


Abstract
Background/Aim: Proteomics technologies provide fundamental insights into the high organizational complexity and diversity of the central nervous system. In the present study, high-resolution mass spectrometry (MS) was applied in order to identify whole-proteome content of anatomically distinct and functionally specific mouse brain regions. Materials and Methods: Brains from eight 8-week-old C57BL/6N normal male mice were separated into seven anatomically district regions. The protein content of each region was analyzed by high-throughput nano-liquid chromatography-MS/MS Orbitrap elite technology. Results: A total of 16,574 proteins were identified: 2,795 in cerebral cortex, 2,311 in olfactory bulb, 2,246 in hippocampus, 2,247 in hypothalamus, 2,250 in mid brain, 2,334 in cerebellum and 2,391 in medulla. Of these proteins, 534 were uniquely expressed in cerebral cortex, 323 in olfactory bulb, 230 in hippocampus, 272 in hypothalamus, 1,326 in mid brain, 320 in cerebellum and 268 in medulla. Conclusion: These data represent the most comprehensive proteomic map of the normal mouse brain and they might further be used in studies related to brain diseases, including cancer and neurodegenerative diseases.
- Proteomics
- proteome
- high resolution mass spectrometry
- nanoLC-MS/MS
- tandem mass spectrometry
- mouse
- brain
- cerebral cortex
- olfactory bulb
- hippocampus
- hypothalamus
- midbrain
- cerebellum
- medulla
- normal mouse brain
- normal adult mouse brain
In higher mammals, the brain, being the center of the nervous system, consists of the cerebrum, the brainstem and the cerebellum. It regulates complex biological functions, including higher activities, such as emotions, cognition and behavior. The brain is divided into sub-compartments, anatomically distinct and functionally specific regions. Numerous large-scale initiatives and projects have been launched aiming to develop a map model and a simulation of the brain. Among them, the Brain Initiative (1), the Human Brain Project regarding the development of a digital simulation model of the human brain (2), the Human Connectome Project concerning the structural and functional brain connections (3), and the Allen Brain, which aims to map gene expression of the human, mouse and pig brains (4, 5).
In the past two decades, although human brain tissue preparations have been shown to be suitable for in vitro or in vivo metabolic, anatomical and functional studies, due to ethical issues, as well as the necessity of extended normal and abnormal subject comparisons, experimental animal studies still remain central to neurochemistry, neuroanatomy and neuroproteomics. Although huge proteomics studies on human brain were performed in abnormal conditions, such as Alzheimer's disease (6), Parkinson's disease (7) and brain tumors (8-10), little work has been carried out on the proteomic analysis of the normal brain (11, 12). The limitations of these analyses are due to ethical issues as well as the necessity for extended normal and abnormal subject comparisons. Therefore, experimental animal studies will remain central to neurochemistry, neuroanatomy and neuroproteomics.
Despite these advances in neurosciences, scientists are still unable to understand numerous normal brain functions to its full extent. Although applications, such as next-generation sequencing (NGS), transcriptomics and serial gene-expression profiling techniques, have provided immense information regarding gene expression in the brain, a comprehensive account of the products of gene expression through protein profiling is more important than genomics information in order to fully comprehend the advanced biological networks of the brain (13). A complex and fairly unpredictable pathway intercedes between mRNA expression and biologically active proteins. Protein levels cannot be extrapolated from mRNA levels, making increasing the sensitivity of proteomics techniques mandatory. Apart from the large numbers of splice variants, the enormous complexity and diversity of proteins is also a result of their post-translational modifications (PTMs), which cannot be assessed using transcriptional studies (14).
In this regard, traditionally, biochemical and biophysical methods have been widely used to study the functions and structures of certain proteins. However, it is still elusive how the proteome behaves as an integrated system and which properties affect this behavior. New technologies, such as powerful mass-spectrometry (MS)-based systems, now provide deeper insights into the composition, structure, function and control of the proteome, allowing the understanding of the complex biological processes and phenotypes (15). This rapid development of proteomic technologies allows scientists to characterize the global protein-expression profiles in specific physical or pathological situations, which are normally achieved through standard molecular technologies (16, 17).
The application of proteomics in the analysis of the central nervous system (CNS) introduces neuroproteomics as a new field, which enables improved understanding of the molecular mechanisms underlying brain function. In neuroproteomics, the different regions of the nervous system are analyzed, so that the dynamics of each sub-proteome can be better understood. By extension, the generation of the proteomic neuroanatomy of individual brain regions of mammals are complimented with the functional neuroanatomical studies. From an evolutionary perspective, the murine brain is a model system for neurological studies including malignancies (for example brain tumors) and degenerative diseases of the human brain. In this regard, the proteomic profile of the mouse brain has been mostly studied by transcriptome analyses or the integration of proteomic and transcriptomic-based methods (5, 18), while certain studies have used pure proteomic techniques (19-21). During work conducted in our previous study, the methodology of 2D electrophoresis coupled to matrix-assisted laser desorption/ionization-time of flight mass spectrometry (MALDI-TOF-MS) was used in order to reveal the whole proteomic profile of seven different regions of the murine brain (20). Moreover, through the development of state-of-the-art MS technologies which enable the identification of a large number of proteins at once, the global protein content of the rodent brain has come to the forefront of neuroproteomics. These technologies include nano-liquid chromatography followed by high-resolution MS (22).
Applying this technology in 2015, Sharma et al. reported a complete mouse cell-type and brain-region proteome that covered approximately 13,000 gene products and provided the first insight into brain-wide profiling methodology. In that study, newborn and 9-week-old wild-type male mice were used, the brains were sliced according to the Mouse Brain Library's instructions and the regions in each slide were detected using the C57BL/6J atlas as reference (18). Recently, Jung et al. presented a mouse brain protein atlas from the analysis of 17 surgically district neuroanatomical regions revealing over 12,000 gene proteins in the whole brain (23).
Bearing in mind that the global proteomic analysis of the normal mouse brain is of great interest, the aim of the current study was to describe the normal mouse brain proteome at a high resolution and determine the differences among seven brain regions. In contrast to related previous studies (18, 23), the brains were dissected into seven anatomically distinct and functionally specific regions (cerebral cortex, olfactory bulb, hippocampus, hypothalamus, midbrain, cerebellum, and medulla) according to the Paxinos and Franklin's mouse brain stereotactic atlas (24). This was followed by global analysis of the protein level using nano-liquid chromatography (nanoLC) coupled to high resolution tandem MS/MS of an early adult (8 weeks old) normal mouse brain, completing our previous study (20).
Materials and Methods
Experimental animal model. Eight male 8-week-old, C57BL/6N inbred mice, were used in the following study. The animals were sacrificed and their brains were separated into seven anatomically distinct regions: the cerebral cortex, olfactory bulb, hippocampus, hypothalamus, midbrain, cerebellum, and medulla. The same type of tissues of the different mice were pooled and the brain regions were kept at −80°C, as previously described (25). Animal treatment and experimental procedures were approved by the European Legal framework that exists for the Protection of animals used for experimental and other scientific purposes (European Convention 123/Council of Europe and Directive 86/609/EEC), and by the Bioethics Committee of the Biomedical Research Foundation of the Academy of Athens (2776/28-4-2016).
Sample preparation and peptide generation. The brain tissues were treated with lysis buffer as previously described (18). Tissue/brain region disruption was performed by mechanical pressure as well as tip-sonication for three cycles of 12 s each, under 38% amplification. Next, the samples were centrifuged for 10 min at 13,000 × g, the pellets containing the organelle debris and insoluble material were discarded and the protein quantification of the supernatant took place using the Bradford assay.
A protein quantity of 200 ng from each sample was treated with 7 M urea buffer and 80 mM triethylamonium bicarbonate. Alkylation of the proteins was carried out using 55 mM iodoacetamide solution. In the final step, digestion of extracted proteins for peptide generation was performed using trypsin (Roche Diagnostics, Mannheim, Germany) at a final concentration of 500 ng/μl applied to all samples in a humidified atmosphere, and the digestion took place overnight. The next day, the peptides were lyophilized in a vacuum concentrator for approximately 2 h and the pellet was dissolved in 0.1% formic acid in double-distilled water for LC-MS/MS analysis.
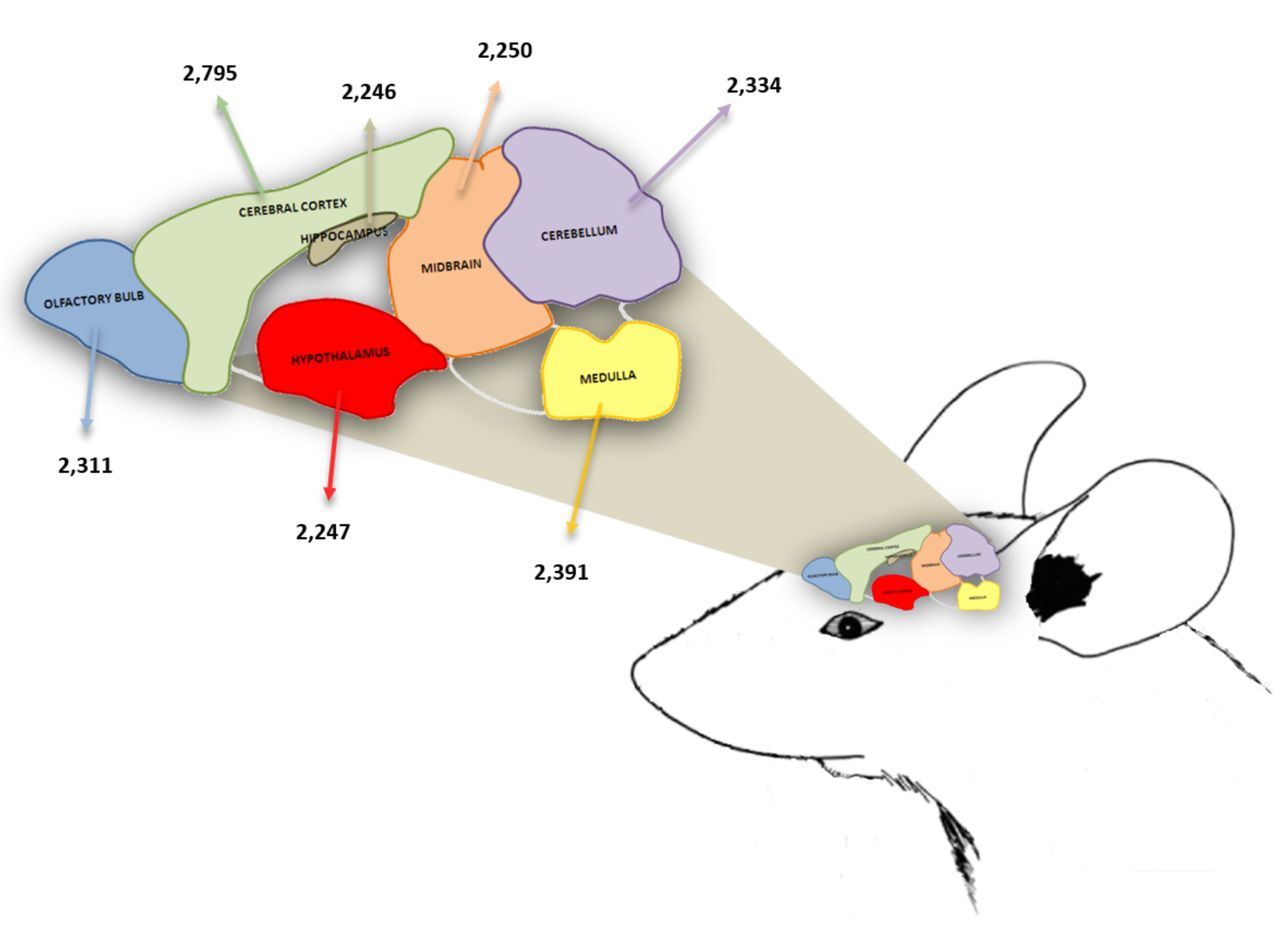
Representation of the mouse brain with the analyzed regions and the number of identified proteins per region.
NanoLC-MS/MS analysis. MS analysis was performed as previously described (22). An LTQ Orbitrap Elite coupled to a Dionex 3000 HPLC system (Thermo Scientific, Rockford, IL, USA) was used for analyzing the digested samples. LC separation of peptides took place on two Thermo Scientific columns (PepMap® RSLC, C18, 100 Å, 3-μm-bead-packed 15-cm column and 2-μm-bead-packed 50-cm column). The mobile phases A and B were 0.1% formic acid in water and 99% acetonitrile in water, respectively, at a flow rate of 3 nl/min. The gradient elution profile was as follows: 2.0% B (98.0% A) for 10 min, 2.0-35.0% B (98.0-65.0% A) for 325 min, 80.0% B (20.0% A) for 10 min, 2.0% B (98.0% A) for 10 min. A standard top-20 method was used in order to collect the data in a data-dependent MS/MS mode. Full-scan data were acquired at a resolving power of 60,000 with a maximum integration time of 250 ms. The scan range was fixed at 250 to 1250 m/z and peptide fragmentation was performed at a high-energy collision dissociation mode with a normalized collision energy of 36%. MS/MS spectra were acquired with 15,000 resolving power and a maximum integration time of 120 ms. Measurements were performed using m/z 445.120025 as lock mass. Dynamic exclusion settings were set to repeat count 1, repeat duration 30 s, exclusion duration 120 s, and exclusion mass width 0.6 m/z (low) and 1.6 m/z (high). The *.raw data files were analyzed using the Proteome Discoverer software (Thermo Scientific), using the Sequest search engine applying the Mus musculus *.fasta databases. MS/MS searches were performed using a 20 ppm parent ion mass tolerance and a 0.05 fragment mass tolerance. Trypsin was selected as the cleavage enzyme with up to two missed cleavage points. Cysteine methylthio modification was selected as a fixed modification and oxidation of methionine was selected as a variable. Peptide identification was considered valid at a 1% false discovery rate (q-value <0.01) (percolator maximum Delta Cn was 0.05). The minimum length of acceptable identified peptides was set as six amino acids. Each sample was analyzed in quadruplicate.
Bioinformatic analysis. The total brain proteins were assigned their gene symbol via the Uniprot Knowledge database (http://www.uniprot.org/). Gene Ontology (GO) regarding molecular function, biological process and subcellular localization was used for the construction of protein classifications, while they were retrieved by DAVID v.6.7 and v.6.8.
Data storage. The data has been stored online and can be accessed at the following link https://bit.ly/3a6Ivrn
Results
Focusing on resolving the proteome of mouse brain under physiological conditions, at a high-resolution scale, eight inbred 8-week old C57BL/6N mice were analyzed (Figure 1). The animals were sacrificed and the brains were removed and divided into regions. In order to obtain sufficient protein quantity, the same brain region of at least eight animals was pooled and the sample was prepared for untargeted tandem mass spectrometry (22). A total of 16,574 proteins, belonging to 6,303 single gene products were identified in the analyzed brain regions (Table I). The highest number of proteins (2,795) were identified in the cerebral cortex, while 2,311 proteins were identified in the olfactory bulb, 2,246 in the hippocampus, 2,247 in the hypothalamus, 2,250 in the mid brain, 2,334 in the cerebellum and 2,391 in the medulla (Table I, Figure 1, Supplementary Tables 1-7). In order to reveal unique region-specific proteins, a comparative computational analysis was performed, indicating that 534 proteins were uniquely expressed in the cerebral cortex, 323 in the olfactory bulb, 230 in the hippocampus, 272 in the hypothalamus, 1,326 in the mid brain, 320 in the cerebellum and 268 in the medulla (Table I and Supplementary Table 8).
Furthermore, for the evaluation of the protein content of each brain region alone and in relation to the other brain regions, a multi-level comprehensive analysis based on GO categorization was performed. As shown in Figure 2, the analysis was based on the cellular component, the biological process, the molecular function and PTMs of the identified proteins. That approach indicated that the majority of the identified proteins belong to cellular components, followed by proteins related to PTMs and then proteins belonging to diverse biological process. The richest brain region was found to be the cerebral cortex, while the poorest region was the midbrain.
The correlation of identified proteins per brain region to cellular components, according to the GO categorization revealed that half of the proteins identified in each studied brain region were located in the cell membranes, including the internal and external ones, followed by a significant number of proteins in the nucleus and the extracellular region (Figure 3A). Cytoplasmic proteins were identified mainly in the cytosol and the mitochondrion, followed by a significant number of cytoskeletal proteins, and proteins of the Golgi apparatus and endoplasmic reticulum, while a smaller number of proteins were found to belong at ribosomes and peroxisomes (Figure 3A). Additionally, a small but significant number of proteins were classified as belonging to neuronal cell structures, i.e. synapse (n=172 proteins in average), neuronal cell body (n=163), dendrite (n=140), axon (n=136) and myelin sheath (n=153), (Figure 3B). Furthermore, the highest number of membranous, nuclear and extracellular region proteins were identified in the cerebral cortex, while the lowest was in the mid brain. In relation to neuroproteins, the highest number of synaptic proteins was found in the cerebral cortex, while the highest number of myelin sheath proteins was found in the medulla.
With regards to the biological processes, according to the GO categorization, a significant number of proteins were found to be implicated in transport, cellular processes, biological regulation, establishment of localization, metabolic processes and responses to stimulus, as well as in the nervous system and its development (Figure 4). The highest number of proteins related to cellular and metabolic processes, biological regulation and the responses to stimulus were founded in the cerebral cortex. In the hypothalamus, the highest number of nervous system development and process proteins were identified, while the highest number of developmental proteins was identified in the olfactory bulb. Finally, the results indicated that the highest number of cell cycle-related proteins was identified in the hippocampus.
Proteins identified in different regions of the normal mouse brain.
Regarding protein classification according to molecular function, the great majority of proteins were found to belong to protein-, nucleotide-, RNA-, calcium- and lipid-binding proteins, while a significant number of proteins had enzymatic activity (Figure 5A). Detailed analysis of the enzymatic proteins revealed them to be hydrolases, kinases and oxidoreductases. Figure 5B indicates that the highest number of hydrolases, kinases and ligases were found in the cerebral cortex, while the highest number of oxidoreductases and lyases were found in the medulla and cerebellum, respectively.
Finally, analysis of the PTMs according to the GO categorization showed that the most frequent PTM was phosphorylation, followed by acetylation and glycosylation (Figure 6). As shown in that figure, the highest number of phosphoproteins were identified in the cerebral cortex, followed by the medulla and the cerebellum, while the lowest number was identified in the mid brain.
Discussion
In-depth investigation of the molecular content of the CNS is of great importance and our knowledge is continuously enriched as state-of-the-art technologies developed. In this regard, the application of proteomics and MS in the investigation of the CNS has established a completely new scientific field named ‘neuroproteomics’. The brain, being the center of the nervous system in mammals, attracts great attention in the scientific community, and in-depth analysis of the brain is the ‘holy grail’ of neuroproteomics. The analysis of the normal brain is crucial for the understanding of brain functions at the molecular level, the functions of particular brain regions and the molecular alterations between brain regions in normal/physiological conditions. It is the center of numerous diseases and it has been studied by high-resolution MS approaches applied to several pathological conditions, such neurodegenerative diseases (Alzheimer's disease, Parkinson's disease), cancer and depression (8, 26, 27). Thus, considering the heightened interest in the proteomic analysis of the normal brain, as well as the difficulties that come with this analysis using human brain samples, neuroproteomics has been used in the analysis of whole brain and brain regions of mice and rats in order to elucidate the general pattern of protein expression in these regions during physiological and pathological conditions (6, 10).

Number of proteins identified in each brain region for general functional categories. PTMs: Post-translational modifications.
Mouse models are extensively used to recapitulate various pathological conditions. In 2010, the proteomic analysis of normal rat brains was announced by Katagiri and colleagues (28). According to their approach, the normal rat brain was divided into six areas and the proteomic analysis of each area was studied by a combination of SDS gel electrophoresis and nanoLC-Q-TOF-MS/MS. They identified 241 proteins in total in the studied normal brain regions and network analysis, and concluded that these proteins were linked to pathways closely related to the biological functions of each specific region.
In line with that study, in 2013 our group identified and published the total protein content of seven different regions of the normal brain of CB7BL/6 mice. Using two-dimensional gel electrophoresis followed by matrix-assisted laser desorption ionization time-of-flight MS, we identified 515 different single gene products leading, for the first time, to the complete characterization of the normal mouse brain proteome as well as the protein-expression profile of the different brain regions (20).
In the current study, we performed a global analysis of the protein level by using high-resolution state-of-the-art mass spectrometry technology revealing the brain proteome of an early-adult mouse, according to the Paxinos and Franklin mouse brain atlas (24). The analysis identified 16,574 proteins in total, belonging to 6,303 single gene products, in the seven specific brain regions analyzed (cerebral cortex, olfactory bulb, hippocampus, hypothalamus, midbrain, cerebellum, and medulla).

Proteins identified according to each brain region. A: Sub-cellular localization of the identified proteins in each brain region. B: Neuroprotein distribution per brain region. Classification of the identified proteins was based on Gene Ontology annotation.
In order to obtain the most representative protein content of the brain we analyzed each brain region individually. In contrast, a study by Sharma et al., attempted to analyze the brain region-specific and cell-specific proteome of adult mice, resulting in the identification of 12,934 different proteins by state-of-the-art MS technology and the integration of sequencing-based transcriptomic analysis (18). Unlike our approach and in order to determine the basis for cellular specialization, brain development and function, this study was performed on 11 brain slices, each one containing more than one anatomical region and different cell types. Thus, in the present study, our results constitute the most complete brain region-specific database, indicating identifying the highest number of proteins in the cerebral cortex, while the highest number of unique region-specific proteins were identified in the mid brain, the least studied brain region.

Biological process classification, based on Gene Ontology annotation of the identified proteins in each brain region.
In the present study, we identified proteins involved in various cellular processes, such as (a) the myelin sheath, i.e. myelin expression factor 2 (MYEF2), myelin basic protein (MBP), myelin-associated glycoprotein (MAG), myelin proteolipid protein (PLP1), myelin transcription factor 1-like protein (MYT1L), myelin-oligodendrocyte glycoprotein (MOG) and oligodendrocyte-myelin glycoprotein (OMG); (b) synapse formation and function, i.e. synapsin-1, 2. 3 (SYN1, SYN2, SYN3), regulating synaptic membrane exocytosis protein 3 (RIMS3), synapse differentiation-inducing gene protein 1-like (SYNDIG1L), synapse-associated protein 1 (SYAP1), synaptotagmin-1 (SYT1), calcium/calmodulin-dependent protein kinase type II subunit alpha (CAMK2A), synaptosomal-associated protein 25 (SNAP25) and syntaxin-1B (STX1B); (c) cytoskeleton structure and regulation, i.e. tubulin alpha 1B chain (TUBA1B), actin, cytoplasmic 1 (ACTB), microtubule-associated protein 1A (MAP1A), microtubule-associated proteins 2, 6 (MAP2, MAP6), spectrin alpha chain, non-erythrocytic 1 (SPTAN1), tubulin beta-3 chain (TUBB3), neurofilament light polypeptide (NEFL), neurofilament heavy polypeptide (NEFH) and glial fibrillary acidic protein (GFAP); and (d) the regulation of glycose uptake and metabolism, i.e. UDP-glucose: glycoprotein glucosyltransferase 1 (UGGT1), glucose 1,6-bisphosphate synthase (PGM2L1), gamma-enolase (ENO2), L-lactase dehydrogenase B chain (LDHB), glucose-6-phosphate 1-dehydrogenase X (G6PDX) and fructose-bisphosphate aldolase A (ALDOA).
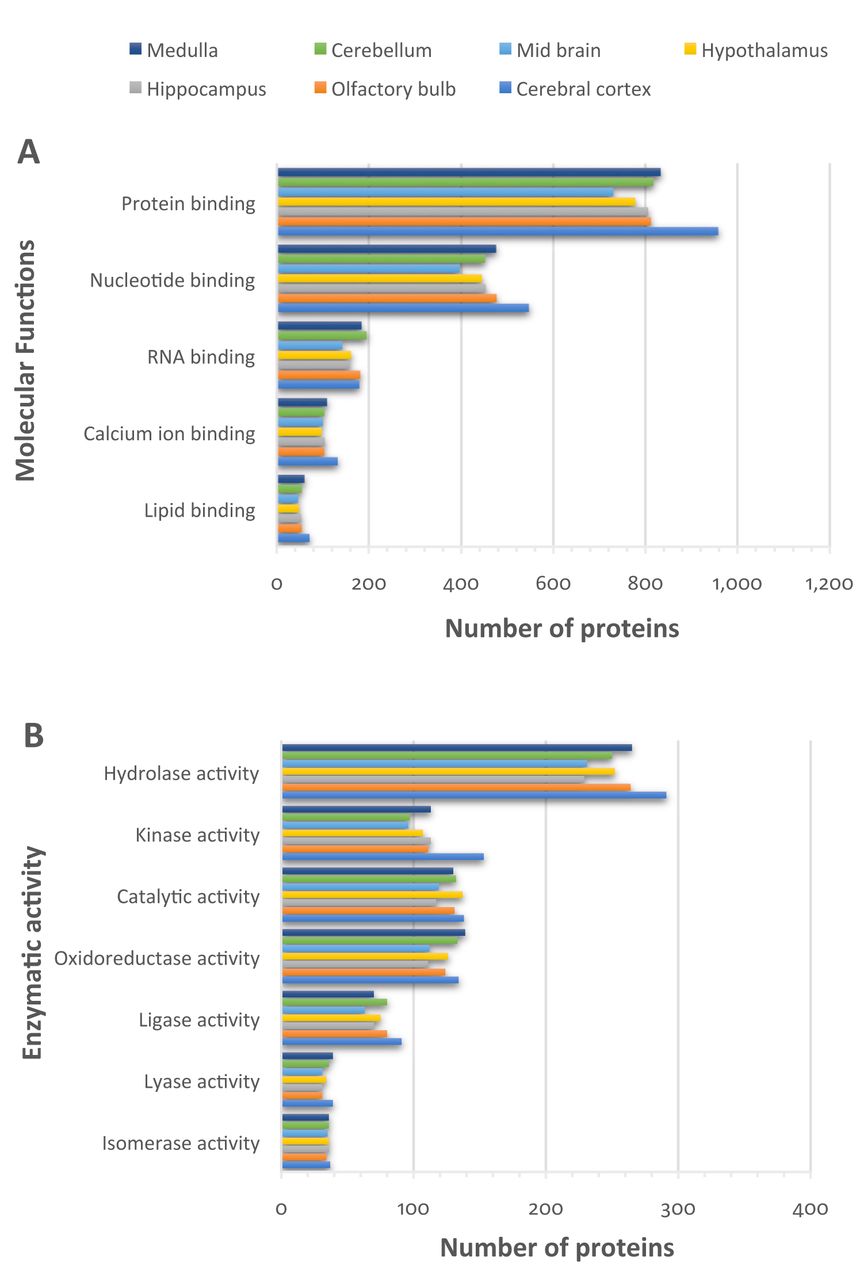
Function classification of the identified proteins, based on Gene Ontology annotation of the identified proteins in each brain region. A: Molecular functions. B: Enzymatic activity.

Post-translation modifications of identified proteins based on Gene Ontology annotation of the proteins identified in each brain region.
Furthermore, as previously described, a number of specific proteins related to the different types of brain cells (neurons and glia) were detected, including well-characterized cell markers, such as (a) astrocytic proteins, i.e. GFAP, glutathione S-transferase Mu 1 (GSTM1), peroxiredoxin-6 (PRDX6), cytosolic 10-formyltetrahydrofolate dehydrogenase (ALDH1L1), and aquaporin-4 (AQP4); (b) the oligodendrocytic proteins, i.e. MBP, PLP1, MAG, MOG, (c) the cerebellar granule neuronal proteins, i.e. the SNAP25, synaptic vesicle glycoprotein 2B (SV2B), G protein-regulated inducer of neurite outgrowth 1 (GPRIN1) and neural cell adhesion molecule L1 (L1CAM); and (d) cortical neuronal proteins, i.e. sodium/potassium-transporting ATPase subunit beta-1 (ATP1B1), dihydropyrimidinase-related protein 1 (CRMP1), neuronal membrane glycoprotein M6-a (GPM6A), neuro-filament medium polypeptide (NEFM), NEFL and NEFH.
Finally, proteins that have previously been associated with region-specific functions were identified here. We identified proteins related to the function of (i) the olfactory bulb, such as olfactory marker protein (OMP), olfactory receptors (OLFR), brain fatty acid-binding protein (FABP7), coiled-coil domain-containing protein 136 (CCDC136), inositol-trisphosphate 3-kinase A (ITPKA), synaptotagmin-2 (SYT2), calbindin-1 (CALB1), purkinje cell protein 4-like protein 1 (PCP4L1), pre-B-cell leukemia transcription factor 1 (PBX1), and homeobox protein Meis2 (MEIS2); (ii) the cerebellum, such as G-protein coupled receptor 37-like-1 (GPR37L1), vimentin (VIM), calbindin-1 (CALB1), cerebellin-3 (CBLN3), Purkinje cell protein 2 (PCP2), regulator of G-protein signaling 8 (RGS8), stromal interaction molecule 1 (STIM1), photocadherin Fat 2 (FAT2), metastasis suppressor protein 1 (MTSS1) and ryanodine receptor 1 (RYR1); (iii) the hypothalamus, such as neuroendocrine protein 7B2 (SCG5), jouberin (AHI1) and galanin peptide (GAL); (iv) the hippocampus, i.e. the neuron-specific calcium-binding protein hippocalcin (HPCA), phosphatidylethanolamine-binding protein 1 (PEBP1), glutamate receptors 1, 2 (GRIA1 and GRIA2), kinase suppressor of Ras 1 (KSR1), glutamate receptor ionotropic, NMDA 2B (GRIN2B); and (v) the cerebral cortex, such as T-box brain protein 1 (TBR1), voltage-gated potassium channel subunit beta-1 (KCNAB1), and synaptopodin (SYNPO). Our findings suggest that quantitative evaluation and co-expression analysis can provide valuable results regarding the protein interaction networks related to specific brain functions. It is worth noting that the complete evaluation of the identified proteins in relation to each brain region-specific function is the next step of our study.
To conclude, in the current study, we detected the global protein content of the early-adult mouse brain, using high-resolution MS. The massive data delivered by our study highlight the important contribution of MS-based techniques to the understanding of the molecular basis of mouse brain physiology and further provides a database for studies in brain physiology and pathology.
Footnotes
Authors' Contributions
AGK and AKA performed the experimental procedures. AGK, VP and GTT performed analysis of data. DJS and GTT designed and supervised the project. AGK and GTT wrote the article. All Authors participated in reviewing the article.
This article is freely accessible online.
Conflicts of Interest
The Authors declare no conflicts of interest regarding this study.
Supplementary Material
The Supplementary Tables 1 to 8 can be accessed online at https://bit.ly/3a6Ivrn
- Received July 22, 2020.
- Revision received August 23, 2020.
- Accepted August 26, 2020.
- Copyright© 2020, International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}