia (Figure 1). There was no mitotic activity nor necrosis. The final diagnosis was spermatic cord leiomyoma.
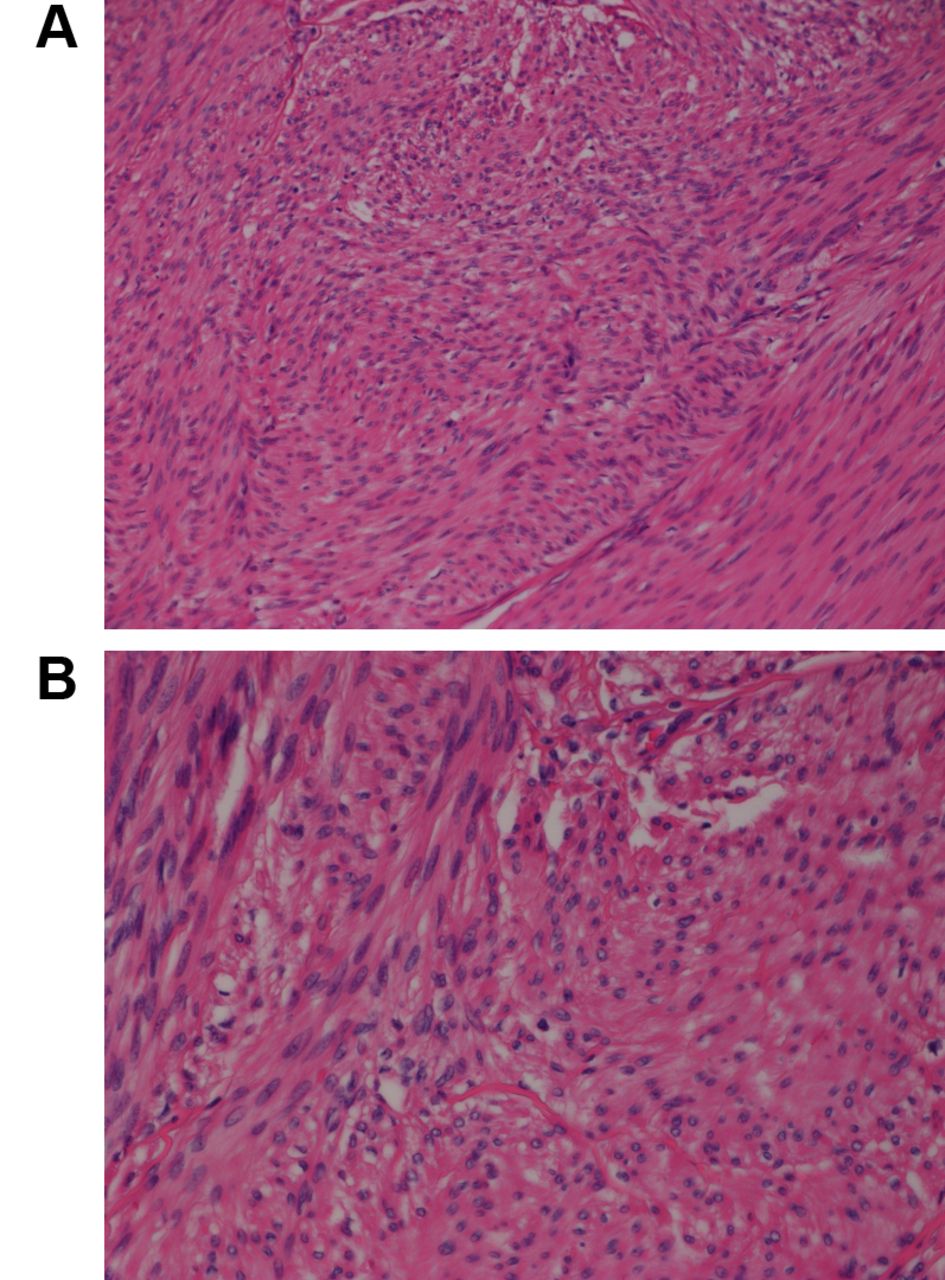
Microscopic examination of the spermatic cord leiomyoma. Hematoxylin and eosin-stained section showing bundles of elongated cells with cigar-shaped nuclei and eosinophilic cytoplasm. Original magnification, A: 20×; B: 40×.
G-Banding karyotyping and array comparative genomic hybridization (aCGH). analyses. Fresh tissue from a representative area of the tumor was analyzed cytogenetically as previously described (25). Genomic DNA was extracted from the tumor using the Maxwell RSC Instrument and Maxwell RSC Tissue DNA Kit (Promega, Madison, WI, USA), the concentration was measured using Quantus Fluorometer and the QuantiFluor ONE dsDNA System (Promega), and aCGH was performed using CytoSure array products (Oxford Gene Technology, Begbroke, Oxfordshire, UK) as previously described (26).
RNA sequencing. Total RNA was extracted from frozen tumor tissue adjacent to that used for cytogenetic analysis and histological examination using miRNeasy Mini Kit (Qiagen, Hilden, Germany). One microgram of total RNA from the tumor was sent to the Genomics Core Facility at the Norwegian Radium Hospital, Oslo University Hospital for high-throughput paired-end RNA-sequencing. Fusion transcripts were found using FusionCatcher software (27, 28).
Reverse transcription PCR (RT-PCR) and Sanger sequencing analyses. The primers used for PCR amplification and Sanger sequencing analyses are shown in Table I. The primer combinations and the corresponding fusion transcripts are presented in Table II. The iScript Advanced cDNA Synthesis Kit for RT-PCR was used to reverse transcribe 1 μg of total RNA according to the manufacturer’s instructions (Bio-Rad, Hercules, CA, USA). cDNA corresponding to 20 ng total RNA was used as template in subsequent PCR assays. BigDye Direct Cycle Sequencing Kit was used to perform both PCR and cycle (Sanger) sequencing according to the company’s recommendations (ThermoFisher Scientific, Waltham, MA, USA). Sequencing was run on an Applied Biosystems SeqStudio Genetic Analyzer system (ThermoFisher Scientific). For computer analysis of sequence data, the basic local alignment search tool (BLAST) software was used (29).
Primers used for polymerase chain reaction amplification and Sanger sequencing analyses. The forward primers (F1 and F2) had at their 5’-ends the M13 forward primer sequence: 5’-TGTAAAACGACGGCCAGT-3’. The reverse primers (R1) had at their 5’-ends the M13 reverse primer sequence: 5’-CAGGAAACAGCTATGACC-3’.
Primer combinations for polymerase chain reaction amplification to detect fusion transcripts.
Results
The G-banding analysis (Figure 2A) yielded the following karyotype: 48~50,XY,add(3)(p21),+4,+7,+8,+9,add(21)(q22)[cp9]/46,XY[2].

Cytogenetic and array comparative genomic hybridization (aCGH) analyses of the spermatic cord leiomyoma. A: Partial karyotype showing the add(3) and add(21) chromosomes with corresponding normal chromosome homologs. B: aCGH showing gains of one copy of each of chromosomes 4, 7, 8, and 9, as well as multiple gains and losses of genomic material from the p arm of chromosome 3 and q arm of chromosome 21. C: aCGH showing multiple gains and losses of genomic material from the p arm of chromosome 3. D: aCGH showing the multiple gain and losses on the q arm of chromosome 21.
aCGH confirmed trisomy for chromosomes 4,7,8, and 9 (Figure 2B). In addition, multiple gains and losses were detected on the p arm of chromosome 3 (Figure 2C) and on the q arm of chromosome 21 (Figure 2D).
RNA sequencing analysis with FusionCatcher detected seven fusion genes (Table III, Figure 3A). One transcript was detected for each of the chimeras inositol 1,4,5-trisphosphate receptor divergent transcript–nuclear receptor subfamily 2 group C member 2 (ITPR1–DT–NR2C2), cytoplasmic linker associated protein 2–interleukin 17 receptor D (CLASP2– IL17RD), nuclear receptor subfamily 2 group C member 2– cilia and flagella-associated protein 410 (NR2C2–CFAP410), zinc finger protein 621–leucyl-tRNA synthetase 2, mitochondrial (ZNF621–LARS2), and contactin 4–ras homolog family member A (CNTN4–RHOA), whereas two chimeric transcripts were detected for the fusion genes Rho guanine nucleotide exchange factor 3–calcium voltage-gated channel auxiliary subunit alpha2delta 2 (ARHGEF3– CACNA2D2) and trafficking kinesin protein 1–TIMP metallopeptidase inhibitor 4 (TRAK1–TIMP4) (Table III). In six of the fusion genes, both the 5’-end and 3’-end fusion partner genes were mapped on chromosome arm 3p (Table III, Figure 3A). In the seventh, the NR2C2-CFAP410 chimera, the 5’-end partner gene, NR2C2, was from 3p25.1, whereas CFAP410 was from 21q22.3 (Tables I and III). All fusion transcripts were verified using RT-PCR and Sanger sequencing analyses (Figure 3B).
Fusion transcripts detected in the spermatic cord leiomyoma using RNA sequencing and FusionCatcher.

The detected chimeras in the spermatic cord leiomyoma. A: Diagram of the p arm of chromosome 3, showing the mapping positions of the genes contactin 4 (CNTN4); inositol 1;4;5-trisphosphate receptor divergent transcript LITPR1-DT); TIMP metallopeptidase inhibitor 4 (TIMP4); nuclear receptor subfamily 2 group C member 2 (NR2C2); cytoplasmic linker associated protein 2 (CLASP2); zinc finger protein 621 (ZNF621); trafficking kinesin protein 1 (TRAK1); leucyl-tRNA synthetase 2, mitochondrial (LARS2); ras homolog family member A (RHOA); calcium voltage-gated channel auxiliary subunit alpha2delta 2 (CACNA2D2); Rho guanine nucleotide exchange factor 3 (ARHGEF3); and interleukin 17 receptor D (IL17RD); as well as the chimeric genes CNTN4−RHOA, ITPR1DT−NR2C2, TRAK1−TIMP4, CLASP2−IL17RD, ZNF621−LARS2 and ARHGEF3−CACNA2D2. The ↑ arrow next to a gene symbol indicates transcription of the gene from centromere to telomere. The ↓ arrow indicates transcription of the gene from telomere to centromere. B: Partial sequence chromatograms showing the junction positions in the chimeric transcripts CNTN4–RHOA, ITPR1-DT–NR2C2, NR2C2−CFAP410, CLASP2−IL17RD, ZNF621−LARS2, TRAK1−TIMP4 and ARHGEF3−CACNA2D2.
Discussion
Prior to the present study, to our knowledge, only a single para-testicular leiomyoma, located beneath the tunica albuginea, had been cytogenetically described, by Gorunova et al. (24). That tumor had an abnormal karyotype: 46,XY,der(5)t(5;14)(q31;q24),der(14)t(12;14)(q15; q24). The net imbalance of the structural rearrangements thus seemed to be the loss of 5q31qter and gain of 12q15qter. Molecular investigations were not performed, but strong nuclear immunostaining was found for the HMGA2 protein in the tumor cells, suggesting that the der(14)t(12;14)(q15;q24) dysregulated the expression of the HMGA2 gene. Because t(12;14)(q15;q24) is the most frequent translocation in uterine leiomyomas and leads to overexpression of HMGA2, the authors concluded that a common pathogenetic pathway exists for both para-testicular (male) and uterine (female) leiomyomas (24).
The present leiomyoma was found in the spermatic cord and had different cytogenetic aberrations from those described by Gorunova et al. (24). Trisomies were found for chromosomes 4, 7, 8, and 9, whereas structural aberrations affected chromosome arms 3p and 21q (Figure 2A). The observed numerical aberrations have also been reported in leiomyomas (30): trisomies for chromosomes 4, 8, and 9 in uterine leiomyomas (16, 31-35) and trisomy 7 in uterine, bladder, and gastric leiomyomas (16, 17, 31, 36).
Rearrangements of the 3p arm were reported in 22 uterine leiomyomas (30). In 12 of them, the 3p aberration was found together with rearrangements of chromosome bands 12q14~15 or band 6p21. Leiomyoma aberrations in the region 12q14~15 target the HMGA2 gene, whereas aberrations in 6p21 target HMGA1 (37, 38). Reports have been published on nine leiomyomas with aberration in the p arm of chromosome 3 without microscopically visible involvement of 12q14~15 or 6p21 (Table IV) (31, 32, 35, 39-42). Structural rearrangement of the 21q arm were reported in 12 uterine leiomyomas, always together with aberrations of 6p (often 6p21) or 12q14~15 (32, 38, 39, 43-46).
Reported leiomyomas carrying a cytogenetic aberration in the p arm of chromosome 3.
The aCGH investigation of the tumor in our case not only confirmed trisomies for chromosomes 4,7, 8, and 9, but also showed multiple gains and losses of material from chromosome arms 3p and 21q (Figure 2B-D), suggesting that the rearrangements detected in 3p and 21q, add(3)(p21) and add(21)(q22) were more complex than those seen in G-banded preparations. RNA sequencing detected six fusion genes, ARHGEF3–CACNA2D2, TRAK1–TIMP4, ITPR1– DT–NR2C2, CLASP2–IL17RD, ZNF621–LARS2, and CNTN4–RHOA, with both 5’- and 3’-end fusion partner genes located on 3p (Table III and Figure 3A), and one fusion gene, NR2C2–CFAP410, with the 5’-end partner (NR2C2) from 3p and the 3’-end partner gene (CFAP410) from 21q (Table III). All of them were verified by RT-PCR/Sanger sequencing methodologies (Tables I and II; Figure 3B).
The data indicated that chromothripsis involving chromosome arms 3p and 21q occurred in the tumor, causing the generation of multiple fusion genes (47, 48). In this phenomenon, through some unusual, cataclysmic event, a chromosome breaks into many fragments, whereupon these fragments, or some of them, are randomly reassembled (47, 48). Some chromosome fragments are lost through this process while some fusion genes with oncogenic potential are generated at the junctions of the assembled fragments (47, 48). Chromothripsis has been detected in uterine leiomyomas which had neither mutations in MED12 or fumarate hydratase genes, nor 12q14~15 or 6p21 chromosomal rearrangements (49-52).
Three types of chimeras were found in our case of para-testicular leiomyoma: The first type, illustrated by the fusions CNTN4−RHOA, ITPR1-DT−NR2C2, and NR2C2−CFAP410, was the promoter-replacement chimera in which 5’-end untranslated regions of the 5’-end fusion partner gene replaced the 5’-end untranslated regions of the 3’-end fusion partner gene. Consequently, the 3’-end fusion partner comes under the control of the 5’-end fusion partner gene promoter. Thus, in the CNTN4–RHOA transcript, the first untranslated exon of CNTN4, which is highly expressed in testis, replaced the first untranslated exon of RHOA, bringing the entire coding part of RHOA under control of the CNTN4 promoter (53). RHOA codes for a member of the Rho family of small GTPases with multiple cellular and biological functions (54-58). Dysregulation of RHOA and other members of the Rho family of small GTPases has been reported in many neoplasias (59, 60).
In ITPR1-DT−NR2C2, the first untranslated exon of ITPR1-DT, which is part of the ITPR1 promoter, replaced the first untranslated exon of NR2C2 (61, 62). Consequently, the coding part of NR2C2 came under the control of the ITPR1 promoter. NR2C2 protein belongs to the nuclear hormone receptor family, acts as a ligand-activated transcription factor and plays a role in many biological processes such as development, cellular differentiation, and homeostasis (63). In the NR2C2–CFAP410 chimeric transcript, the first untranslated exon of NR2C2 fused to exon 4 of CFAP410. In exon 4 of CFAP410, there is an ATG which could act as a starting codon (NM_004928.3; position 521-523; XM_017028472.1; 421-423). In NR2C2-CFAP410, the part of CFAP410 coding for the last 147 amino acids (positions 109-256 in the sequence with accession number NP_0049 19.1; protein with accession number XP_016883961.1) comes under the control of the NR2C2 promoter. Four alternatively spliced transcript variants have been found for the CFAP410 gene. Three of the transcript variants code for mitochondrial proteins. The fourth transcript (accession number NM_001271442.1) codes for a cytoplasmic protein lacking the mitochondrion-target peptide (NP_001258371.1; https://www.ncbi.nlm.nih.gov/gene/755). CFAP410 was found to form a complex with NIMA-related serine/threonine kinase 1 (NEK1) protein and is involved in DNA-damage repair (64). Similarly to the fourth transcript variant of CFAP410, the putative CFAP410 translated from the NR2C2– CFAP410 chimera of this tumor would also be expected to lack the mitochondrion-target peptide and might interfere with the CFAP410/NEK1 complex and DNA-damage repair.
The second type of chimera involves out-of-frame transcripts. In the two detected ARHGEF3–CACNA2D2 transcripts, exon 1 or 2 of ARHGEF3 fused with exon 2 of CACNA2D2, introducing a stop codon shortly after the junctions. ARHGEF3 (also known as XPLN) codes for guanine nucleotide exchange factor 3, which specifically activates Rho GTPase family members RHOA and RHOB (65, 66). The putative truncated form of ARHGEF3 would lack the carboxyl terminal part of the ARHGEF3 protein responsible for guanine nucleotide exchange factor activity but retain the amino terminal part of the protein which interacts with mammalian target of rapamycin complex 2 (mTORC2), inhibiting mTORC2 kinase activity (66, 67). The presence of the truncated ARGEF3 protein and its possible functions cannot be determined without further cellular studies. Nevertheless, the ARHGEF3–CACNA2D2 transcript indicated further the dysfunction of RHOA signal transduction in this tumor.
The third type of chimera resulted in in-frame transcripts translated into chimeric proteins. In the ZNF621–LARS2 transcript, exon 2 of ZNF621 fused in-frame to exon 5 of LARS2. In the chimeric ZNF621–LARS2, the first 121 amino acids of LARS2 are replaced by the first eight amino acids of ZNF621. The LARS2 gene codes for a mitochondrial leucyl-tRNA synthetase with a mitochondrial targeting signal within its first 50 amino acids (68). Thus, since the mitochondrial targeting signal is absent from the chimeric ZNF621–LARS2 protein, the protein would probably be unable to enter the mitochondrion from the cytosol.
The two in-frame TRAK1–TIMP4 fusion transcripts would also give rise to chimeric proteins. The ubiquitously expressed TRAK1 gene codes for trafficking kinesin-binding protein 1 which is involved in mitochondrial and endosome-to-lysosome trafficking (69-74). Based on the reference sequence NP_001036111.1, the 953 amino acids-long TRAK1 protein has the following regions: A huntingtin-associated protein 1 conserved region between amino acids 50-353 binding to huntingtin in a polyglutamine repeat-length-dependent manner; a region between amino acids 359-509 which interacts with hepatocyte growth factor-regulated tyrosine kinase substrate protein; a region named milton between 416-583 which recruits the heavy chain of kinesin to mitochondria enabling the motor movement function of kinesin; and a region between amino acids 658-672 which interacts with O-linked N-acetylglucosamine (GlcNAc) transferase protein (70-72, 75, 76).
TIMP4 which codes for tissue inhibitor metalloproteinase 4, is weakly expressed in testis (77) and inhibits many metalloproteinases (78-80). The inhibitory property of TIMP4 was shown to depend on its N-terminal part, between residues 1-127, which contains a site of interaction with the catalytic domain of metalloproteinases (79, 80). In the tumor investigated here, the two translated TRAK1–TIMP4 chimeric proteins would lack all the above-mentioned functional regions of TRAK1 and residues 1-127 of TIMP4 which interact with and inhibit metalloproteinases. Their role in tumorigenesis is difficult to assess.
Based on the reference sequences NM_015097.2/NP_055912.2 for CLASP2 and NM_017563.4/NP_060033.3 for IL17RD, the CLASP2–IL17RD chimeric transcript would be expected to consist of exons 1 to 34 of CLASP2 and exons 2 to 13 of IL17RD, be 12317 bp-long, and code for an 1,892 amino acid protein composed of amino acids 1 to 1,195 from CLASP2 and 43 to 739 from IL17RD. The CLASP2 gene codes for cytoplasmic linker protein-associating protein that is a microtubule plus end tracking protein (81, 82). CLASP2 is localized to the distal ends of microtubules and is involved in the regulation of microtubule dynamics (83). Microtubules switch between phases of growth and shrinkage through transitions known as microtubule catastrophe and rescue (84). CLASP2 was found to specifically suppress microtubule catastrophe and promote rescue without affecting the rates of microtubule growth or shrinkage (85, 86). CLASP2 functions in various microtubule-dependent processes including cell division, cytoskeletal remodeling for cell migration, and vesicle transport between intracellular structures and the plasma membrane (87). CLASP2 protein has multiple HEAT repeats, a structural motif composed of two alpha helices linked by a short loop (88), two domains from tumour overexpressed gene that interact with αβ-tubulin, contributing to microtubule dynamics (89, 90), a region for interaction of microtubule-associated protein RP/EB family member 1 (MAPRE1, also known as EB1) (85, 91), and a carboxyl terminal region which interacts with CLIP1 protein and is required for the localization of CLASP2 protein to the Golgi apparatus and kinetochores (92).
The IL17RD gene codes for a single pass transmembrane protein mainly located in plasma membrane and regulating various signal pathways such as fibroblast growth factor and mitogen-activated protein kinase/extracellular signal-regulated kinase signal pathways (93-95). Murine models and expression patterns indicate that IL17RD is a tumor-suppressor gene (96-100). Thus, CLASP2–IL17RD chimeric transcript would produce a chimeric protein affecting microtubules and signaling pathways.
In summary, our data, together with those previously published, indicate the existence of a group of leiomyomas cytogenetically characterized by aberration of the p arm of chromosome 3 and the formation of fusion genes. A chromothripsis event seems to lie behind the pathogenetic changes that occurred in this para-testicular leiomyoma, resulting in multiple fusion genes.
Acknowledgements
This work was supported by grants from Radiumhospitalets Legater.
Footnotes
Authors’ Contributions
IP designed and supervised the research, performed molecular genetic and bioinformatic analyses, and wrote the article. LG performed cytogenetic analyses. KA performed molecular genetic analyses and evaluated the data. IL performed the pathological examination. SH assisted with experimental design and writing of the article. All Authors read and approved of the final article.
This article is freely accessible online.
Conflicts of Interest
No potential conflicts of interest exist.
- Received April 18, 2021.
- Revision received May 11, 2021.
- Accepted May 18, 2021.
- Copyright© 2021, International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved




{kind=link}
{kind=link}
{kind=link}