


Abstract
Background/Aim: The entire mechanisms by which epigenetic modifiers contribute to the development of pancreatic cancer remain unknown. Although the histone methyltransferase G9a is a promising target in human cancers, its role in pancreatic carcinogenesis has been under-studied. The aim of the study was to examine the role of G9a in pancreatic carcinogenesis by a gene-targeting mouse model. Materials and Methods: We established pancreas-specific G9aflox/flox mice and crossed them with Ptf1aCre/; KrasG12D/+ (KC) mice, which spontaneously develop pancreatic cancer. The phenotypes of the resulting KC mice with G9a deletion were examined. We analyzed transcriptomic data by microarray and genome-wide chromatin accessibility by transposase-accessible chromatin using sequencing. We established pancreatic organoids from KC mice. Results: G9a deficiency impaired the progression of pancreatic intraepithelial neoplasia (PanIN) and prolonged the survival of KC mice. The number of phosphorylated Erk-positive cells and Dclk1-positive cells, which are reported to be essential for the progression of PanIN, were decreased by G9a deletion. UNC0638, an inhibitor of G9a, suppressed the growth of organoids and increased global chromatin accessibility, especially around the regions including the protein phosphatase 2A genes. Conclusion: Thus, our study suggested the functional interaction of G9a, Dclk1 and Mapk pathway in the Kras-driven pancreatic carcinogenesis. The inhibition of G9a may suppress the initiation of oncogenic Kras-driven pancreatic carcinogenesis.
Pancreatic cancer is the most common cause of cancer-related mortality, with pancreatic ductal adenocarcinoma (PDAC) representing the most typical histological subtype. The global incidence of pancreatic cancer is predicted to be greater than 400,000; however, early diagnostic markers and therapeutic options still need to be developed (1). Genomic abnormalities, especially mutations or deletions in KRAS, TP53, SMAD4, and CDKN2A genes play essential roles in the development of PDAC. These mutations are already present in pancreatic intraepithelial neoplasms (PanINs), which are precursor lesions of PDAC (2), offering clues to the mechanism of pancreatic carcinogenesis (3).
To explain the evolution of pancreatic tumors, a gradual model has been proposed in which the independent accumulation of driver mutations and other oncogenic events accumulate during the long tumorigenic period (4). Additionally, a punctuated equilibrium model has been proposed in which pancreatic transformation is rapidly induced due to genetic instability (5). To date, many studies have elucidated the impact of these genetic events using genetically engineered mice harboring oncogenic KRAS mutations (6, 7).
In addition to genomic events, the involvement of epigenomic factors in determining the malignant characteristics of pancreatic cancers has received much attention (8, 9). Although previous reports have documented the roles of various epigenetic modifiers using mutant Kras-driven mouse models (10-13), the significance of epigenetic factors in pancreatic tumor evolution remains obscure.
G9a is a specific methyltransferase responsible for H3 lysine 9 dimethylation (H3K9me2), which is linked to transcriptional suppression. In addition, G9a plays a pivotal role in chromatin organization, for example, to adapt to the specific lineage commitment during normal developmental processes (14). In addition to its role in diverse biological processes in normal cells, the functions of G9a have been implicated in several types of cancers, including pancreatic cancer cells (15-17). However, its role in the initiation of pancreatic tumorigenesis remains to be elucidated.
For genomic or epigenomic analyses of tumor cells, the efficient isolation of cancer cells is important for avoiding the contamination of other stromal or immune cells. In particular, the abundant stromal components of PDAC are considered troublesome for various molecular studies. Organoids, which are recently developed culture models (18), are able to enrich cancer epithelial cells. In the research of PDAC, this model has been reported to be valuable for maintaining the original histological, biological, and molecular features of primary tumors (18-20).
In this report, we established a pancreas-specific G9a-knockout mouse model and examined the effects of this genetic manipulation on pancreatic carcinogenesis. Additionally, we examined the effects of G9a deletion on the chromatin accessibility profile of organoids propagated from oncogenic Kras-harboring pancreatic cells.
Materials and Methods
Animals. All experiments were performed in accordance with protocols approved by the Animal Ethics Committee of the University of Tokyo, in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publication No. 8023, revised 1978). Floxed G9a (G9aflox/flox) mice were generated as previously described (21). Ptf1aCre/+ mice and LSL-KrasG12D/+ mice have previously been described (22, 23). The mice were maintained in a temperature- and light-controlled facility and permitted ad libitum access to a regular chow diet and autoclaved water. All mice were backcrossed with the C57BL/6 strain, and both sexes were analyzed.
Histology and immunohistochemistry. Pancreatic tissues were fixed in 4% paraformaldehyde and embedded in paraffin. Tissue sections were stained with hematoxylin and eosin (H&E) for pathological evaluation. The PanIN lesion grade was defined based on previously described criteria (24). For immunohistochemistry, mouse tissue slides were subjected to heat-induced antigen retrieval using 10 mM citrate buffer (pH 6.0), and incubated with the indicated primary antibodies overnight at 4°C. Primary antibodies recognizing H3K9me2 (ab1220; Abcam, Cambridge, UK), G9a (PP-A8320A-00; R&D Systems, Minneapolis, MN, USA), Cpa1 (AF2765, R&D Systems), Krt19 (A-3; Santa Cruz Biotechnology, Dallas, TX, USA), Dclk1 (NBP1-96746; Novus Biologicals, Centennial, CO, USA), and phospho-Erk (9101; Cell Signaling Technology, Danvers, MA, USA) were used for immunohistochemical analysis. The sections were then incubated with biotinylated secondary antibodies and avidin-biotin-peroxidase (VECTASTAIN Elite ABC kit; Vector Laboratories, Burlingame, CA, USA), and were developed using diaminobenzidine as the chromogenic substrate. The slides were then briefly counterstained with hematoxylin. Negative control sections were not incubated with primary antibodies.
Quantitative reverse transcription PCR (qRT-PCR) analysis. Total RNA was extracted from tissues using an RNeasy Mini Kit (Qiagen, Hilden, Germany) and quantified using a NanoDrop spectrophotometer. Total RNA (1 μg) was reverse transcribed using the ImProm-II Reverse Transcription System (Promega, Madison, WI, USA). qRT-PCR analysis was performed using the StepOnePlus Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) according to the manufacturer's instructions. The test transcript levels were internally normalized against Actb transcript levels. The primers used for qRT-PCR were as follows: Dclk1 forward, 5’-GCTGGGGACTTGACACATTT-3’; Dclk1 reverse, 5’-CAAGGGCAGACTCTCCAAAG-3’; Actb forward, 5’-CTGGAACGGTGAAGGTGACA-3’; Actb reverse, 5’-AAGGGACTTCCTGTAACAACGCA-3’.
Microarray analysis. Microarray analyses were performed using the GeneChip Mouse Gene 2.0 ST Assay according to the manufacturer's protocol (Affymetrix, Santa Clara, CA, USA). Briefly, total RNA was extracted from murine pancreata obtained from Ptf1aCre/+; KrasG12D/+ (KC) and Ptf1aCre/+; KrasG12D/+; G9aflox/flox (GGKC) mice approximately 12 months after birth (n=2 each). RNA quality was determined using the Agilent 2100 Bioanalyzer (Agilent, Santa Clara, CA, USA). Fragmented, labeled cDNA was constructed using the GeneChip WT PLUS Reagent Kit and hybridized in a GeneChip Hybridization Oven 645 at 45°C for 16 h. Array scanning was performed using a GeneChip Scanner 3000 7G instrument. These experiments and the associated data processing were outsourced to Filgen Biosciences (Nagoya, Japan). Data analysis complied with the Minimum Information About Microarray Experiments standard. Prior to performing data analysis, chrX,Y and non-annotated probes were excluded, resulting in 19,166 probes included in the analysis. The probe design files and microarray data were submitted to the National Center for Biotechnology Information Gene Expression Omnibus database, with the accession number GSE149093.
Organoid culture. Organoids were established from murine pancreata using published protocols (18). Resected mouse pancreata were minced under sterile conditions and digested using 1 mg/ml collagenase type IV (Invitrogen, Carlsbad, CA, USA) in PBS for 10 min. The suspension was filtered through a cell strainer (100 μm) and centrifuged to sediment. The resulting pellet was mixed with Matrigel (BD Biosciences, Franklin Lakes, NJ, USA) and the tissue was cultured in vitro, as previously described (18), in AdDMEM/F12 medium (Invitrogen) supplemented with B27 (Invitrogen), 1.25 μM N-acetylcysteine (Wako, Osaka, Japan), 50 ng/ml EGF (Wako), 100 ng/ml Fgf10 (Wako), 10% Wnt3a-conditioned medium, 10% RSPO1-conditioned medium, 10 nM gastrin (Sigma-Aldrich), and 10 mM nicotinamide (Sigma-Aldrich). Additionally, for 4 days after seeding, the cells were supplemented with 100 ng/ml Noggin (PeproTech, Cranbury, NJ, USA). For transposase-accessible chromatin using sequencing (ATAC-seq) experiments, the organoids were treated with DMSO or UNC0638 (Selleck Chemicals, Houston, TX, USA) for 24 h.
ATAC-seq. ATAC-seq experiments were performed on 50,000 cells from the organoid samples, following published protocols (25). ATAC-seq reads were processed as follows. Sequence reads were trimmed and aligned to the mouse genome (mm10) using BWA (26). PCR duplicates were removed using Picard's MarkDuplicates tool, and peaks were called using MACS2, with the parameter - q=0.05 (27). All peaks were merged to obtain consensus peak sets. Reads for each peak were extracted using featureCounts (28) and were normalized by computing log2CPM using edgeR, before being used for downstream analysis (29). The peaks were annotated to the nearby genes by GREAT software (30). These data have been submitted to the National Center for Biotechnology Information Gene Expression Omnibus database, with the accession number GSE151434.
Statistical analysis. The data are presented as means±standard errors of the mean. Statistical analyses were performed using a two-tailed Student' s t-test.
Results
G9a deficiency attenuated PanIN progression in KC mice. KC mouse pancreata spontaneously developed acinar ductal metaplasia (ADM), murine PanINs (mPanINs), and invasive tumors (6). To assess the role of G9a in pancreatic tumorigenesis, we analyzed the phenotypic effects of G9a deficiency in KC mice. The loss of G9a expression was confirmed in the GGKC mouse pancreas. The pancreatic tissues of KC mice developed ADM and various stages of mPanINs several months after birth, while the mPanINs were less prominent in the GGKC mouse pancreas (Figure 1A, left). Alcian blue staining confirmed a limited distribution of mPanIN lesions with mucin production in the GGKC compared with KC mice (Figure 1A, middle). Moreover, Masson's trichrome staining suggested more fibrotic changes in the KC mouse pancreas (Figure 1A, right). To estimate the degree of mPanIN progression, we counted the number of respective grades of mPanINs. At 2 months after birth, the development of mPanIN1 was similar in both groups (Figure 1B). In contrast, up to ~1 year of age, the number of PanIN2 and PanIN3 lesions gradually increased in the KC mouse pancreas, but they were rarely found in GGKC mice (Figure 1B). KC mice occasionally developed pancreatic cancer (6), but GGKC mice did not (Figure 1C). Finally, the median survival time of the GGKC mice was significantly longer than that of the KC mice [627 days (n=18) vs. 419 days (n=17), *p<0.0001, Figure 1D]. No association with sex was observed in these results. Taken together, these data suggested that G9a played an important role in pancreatic oncogenesis in the presence of Kras mutations.
G9a deletion retained the molecular transition in ADM. The induction of ADM, which involves the degranulation of acinar cells and the emergence of cuboidal to columnar duct-like structures, is often observed prior to mPanIN formation in KC mice. G9a was expressed in the ADM lesions in KC mice, but not in GGKC mice (Figure 2A). Consistent with the ability of G9a to introduce di-methylation of H3K9 (21), H3K9me2 was absent in the ADM lesions of GGKC mice (Figure 2B). To assess the metaplastic status of ADM lesions in GGKC mice, we analyzed the expression of the acinar and ductal lineage markers, carboxypeptidase A1 (Cpa1) and cytokeratin 19 (Krt19), respectively, in serially sectioned specimens. Immunohistochemical analysis of these proteins indicated more ADM lesions in the KC pancreas than in the GGKC pancreas (Figure 2C and D). Cells positive for Cpa1 or Krt19 existed in a mutually exclusive pattern, coinciding with the cell lineages in the ADM lesions of KC mice (Figure 2E and F, upper). This mutually exclusive expression of Cpa1 and Krt19 was conserved in the ADM lesions of GGKC mice (Figure 2E and F, lower), indicating that the emergence of ADM was less frequent, but still occurred, in the GGKC pancreas (Figure 1B). Therefore, we suspected that G9a may be more critical in the progression of mPanIN than the development of ADM (Figure 1B).
G9a deficiency reduced the number of Dclk1-positive tuft-like cells and Erk phosphorylation in mPanIN lesions. To identify the genes responsible for the impaired progression of mPanIN in the GGKC pancreas, gene expression profiles were analyzed in whole pancreata of KC and GGKC mice. Among the 19,166 probes analyzed, 147 genes were found to be significantly up-regulated and 178 genes were down-regulated in the pancreata of GGKC mice compared to the pancreata of KC mice. Genes encoding pancreatic enzymes were significantly enriched in the GGKC mouse pancreas (Figure 3A), concurrent with the preservation of acinar cells in these mice (Figure 1). Down-regulated genes included epithelial genes, such as Krt20 (Figure 3A), which was likely due to fewer metaplastic duct cells. Furthermore, we found decreased expression levels of doublecortin-like kinase-1 (Dclk1) in GGKC mice (Figure 3A). Dclk1 was reported to mark a population of pancreatic cancer-initiating cells (31, 32). In addition, Dclk1 was suggested to be a marker of normal pancreatic stem/progenitors (32, 33). The down-regulation of Dclk1 in GGKC mice was confirmed in the validation cohorts (Figure 3B). As previously reported, Dclk1-positive cells presented the typical morphological features of intestinal tuft cells, with a narrow apical side and a wide basal surface in KC mice (asterisk in Figure 3C, upper right) (31, 34). However, the number of Dclk1-positive cells was clearly lower in the GGKC mouse pancreas (40.0% vs. 11.9%, **p<0.01, Figure 3C and 3D) and, if present, they did not always show the classic tuft cell morphology (Figure 3C, lower right). Given the reported critical function of Dclk1-positive cells in the expansion of PanINs (32, 35), we suspected that the absence of these cells led to the impaired progression of mPanINs in GGKC mice. Dclk1 has been reported to function as an effector of the mitogen-activated protein kinase (Mapk) pathway in the presence of Kras mutations in pancreatic carcinogenesis (32). The Mapk pathway mediates the de-differentiation of acinar cells into duct-like cells and the subsequent initiation of tumorigenesis (36, 37). Therefore, we next examined the activation status of Erk in KC and GGKC mouse pancreata. Erk phosphorylation levels were clearly decreased in GGKC mice compared with KC mice (38.3% vs. 12.8%, *p<0.05, Figure 3E and F). Of note, the Dclk1-deficient KrasG12D/+ mice demonstrated a phenotype similar to that of GGKC mice, namely, suppressed activation of the Mapk pathway and impaired development of high-grade PanIN lesions (32). Taken together, these data indicated that the impaired activation of Erk may contribute to the decreased progression of mPanINs in GGKC mice.
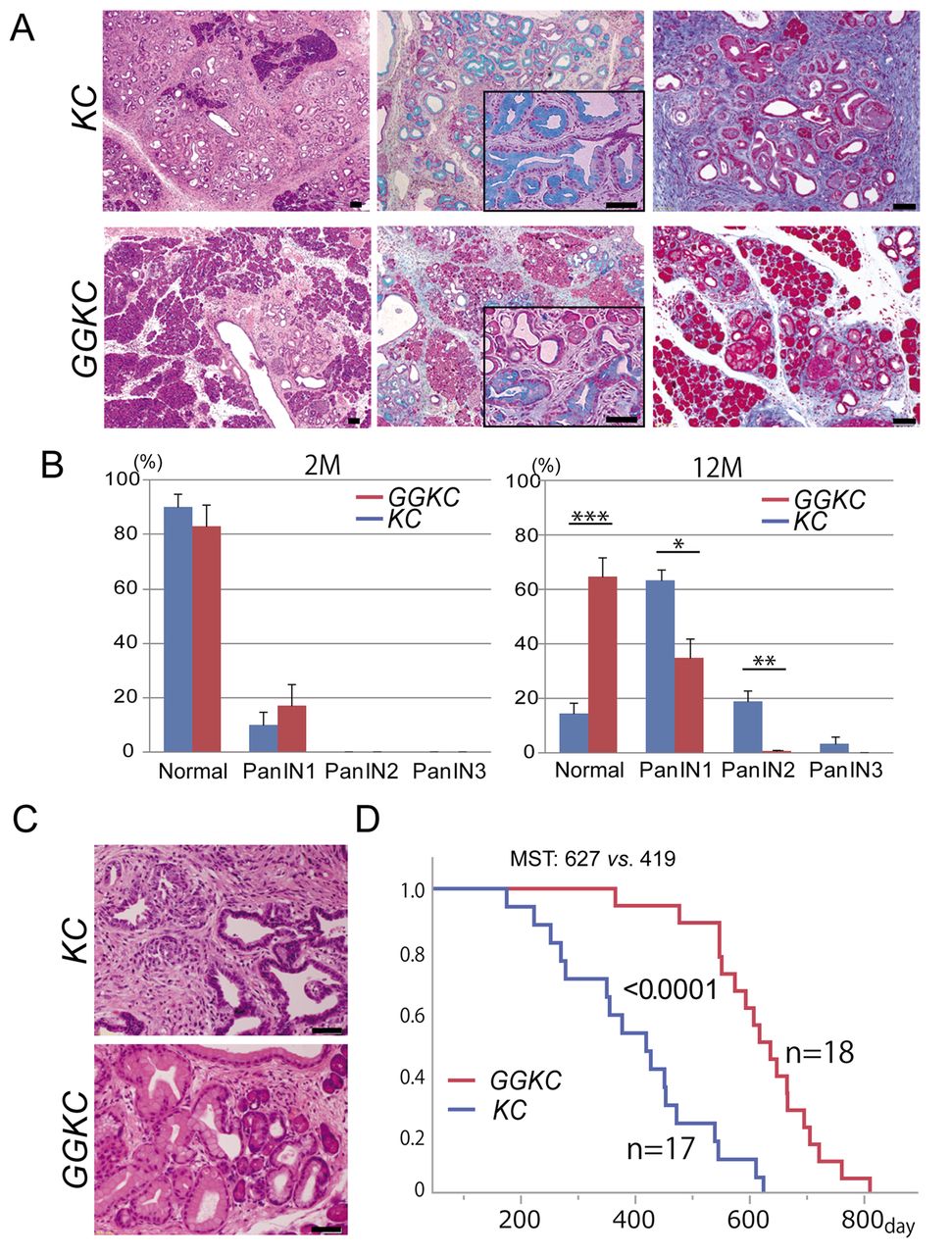
Depletion of G9a impaired the progression of PanIN in P48-Cre; KrasG12D (KC) mice. (A) Representative images of pancreatic tissue from the KC and GGKC mice. Left: H&E staining at 6 months. Center: Alcian blue staining at 11 months. Right: Masson's trichrome staining at 11 months. Scale bars: 50 μm. (B) Percentages of normal and neoplastic ducts in mice with an average age of 2 months (n=8 lesions from 4 mice each) and 12 months (n=10 lesions from 5 mice each). *p<0.01, **p<0.001, ***p<0.0001. (C) Representative images of pancreatic tumors in KC mice and PanIN lesions in the pancreata of GGKC mice at 13 months. (D) Survival duration of KC and GGKC mice. p<0.0001. MST: median survival time.
G9a was essential for the viability of mutant Kras-harboring pancreatic organoids. To confirm the in vivo findings, we next aimed to culture pancreatic organoids established from KC and GGKC mouse pancreata. The pancreatic organoids from KC mice continuously grew, as previously reported (18); however, those from GGKC mice did not survive in the culture (Figure 4A). Additionally, when the pancreatic organoids from KC mice were treated with UNC0638, a G9a chemical inhibitor, the growth of the organoids was clearly inhibited (Figure 4B). These data indicated that G9a was essential for the survival of mutant Kras-harboring pancreatic cells.
Inhibition of G9a affected chromatin accessibility in mutant Kras-harboring pancreatic cells. Finally, we analyzed the epigenetic status of pancreatic organoid cells. Given the function of G9a in regulating chromatin organization (38), chromatin accessibility in pancreatic organoid cells was examined using an ATAC-seq assay (25). Treatment with UNC0638 induced a greater number of open chromatin lesions in the organoids from KC mice (KC vs. KC+UNC0638, p<10-300, Figure 5A). Notably, the open chromatin lesions induced by UNC0638 treatment were enriched in genes of the protein phosphatase 2A (PP2A)- and Mapk-related categories (Figure 5B). PP2A regulates various oncogenic signals, including Mapk (39), and has been reported to inhibit Kras-driven lung tumor growth (40). The higher chromatin accessibility around PP2A-related genes was found in the UNC0638-treated organoids (Figure 5C and D). Despite the technical difficulties in validating the altered chromatin accessibility in the pancreas in vivo, it is possible that altered chromatin status may be involved in the phenotype observed in the GGKC mouse pancreas. In conclusion, G9a is an epigenetic modifier essential for the expansion of pancreatic cells harboring oncogenic KRAS mutations.
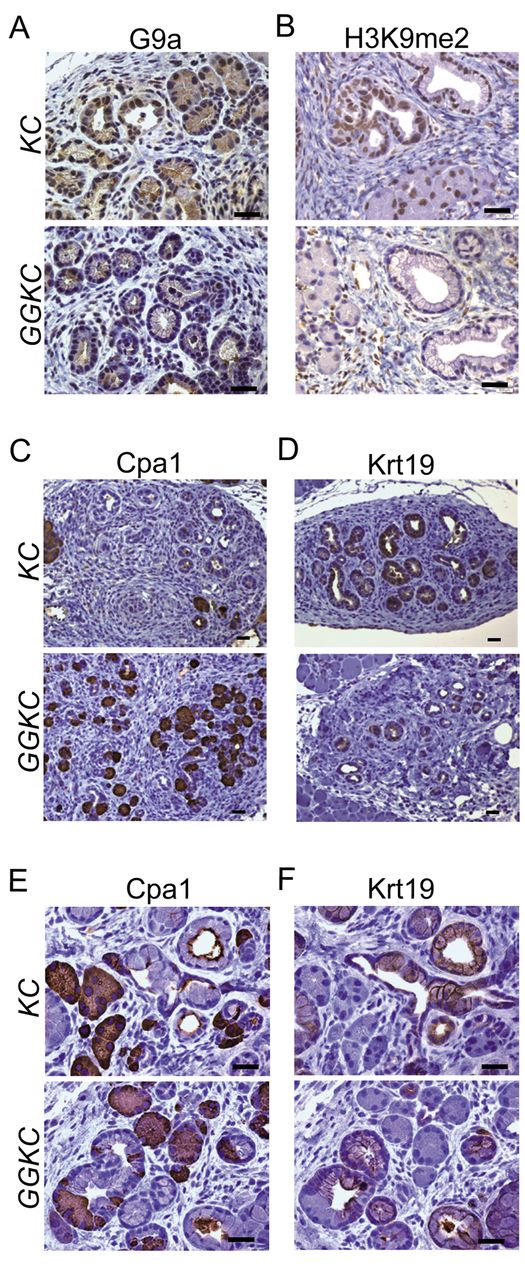
Spared acinar ductal transition shown by marker expression in the GGKC mouse pancreas. Representative staining images for G9a (A) and H3K9me2 (B) in pancreatic tissues obtained from KC or GGKC mice. Scale bars: 50 μm. (C-F) Representative staining images for Cpa1 (C, E) and Krt19 (D, F) in ADM tissues obtained from KC and GGKC mice. Scale bars: 50 μm.
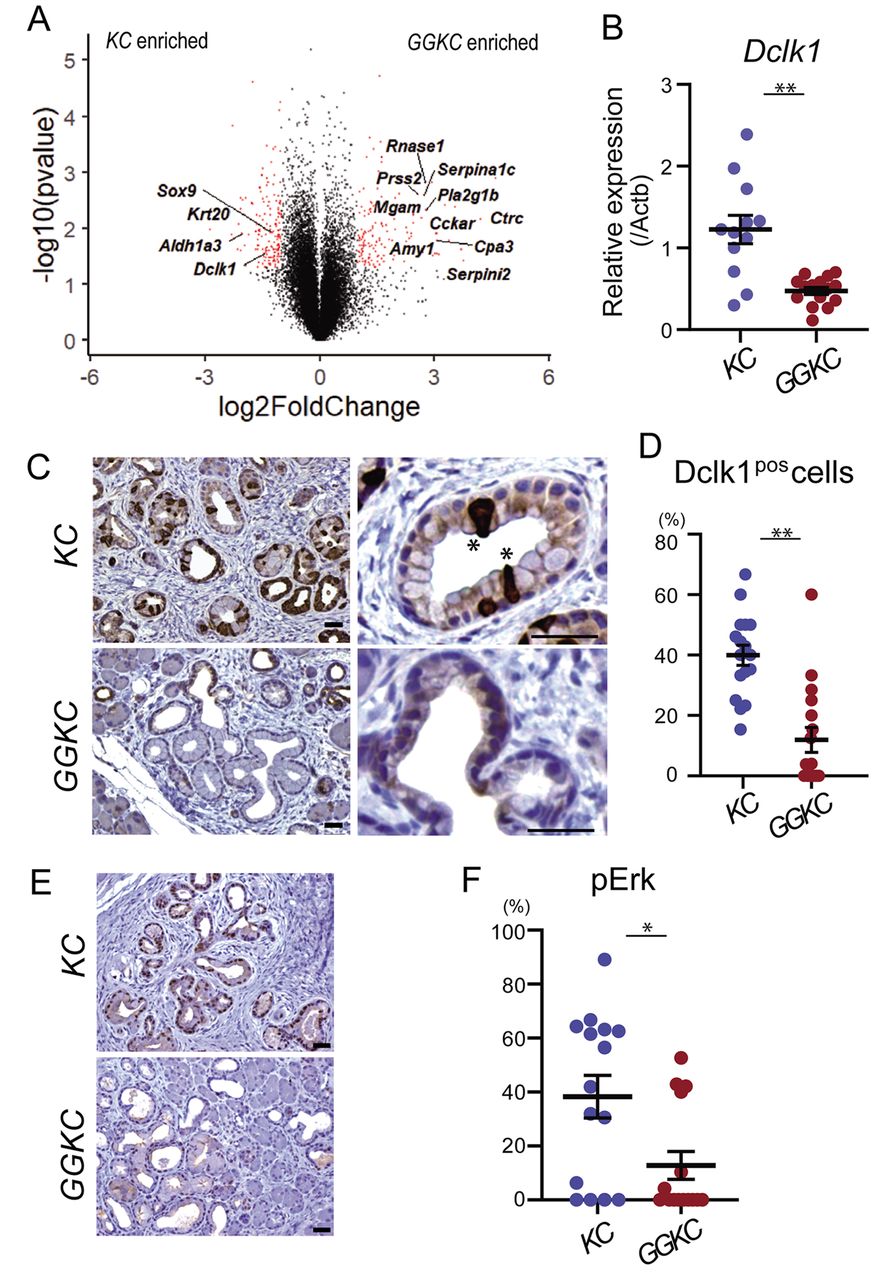
G9a deficiency reduced the number of Dclk1-positive cells and inhibited Erk phosphorylation in mPanIN lesions. (A) Comparison of gene expression between KC and GGKC pancreata. Red dots denote differentially expressed genes (p<0.05, log2FC <-1, >1). (B) qRT-PCR analysis of relative Dclk1 expression in KC and GGKC mice. **p<0.01. (C) Dclk1 staining of the pancreata of KC and GGKC mice. Asterisk indicates the tuft-like structure. Scale bars: 50 μm (D) Ratio of Dclk1-positive cells in mPanIN lesions (n=17 lesions from 2 mice each). **p<0.01. Scale bars: 50 μm. (E) Staining of phospho-Erk in the pancreata of KC and GGKC mice. Scale bars: 50 μm. (F) Ratio of phospho-Erk-positive cells in mPanIN lesions (n=15 lesions from 3 mice each). *p<0.05.
Discussion
In this study, we established a pancreas-specific G9a-knockout mouse model and analyzed the effects on mutant-Kras-driven pancreatic tumorigenesis using this model. While the role of G9a in pancreatic cancer cells has previously been reported (15-17), the effects on chromatin accessibility by G9a inhibition were not analyzed. This study demonstrated the significance of G9a in the initiation of pancreatic cancer development in vivo.
In the GGKC mouse pancreas, low-grade mPanINs and ADM lesions were fewer in number, but still observed. In contrast, high-grade mPanINs were barely detected (Figures 1 and 2), implying the role of G9a in the malignant transition of low-grade mPanIN lesions. Expression analysis to address the genes responsible for the impaired mPanIN progression in the GGKC mouse pancreas revealed decreased expression levels of Dclk1 (Figure 3). Of note, the pancreas-specific Dclk1-knockout mouse demonstrates delayed progression of mPanIN lesions (32), similar to the situation seen in GGKC mice. Therefore, we considered that the decrease in the number of Dclk1-positive cells may be related to the phenotype of the GGKC mouse pancreas. However, the exact mechanism linking the loss of G9a to the number of Dclk1-positive cells remains unclear.
The activation of Mapk downstream of Kras leads to multiple cellular effects including transcription, cell cycle regulation and survival (41). The inhibition of Mapk pathway resulted in regression of mPanIN regions induced by oncogenic Kras (36), indicating the pivotal role in the maintenance of mPanIN. In addition, Mapk mediates the dedifferentiation of oncogenic Kras harboring acinar cells to duct-like cells. The mechanism is considered to be mediated by the altered expression of key transcription factors including Mist1, Sox9, Hes1 and Pdx1 (36, 37). Phosphorylation of Erk indicates the activation of the Mapk pathway. Functional relationships between G9a and ERK have previously been reported in other models (42, 43). Although the relationship between G9a and ERK is unknown in pancreatic cells, GGKC mice showed a decreased number of phospho-Erk-positive PanIN cells (Figure 4). Dclk1 has been reported to be a critical factor for Mapk activation in pancreatic cancer cells, through direct binding to Kras protein (32). In addition, Dclk1-positive cells have clonogenic potential, sustaining the growth of pancreatic cancer organoids (31, 32). Taken together with our data, these findings indicate that G9a may be related to Mapk activation and the clonogenic ability of Dclk1-positive cells in the pancreata of KC mice.
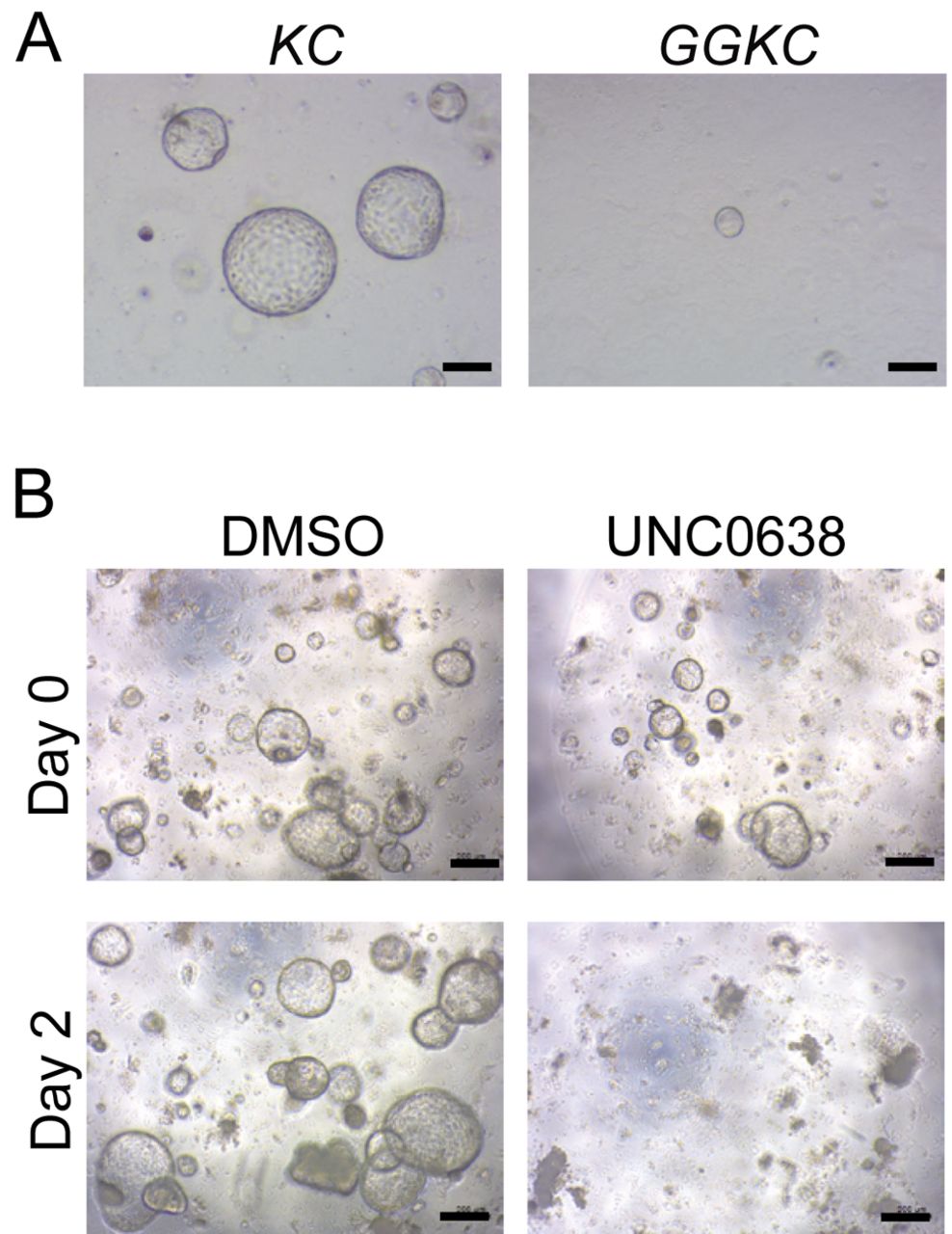
Treatment with UNC0638 affected the growth of pancreatic organoids from KC mice. (A) Representative images of organoids established from KC and GGKC mouse pancreata. Scale bar: 200 μm. (B) Representative images of organoids derived from KC mice at day 2 after treatment with DMSO or 10 μM UNC0638 for 24 h. Scale bar: 200 μm.
Altered chromatin accessibility due to G9a inhibition has been shown in a previous study (38, 44). Consistent with these findings, G9a deletion affected the global level of chromatin accessibility in pancreatic organoids derived from KC mice (Figure 5A). In addition, PP2A-related genes, which are negative regulators of the Mapk pathway, were identified as the most affected gene category (Figure 5B). Considering that the functional relationship of G9a and PP2A phosphatase activity has been previously reported (45), PP2A may be involved in the impaired activation of Mapk in the GGKC mouse pancreas. However, in general, the extent to which the altered chromatin structure is directly linked to the biological outcome remains obscure.
H3K9 dimethylation status can be demethylated by a histone demethylase KDM3A/JHDM2A/JMJD1A (46). A recent study showed that KDM3A knockdown suppresses DCLK1 expression in human pancreatic cancer cell (47). While our data contradicted these previous findings, there was a clear difference in the model used in the two studies. Our study focused on the initiation of pancreatic tumorigenesis in an in vivo mouse model, whereas, the previous study analyzed established human pancreatic cancer cells. Indeed, because the tuft-like Dclk1-positive cells observed in KC mice are rarely seen in established pancreatic cancer tissues (32, 35), the Dclk1-positive cells analyzed in the two studies may be distinct cell populations.

G9a affected chromatin accessibility in pancreatic organoids derived from KC mice. (A) Histogram of global chromatin accessibility in KC and KC+UNC0638 organoids. p<10-300. (B) Gene ontology molecular function categories of ATAC-enriched peaks in KC+UNC0638 organoids, compared to KC organoids. (C) PP2A-related genes were enriched in ATAC peaks in KC+UNC0638 organoids, as analyzed using GREAT software. (D) Representative ATAC tracks at the Ppp2r3a and Ppp2r5d loci in normal (K), KC, and KC+UNC0638 organoids. Grayed box in each locus denotes the differential ATAC peaks.
In summary, our data suggested a possible effect of G9a inhibition on pancreatic carcinogenesis. To date, some G9a inhibitors have been developed as potential anti-cancer agents. However, a recent study indicated that long-term G9a inhibitor administration induced selection for aggressive Trp53-mutant tumors (38). That study suggested the possibility that the outcomes of inhibiting G9a may be affected by the features or stage of the tumor, such as Trp53 mutation status. The efficacy of epigenetic therapy may depend on the genomic profile, in a manner similar to the current molecular targeting agents.
Acknowledgements
This study was partially supported by Grants from JSPS KAKENHI (Grant Number 23591009, 18K07962) and a grant from the International Joint Usage/Research Center, the Institute of Medical Science, the University of Tokyo. We thank Mitsuko Tsubouchi and Sayaka Ito for their technical assistance.
Footnotes
Authors' Contributions
Conception and design: KT, HK. Development of methodology: KT, HK, KY, TN, YK, and HI. Acquisition of data and management of animal samples: KT, HK, and HF. Resources: MT, YS. Writing and review: MK, MS, YH, HN, YT, MO, YH, and KK. Final approval of article: All Authors.
This article is freely accessible online.
Conflicts of Interest
The Authors declare that they have no competing interests.
- Received August 5, 2020.
- Revision received August 28, 2020.
- Accepted August 31, 2020.
- Copyright© 2020, International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved
References
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.