


Abstract
Background: Classification of splicing variants (SVs) in genes associated with hereditary cancer is often challenging. The aim of this study was to investigate the occurrence of SVs in hereditary cancer genes and the clinical utility of RNA analysis. Material and Methods: 1518 individuals were tested for cancer predisposition, using a Next Generation Sequencing (NGS) panel of 36 genes. Splicing variant analysis was performed using RT-PCR and Sanger Sequencing. Results: In total, 34 different SVs were identified, 53% of which were classified as pathogenic or likely pathogenic. The remaining 16 variants were initially classified as Variant of Uncertain Significance (VUS). RNA analysis was performed for 3 novel variants. Conclusion: The RNA analysis assisted in the reclassification of 20% of splicing variants from VUS to pathogenic. RNA analysis is essential in the case of uncharacterized splicing variants, for proper classification and personalized management of these patients.
The advancement of next-generation sequencing (NGS) technologies has contributed to multi-gene panel analysis used in clinical practice; the interpretation of such variants remains a challenge. Variants of uncertain significant (VUS) are identified in high rates (35-48%) using multi-gene panels and these variants do not allow for management decisions (1-4).
Among the variants which are identified in genes associated with hereditary cancer, splicing variants (SVs) account for 15-25% (5). More frequently, the exon/intron boundary sequences contain GT and AG motifs at the 5’ and 3’ ends of the intron, respectively (6). Some of the variants in the splice sites lead to incorrect intron removal and alterations of the open reading frame (7). Furthermore, most of SVs are classified as VUS due to lack of functional studies such as RNA analysis (8).
The detection of such splicing variants provides strong evidence of pathogenicity according to the guidelines of the American College of Medical Genetics and Genomics (ACMG), however their effect could be also investigated by in silico analysis confirmed either at mRNA or protein level (9). The importance of immediate reclassification of SVs classified as VUS lies in the fact that the patient needs to follow precise medical and therapeutic management, in accordance with the international guidelines. In our study we investigated the occurrence of SVs in hereditary cancer genes and the utility of RNA analysis in clinical diagnostics.
Materials and Methods
Individuals who were referred to Genekor’s Medical S.A (Athens, Greece) laboratory for genetic testing using a hereditary cancer panel were evaluated. All individuals signed an informed consent form prior to molecular genetic testing and permission for the anonymous use of their data for research purposes and/or scientific publications was provided. Moreover, information on demographics, clinical and family history of cancer was collected and pedigrees were ordered by clinicians at the time of testing.
Gene testing. Genomic DNA was extracted from peripheral blood leukocytes using MagCore® Genomic DNA Whole Blood Kit (RBC Bioscience) according to the manufacturer’s instructions.
The analysis of genes involved in hereditary cancer predisposition was performed using a solution-based capture approach. Targeted NGS was performed with a panel of 36 genes (Roche NimbleGen SeqCap EZ Choice), consisting of: APC (NM_000038), ATM (NM_000051), BARD1 (NM_000465), BMPR1A (NM_004329), BRCA1 (NM_007294), BRCA2 (NM_000059), BRIP1 (NM_032043), CDH1 (NM_004360), CDK4 (NM_000075), CDKN2A (NM_000077), CHEK2 (NM_007194), EPCAM (NM_002354), FANCA (NM_000135), FANCM (NM_020937), HOXB13:c.251G>A p.(G84E) (NM_006361), MEN1 (NM_000244), MLH1 (NM_000249), MRE11 (NM_005591), MSH2 (NM_000251), MSH6 (NM_000179), MUTYH (NM_001128425), NBN (NM_002485), NF1 (NM_000267), PALB2 (NM_024675), PMS2 (NM_000535), POLD1 (Exons 7-12) (NM_001256849), POLE (Exons 35-48) (NM_006231), PTEN (NM_000314), RAD50 (NM_005732), RAD51C (NM_058216.2), RAD51D (NM_002878.3), RET (NM_020975), SMAD4 (NM_005359), STK11 (NM_000455), TP53 (NM_000546) and VHL (NM_000551). The sample preparation was performed according to the manufacturer’s instructions in the SeqCap EZ Choice Library User’s Guide (Roche NimbleGen, Pleasanton, CA, USA). Sequencing was carried out using the Miseq Illumina NGS (Illumina, San Diego, CA, USA) technology and sequence changes were identified and interpreted in the context of a single clinically relevant transcript using the commercially available software suite SeqNext version 4.4.0 (JSI Medical Systems GmbH, Ettenheim, Germany).
The presence of large genomic rearrangements (LGRs) was investigated using the commercial computational algorithm SeqPilot (JSI medical systems GmbH, Germany). In addition, the computational algorithm panelcn MOPS (10) was also used in the BRCA1 and BRCA2 genes. The presence of LGRs was verified by the use of the MLPA method (Multiplex Ligation-dependent Probe Amplification, MRC Holland) (11).
Variant classification and bioinformatics analysis. The clinical significance of variants was further examined using the standards and guidelines for the interpretation of sequence variants recommended by the ACMG Laboratory Quality Assurance Committee and the Association for Molecular Pathology (AMP) (9). The impact of missense substitutions on protein function or structure was analyzed using MetaSVM (12) which combines computational (in silico) predictive algorithms under an ensemble mutational impact score. The effect on splicing was computationally examined using the VarSeak program (13).
Tissue selection, nucleic acid isolation, MSI and BRAF. Genomic DNA and RNA were isolated from formalin-fixed and paraffin-embedded (FFPE) tumor biopsies using the MagMAX™ Total Nucleic Acid Isolation Kit (Thermo Fischer Scientific, Waltham, MA, USA) according to the manufacturer’s instructions. The nucleic acid isolation was conducted in the areas of the FFPE block with the majority of tumor cell content (TCC), as indicated by experienced pathologists in hematoxylin and eosin stained sections. Minimum required TCC was over 20%, in a tumor area of > 4mm2.
Microsatellite analysis was conducted using the Ion AmpliSeq™, as indicated by the manufacturer. Microsatellite Instability Panel (Thermo Fischer Scientific, Waltham, MA, USA) is an NGS based assay analyzing 76 markers for assessing Microsatellite Instability (MSI) status in tumor-only and tumor-normal samples. Analysis of the sequencing output from this panel was carried out using the “MSICall” plugin in the Torrent Suite.
A targeted resequencing assay (Ion AmpliSeq NGS Panel, Thermo Fisher Scientific) was used for mutation detection in exons 11 and 15 of the BRAF gene. The panel analyzes hotspot mutations in 24 genes related to colorectal and lung cancer. Details about the target regions of the 24-gene panel are available upon request. Sequencing was carried out using the Next Generation Sequencing Platform Ion Gene Studio S5 Prime System (Thermo Fisher Scientific).
RNA analysis. Total RNA was extracted from peripheral blood lymphocytes using Trizol (Invitrogen, Paisley, UK) following a standard protocol. cDNA was synthesized using the SuperScript™ VILO™ cDNA Synthesis Kit (Thermo Fisher Scientific) as described by the manufacturer. The resulting cDNA was amplified by specific primers around the region of each variant and positioned at least two exons up- and down-stream of the target region. The PCR-products were purified using the NucleoFast® 96 PCR Clean-up kit (Macherey-Nagel GmbH and Co., Düren, Germany). The purified PCR product was used for each sequencing reaction, performed using the BigDye® Terminator v1.1 Cycle Sequencing kit (Applied Biosystems, Foster City, CA, USA). Sequencing reaction products were purified prior to electrophoresis using the Montage™ SEQ96 Sequencing Reaction kit (EMD Millipore Corp., Billerica, MA, USA) and sequenced using an Applied Biosystems 3130 Genetic Analyzer (Applied Biosystems).
Results
A total of 1,518 individuals were referred to our laboratory for genetic testing: 1,001 individuals from Greece (65.9%), 284 from Turkey (18.7%) and 233 from Romania (15.4%). The median age of individuals in our cohort was 49.5 years old (20-86 years old). The majority of individuals tested were female (90.5%, 1,374/1,518), while only 9.5% (144/1,518) were male. Among the 1518 cases referred for testing, 83.9% (1,274/1,518) had a personal history of cancer, 11.8% (179/1,518) were unaffected at the time of testing, while no clinical data was available for 4.3% (65/1,518) of individuals. The majority (70%) (890/1,274) of affected individuals had a personal history of breast cancer. The subgroup of affected individuals with other cancers included patients with personal history of the following cancer types: colorectal, endometrium, gastric, gynecological, leukemia, liver, melanoma, pancreas, prostate, stomach, thyroid and cancer of unknown origin.
DNA from a total of 1,518 individuals was analyzed. At least one pathogenic/likely pathogenic variant was identified in 285 samples (19%). Frameshift, nonsense, missense and frame insertion/deletion mutations accounted for 84.2% (240/285) of the variants identified, while large genomic rearrangements (LGR) accounted for 9.1% (26/285).
A splicing variant was identified in 36 out of the 1518 individuals (2.4%). In total, 34 different SVs were identified, 10 of which (29%) were classified as pathogenic and 8 (24%) as likely pathogenic. The remaining 16 variants were initially classified as VUS (47%) (Figure 1A, Table I). 18 (52.9%) splicing variants were located within the donor splice site or splice region (defined by sequence ontology as extending from the third last base of the exon up to the eight base of intron) and 16 (47.1%) were located within the acceptor splice site or splice region (from the eight-last base of the intron up to the third base of exon). 25 SVs were present in the ClinVar mutation database while the remaining nine variants were novel. In silico analysis predicted that most of these SVs have an impact on splicing events (28/34) but functional studies were available only for 57% (16/28) of them (Table I). Spicing variants were detected in 17 of 36 genes and the frequency of splicing variants among genes is represented in Figure 1B.

Variant classification results for splicing variants identified in the 36 genes tested in this study. A. Classification of splicing variants according to their clinical significance. B. The number and frequency of Pathogenic/Likely Pathogenic and VUS splicing variants among genes.
The 34 splicing variants detected in patients with the in silico exon splicing analysis and functional analysis results.
For those individuals in whom VUS or a probable pathogenic splice variant was identified, we requested a new blood sample for RNA extraction. In our study, RNA analysis was performed for the following 3 novel variants: ATM (c.2125-1G>T), ATM (c.2464_2466+2delTTAGT) and MSH2 (c.1510+1_1510+2dupGT).
RNA analysis of a variant in ATM (c.2125-1G>T) revealed the deletion of 11 bases (r.2125del), resulting in a frameshift deletion, p.(Ile709Phefs*22). In a 33-old year-old woman who was diagnosed with breast cancer at 33 years of age the splicing variant c.2125-1G>T in the ATM gene was identified, in heterozygosity (Figure 2A). ATMc.2125-1G>T was predicted to affect the acceptor splice site (Table I). This prediction was confirmed by RNA analysis and shows that the c.2125-1G>T variant affects splicing by creating an alternative splice site, 11 bp downstream (r.2125del), and generating a premature translation stop signal 22 amino acid residues later, predicted to result in a truncated protein (p.(Ile709Phefs*22)) (Figure 2B). This variant has not been reported in the literature and is not present in the mutation database ClinVar. Moreover, a variant occurring at the same position c.2125-1G>A with the same expected result on the protein product has been described as likely pathogenic in the mutation database ClinVar (Variation ID:551858). The RNA analysis confirmed the disrupting impact of the splice site variant c.2125-1G>T in the ATM mRNA, leading to the definite classification of this variant as pathogenic. The truncated protein products caused by alternative splicing may be associated with an increased risk of breast cancer.
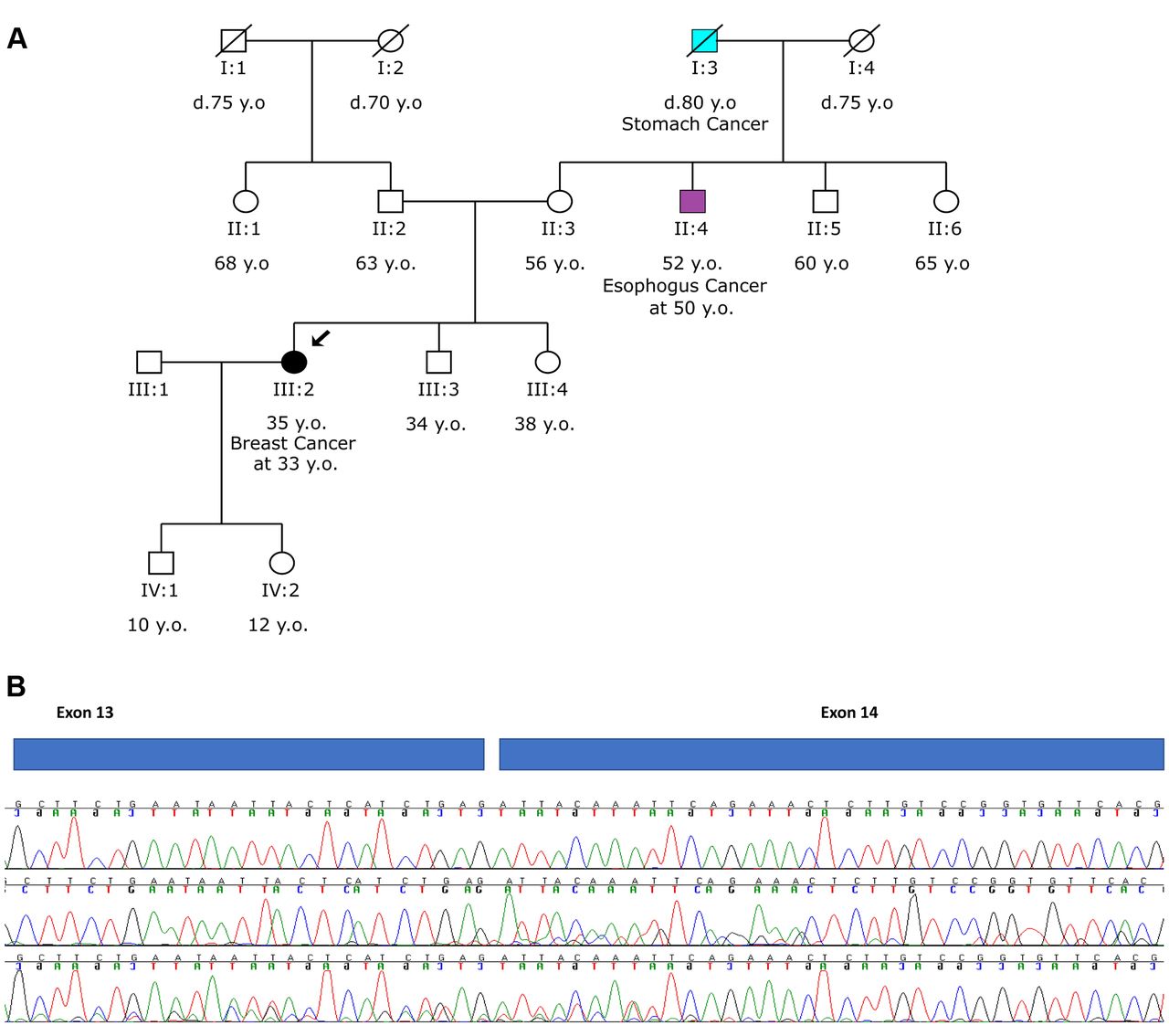
Analysis of ATM c.2125-1G>T. A. Pedigree of the proband with the c.2125-1G>T variant in ATM. B. RNA analysis of the variant revealed the deletion of 11 bases (r.2125del), resulting in a frameshift deletion, p.(Ile709Phefs*22) (the second and third chromatograms) compared to the analysis of a wild type sample (top chromatogram).
RNA analysis of a variant in ATM (c.2464_2466+2delTTAGT) revealed the deletion of 5 bases (r.2466del), thus resulting in a frameshift, p.(Ser821Serfs*8). The ATMc.2464_2466+ 2delTTAGT variant was identified in heterozygosity, in a 52-old year-old woman who had developed polyps at 52 years of age. The proband’s twin sister was diagnosed with colorectal cancer at 42 y.o (Figure 3A). In silico analysis predicted that this change is disrupting the wild type of donor site (Table I). Using RNA analysis the c.2464_2466+2delTTAGT variant was shown to affect splicing by generating an alternative splice site 5 bp upstream (r.2466del) and a premature translation stop signal 8 amino acid residues later p.(Ser821Serfs*8) (Figure 3B). This variant was predicted to truncate the ATM protein, leading to the unequivocal classification of this variant as pathogenic.

Analysis of ATM c.2464_2466+2delTTAGT. A. Pedigree of the proband with the c.2464_2466+2delTTAGT variant in ATM. B. RNA analysis of the variant revealed a deletion of 5 bases (r.2466del), resulting in a frameshift deletion, p.(Ser821Serfs*8) (bottom panel) compared to the analysis of a wild type sample (top panel).
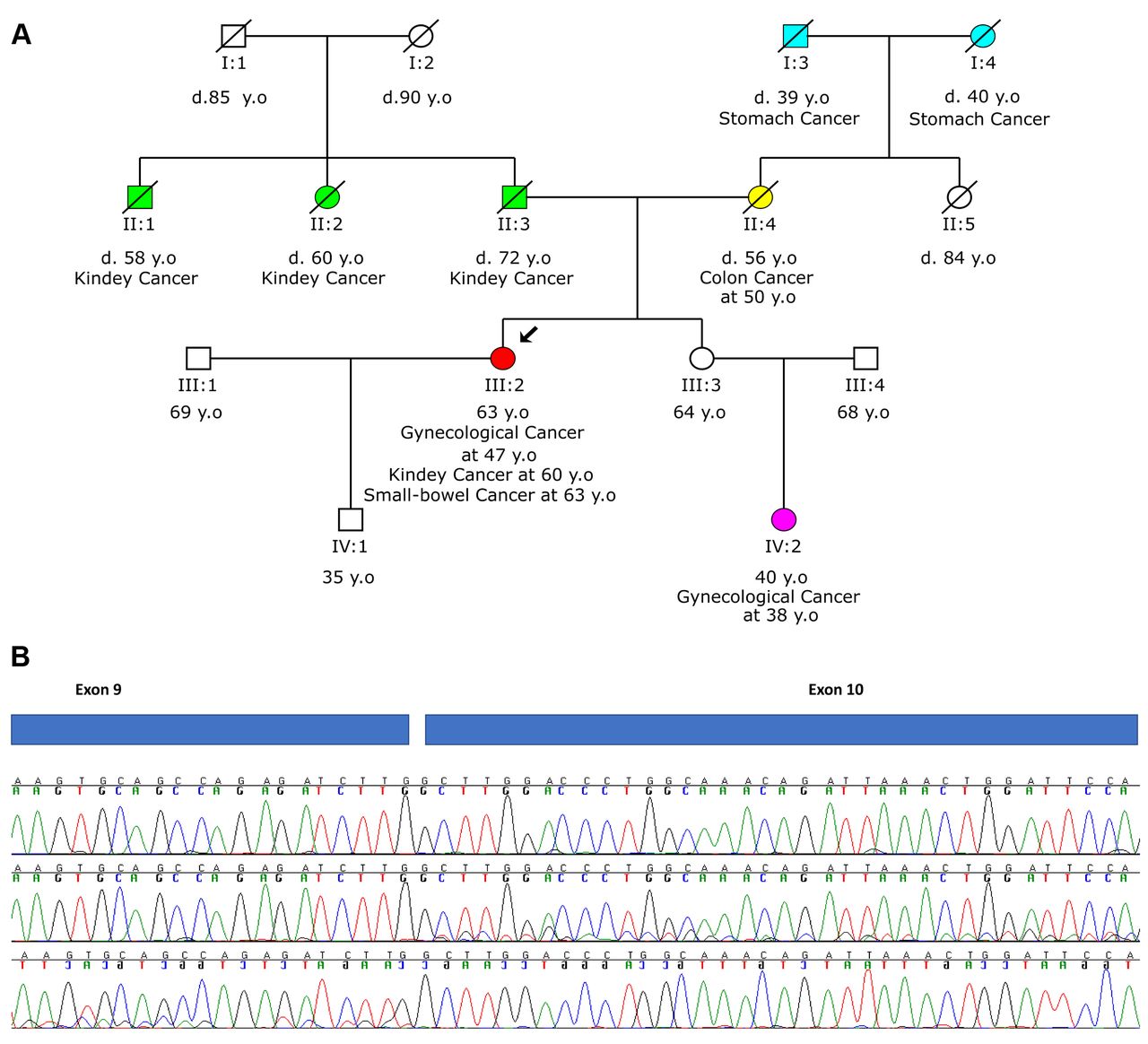
RNA analysis of a variant in MSH2 (c.1510+ 1_1510+2dupGT) revealed the insertion of 2 bases (r.1510dup) causing a frameshift insertion, p.(Gly5040 Glyfs*39). A 63-old year-old woman was referred for genetic testing after a diagnosis of gynecological cancer at 47, kidney cancer at 60 and small-bowel cancer at 63 years of age. Her mother was diagnosed with colon cancer at 50 years of age. Multigene genetic testing revealed the spicing variant c.1510+1_1510+2dupGT in the MSH2 gene in heterozygosity (Figure 4A). In silico analysis for MSH2c.1510+ 1_1510+2dupGT predicted that the donor splice site may be disrupted (Table I). Functional analysis revealed that this variant induces aberrant splicing via an insertion of 2 bases (r.1510dup) that causes a frameshift insertion and generation of a premature translation stop signal 39 amino acid residues later, predicted to result in a truncated protein [p.(Gly5040Glyfs*39)] (Figure 4B). The disrupting impact of the splice site variant c.1510+1_1510+2dupGT in the MSH2 mRNA was confirmed by RNA analysis, leading to the classification of this variant as pathogenic. Moreover, microsatellite instability analysis of the tumor sample (bowel tissue) by NGS revealed the microsatellite unstable status of the tumor (MSI-High), in the absence of a BRAF mutation. These results further support the germline origin of the variant c.1510+1_1510+2dupGT in the MSH2 gene.

Analysis of MSH2 c.1510+1_1510+2dupGT. A. Pedigree of the proband with the c.1510+1_1510+2dupGT variant in MSH2. B. RNA analysis of the variant revealed the insertion of 2 bases (r.1510dup) causing a frameshift insertion, p.(Gly5040Glyfs*39) (second and third chromatograms) compared to the analysis of a wild type sample (top chromatogram).
These three novel variants were initially classified as VUS while the RNA analysis reclassified them as pathogenic. In our study, RNA analysis assisted in the reclassification of 20% of VUS implicated in splicing.
Discussion
Genetic test results are used to guide clinical decisions for cancer treatment, surveillance and prevention. Nevertheless, the interpretation of VUS is a challenge for the laboratories performing the molecular diagnosis of hereditary cancer. A significant proportion of VUS classification arises from the detection of variants predicted to generate abnormal mRNA transcripts but lack functional evidence. Our results indicate that RNA splicing analysis, using RT-PCR, can produce data that help clarify the interpretation of variants.
VUS reclassification efforts have shown that 97.2% of VUS can be reclassified in the future and the majority of variants in this class (82.1%) are likely benign (14). In our study, RNA analysis contributed to the reclassification of 20% of splicing VUS to pathogenic. The significance of reclassifications of VUS to pathogenic variants, using RNA analysis, lies in the management of both patients and their family members, leading to increased screening and/or more aggressive risk-reducing interventions.
In this study, we reclassified two novel splicing variants (c.2125-1G>T, c.2464_2466+2delTTAGT) from VUS to pathogenic in the ATM gene. The ATM gene is implicated in the homologous recombination complex (HR) and is associated with an increased risk for autosomal dominant breast cancer (15-17) and possibly for other types of cancer (18-19) in individuals who carry a single pathogenic ATM variant. Additionally, this gene is associated with autosomal recessive ataxia-telangiestasia (A-T). According to the National Comprehensive Cancer Network (NCCN) Guidelines (Version 2.2021), for carriers of ATM pathogenic/likely pathogenic variants there is strong evidence for increased risk of breast cancer with an absolute risk of 15-40% (20). The proposed management includes an annual mammogram and consideration of tomosynthesis and breast MRI with contrast, starting at the age of 40. Concerning the ovarian and pancreatic cancer risk, the evidence for increased risk is strong with absolute risk <3% and 5-10%, respectively.
Likewise, we reclassified a novel variant (c.1510+1 _1510+2dupGT) in the MSH2 gene from VUS to pathogenic. The MSH2 gene is associated with autosomal dominant Lynch syndrome (also called hereditary nonpolyposis colorectal cancer syndrome or HNPCC) and autosomal recessive constitutional mismatch repair deficiency syndrome (CMMR-D). MSH2 gene has been associated with an increased risk of colorectal (52-82%), endometrium (25-60%), prostate (~30%), ovarian (15-24%), stomach (6-13%), hepatobiliary tract (1-4%), urinary tract (1-7%), small bowel (3-6%), brain (1-3%) and pancreatic (1-6%) cancer in carriers of a single pathogenic mutation (21, 22). Based on the NCCN Guidelines (Version 2.2019, Genetic/Familial High-Risk Assessment: Colorectal), surveillance/prevention strategies for colon cancer recommend high-quality colonoscopy at the age 20-25 and specific follow-up (such as proctocolectomy) depending on colonoscopy findings (23). There are also data demonstrating that the daily intake of 600 mg of aspirin for at least 2 years decreases colorectal cancer risk. Concerning endometrial cancer, hysterectomy is a risk-reducing option that can be considered, while bilateral salpingo-oophorectomy may reduce the incidence of ovarian cancer. Concerning cascade family testing, patients are advised to inform their relatives about the possible inherited cancer risk, the options for risk assessment, as well as its management. Genetic counseling and consideration of genetic testing is recommended for all at-risk relatives.
Although the routine use of RNA analysis in genetic diagnostics requires additional experiments to consolidate its role, we suggest that RNA splicing analysis could be considered in genetic disease variant interpretation for improving diagnostic testing. Patient education and counseling could assist in identifying at-risk individuals that could benefit from RNA analysis in families with splicing variants. RNA analysis should be encouraged in cases where splicing variants are identified, so as to finalize their classification (24, 25). Our data suggest that approximately only 14% of patients with SV continue with RNA analysis when this is requested at no cost. Alternatively, before the release of the report, the laboratory could request fresh blood when these findings are identified to perform RNA analysis instead of releasing a VUS report. Furthermore, RNA analysis could be carried out by keeping fresh blood from the initial sample in –20°C for RNA extraction when applicable. In conclusion, this study establishes the potential of RNA analysis in the classification of splicing VUS and the clinical utility in routine genetic testing.
Footnotes
Authors’ Contributions
KA drafted the manuscript. KA and EP designed the study. KA, GP, SK and SM carried out the DNA extraction, sequencing and contributed to the analysis and interpretation of the variant data. GNT performed the bio-informatics analysis. AF, CC, RI, SK, CM, IN, KP, MVA, VV, VO, ST, KK, DTE and AC provided the materials, demographic data and family history. GN conceived of the study and participated in its design and coordination. All authors read and approved the final manuscript.
This article is freely accessible online.
Conflicts of Interest
There are no conflicts to declare.
- Received March 11, 2021.
- Revision received March 24, 2021.
- Accepted March 29, 2021.
- Copyright© 2021, International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved




{kind=link}
{kind=link}
{kind=link}
{kind=link}