


Abstract
Background/Aim: Fusions of the ABL proto-oncogene 1 gene (ABL1 in 9q34) are common in leukemias but rare in solid tumors. The most notable is the t(9;22)(q34;q11)/BCR-ABL1 coding for a chimeric tyrosine kinase. We herein report an ABL1-fusion in a pediatric tumor. Materials and Methods: G-banding, fluorescence in situ hybridization, reverse transcription polymerase chain reaction and Sanger sequencing were performed on a soft tissue perineurioma found in the left musculus erector spinae of a child. Results: A der(4)t(4;9)(q31;q34) and a fusion of the GRB2 associated binding protein 1 (GAB1 in 4q31) gene with ABL1 were found. A literature search revealed 3 more cases with similar genetic and clinicopathological characteristics: a soft tissue perineurioma with t(2;9;4)(p23;q34;q31) and ABL1 rearrangement, a soft tissue angiofibroma with a GAB1-ABL1 chimeric gene, and a solitary fibrous tumor carrying a der(4)t(4;9)(q31.1;q34). Conclusion: GAB1-ABL1 is a recurrent fusion gene in benign pediatric tumors.
- Benign pediatric soft tissue tumors
- pediatric
- soft tissue perineurioma
- chromosome translocation
- GAB1
- ABL1
- GAB1-ABL1 fusion gene
- der(4)t(4;9)(q31;q34)
The ABL proto-oncogene 1 gene (ABL1, previous symbol ABL) in chromosome band 9q34 is ubiquitously expressed and codes for a non-receptor tyrosine kinase which is localized at many subcellular sites including the nucleus, cytoplasm, mitochondria, and endoplasmic reticulum. It is involved in a variety of cellular processes such as cell division, adhesion, differentiation, and response to stress (1-3). ABL1 together with ABL2 (the proto-oncogene 2 gene which encodes a non-receptor tyrosine kinase and maps to chromosome band 1q25) constitute the ABL family of kinase genes (2, 4-6). Both ABL1 and ABL2 fuse with a variety of translocation partner genes in various hematological malignancies (7-10). The most notable fusion is between ABL1 and the 5’ end of the breakpoint cluster region gene (BCR), located in 22q11, through the t(9;22)(q34;q11) chromosome translocation that gives rise to the Philadelphia chromosome in chronic myeloid leukemia (CML) (11, 12). The BCR-ABL1 fusion gene codes for a leukemogenic, constitutively active tyrosine kinase (11, 12). The discovery that 2-phenylaminopyrimidines inhibit the ABL protein kinase both in vitro and in vivo, led to the development of imatinib mesylate that now constitutes the first-line treatment of CML, as well as to introduction of other protein kinase inhibitors into cancer therapy (13-19).
Rearrangements of the ABL1 and ABL2 genes in solid tumors have also been documented (3-6, 20). Phosphorylation and activation of ABL kinases were reported in various tumors such as breast and lung adenocarcinomas, melanomas, and cancers of the brain (3-6, 20). The mechanisms for activation of ABL1 and ABL2 kinases are in these settings not chromosome translocations/fusion genes but rather genomic amplification, increased expression of mRNA, enhanced protein expression, and increased catalytic activity (3-6, 20).
Perineurioma is a tumor composed entirely of neoplastic perineurial cells with ultrastructural and immunohistochemical features similar to those of their normal counterparts (21). According to the latest WHO Tumors of Soft Tissue and Bones, perineuriomas are nearly always benign, although rare malignant variants have been reported (22). There are two main types: intraneural perineuriomas are confined within peripheral nerve boundaries whereas extraneural perineuriomas are found in soft tissue and skin (21, 23-25). Based on clinicopathological characteristics, the extraneural tumors are further subdivided into soft tissue, sclerosing, and reticular lesions (21, 23, 26-33).
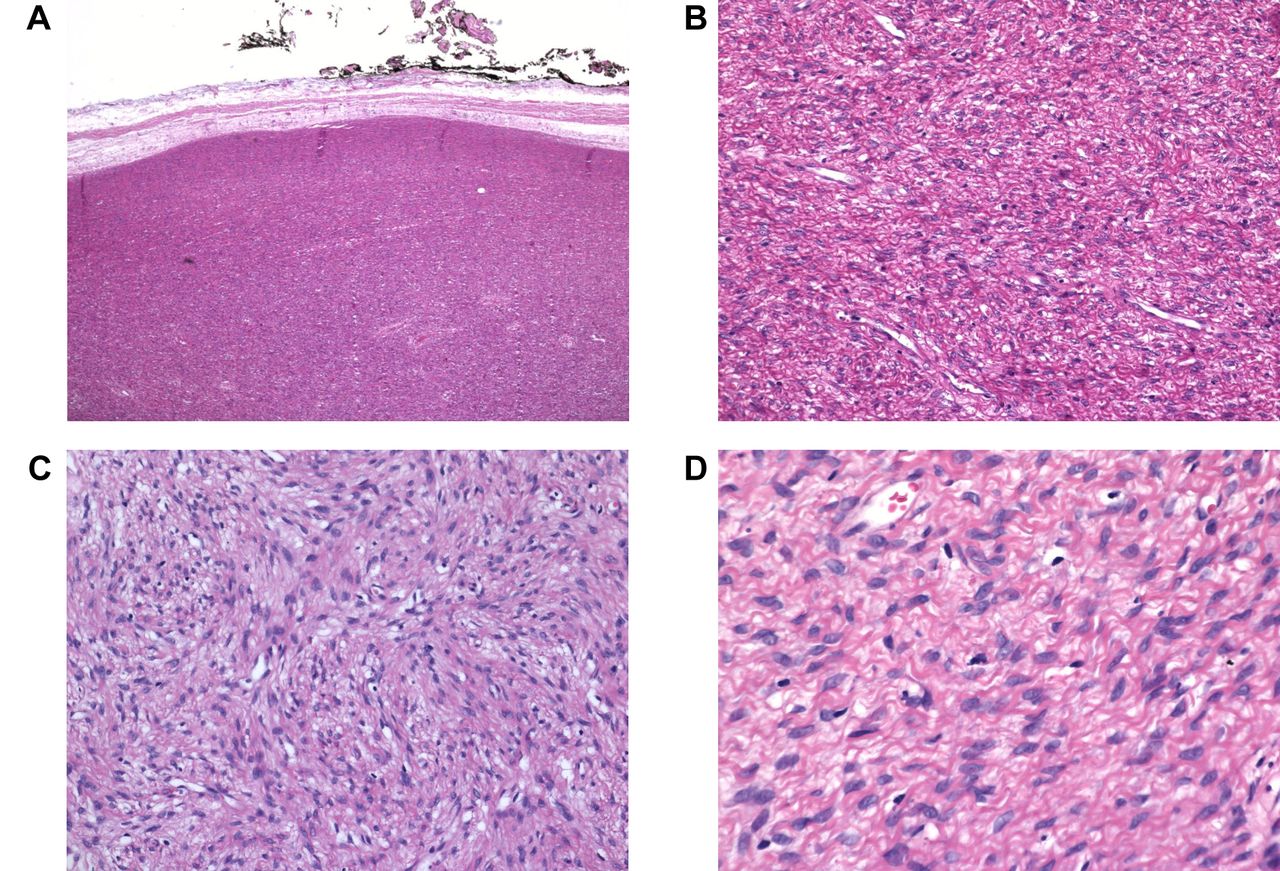
Hematoxylin and eosin (H&E) staining of a pediatric soft tissue perineurioma. A) H&E stained section showing well demarcated, unencapsulated, highly cellular solid tumor tissue, magnification 1×. B) H&E stained section (low magnification) showing ovoid to spindle cells, magnification 4×. C) H&E-stained section showing a whorled to storiform growth pattern, magnification 10×. D) H&E-stained section showing relatively uniform cells with oval-shaped to slender, tapering nuclei, magnification 20×.
In the present study, we report the finding of fusion of the GRB2 associated binding protein 1 (GAB1) gene with ABL1 in a pediatric soft tissue perineurioma. We review the literature and conclude that GAB1-ABL1 is a recurrent fusion which appears to characterize a benign, pediatric tumor type.
Materials and Methods
Ethics statement. The study was approved by the Regional Ethics Committee (Regional komité for medisinsk forskningsetikk Sør-Øst, Norge, http://helseforskning.etikkom.no; 2010/1389/REK sør-øst A). Written informed consent was obtained from the patient's parents. The Ethics Committee's approval included a review of the consent procedure. All patient information has been de-identified.
Case description. The patient was a 12-year-old boy with a tumor in the left erector spinae musculature. It measured 65×45×20 mm, was circumscribed but unencapsulated, and showed small areas of infiltration into the skeletal muscle. Microscopically, a whorled to storiform growth pattern was seen (Figure 1). The tumor cells were relatively uniform with oval, slender, relatively uniform nuclei. The cytoplasm showed elongated extensions within a collagenous stroma. Mitotic figures were rarely seen. Immunohistochemistry performed at the primary lab showed negative results for epithelial membrane antigen (EMA), S100 protein, mucin 4 (MUC4), and cluster of differentiation 34 (CD34). After consulting Professor Jason Hornick, Department of Pathology, Brigham and Women's Hospital, Boston, USA, the patient was diagnosed as having a soft tissue perineurioma. Repeated EMA immunohistochemistry at Brigham & Women's Hospital was focally positive.
G-Banding, karyotyping, and fluorescence in situ hybridization (FISH). Cells from a representative area of the tumor were short-term cultured and analyzed cytogenetically as previously described (34). The karyotype was written according to the International System for Human Cytogenomic Nomenclature (35). FISH experiments were performed on interphase nuclei using the ZytoLight SPEC ABL1 Dual Color Break Apart Probe (ZytoVision, Bremerhaven, Germany) following the company's recommendations.

Cytogenetic analysis of a pediatric soft tissue perineurioma. A karyogram showing the abnormal chromosomes der(1)(4qter->4q31::9q34->9q34::1p34~35->1qter), der(4)t(4;9)(q31;q34), and der(9)t(1;9)(p34~35;q34). Breakpoint positions are indicated by arrows. tas(17;19)(qter;qter) is not clonal.
Reverse transcription (RT) polymerase chain reaction (PCR) and Sanger sequencing. Total RNA was extracted from frozen (-80°C) tumor tissue adjacent to that used for cytogenetic analysis and histological examination using miRNeasy Mini Kit (Qiagen, Hilden, Germany). For cDNA synthesis, the iScript Advanced cDNA Synthesis Kit for RT-qPCR was used to reverse transcribe one μg of total RNA according to the manufacturer's instructions (Bio-Rad, Hercules, CA, USA). cDNA corresponding to 20 ng total RNA was used as template in subsequent PCR assays. The BigDye Direct Cycle Sequencing Kit was used for PCR/cycle (Sanger) sequencing according to the company's recommendations (ThermoFisher Scientific, Waltham, MA, USA). The primers were M13For-GAB1-1676F1: TGTAAAACGACGGCCAGTCCACCACGACAACATTCCAGCAGTT and M13Rev-ABL1-167R1: CAGGAAACAGCTATGACCGGTCATTTTCACTGGGTCCAGCGA. Sequencing was run on the Applied Biosystems SeqStudio Genetic Analyzer system (ThermoFisher Scientific). For computer analysis of sequence data, the basic local alignment search tool (BLAST) software (https://blast.ncbi.nlm.nih.gov/Blast.cgi) was used (36). The web version of Open Reading Frame Finder at NCBI (https://www.ncbi.nlm.nih.gov/orffinder/) was used to search for open reading frames of the sequence.
Results
The initial G-banding analysis yielded a karyotype with a three-way chromosomal translocation as the only cytogenetic aberration: 46,XY,t(1;9;4)(p34~35;q34;q31)[7]/46,XY[4] (Figure 2). FISH with the ZytoLight SPEC ABL1 Dual Color Break Apart Probe showed the following signal pattern in 92 out of 246 (37%) examined interphase nuclei: one red/green (corresponding to normal ABL1), one red (distal part of the probe), and two green signals (proximal part of the probe) (Figure 3A). The results indicated rearrangements of both ABL1 and the region immediately proximal to this gene which hybridized to the 710 Kbp green-labelled part of the probe (Figure 3B). Taking into consideration the initial karyotype as well as the FISH results, we concluded that the observed rearrangement was more complex than it appeared and included at least one cryptic change. A likely explanation is that two cytogenetic events had taken place (Figure 3C): a translocation between chromosomes 4 and 9 which generated a der(4)t(4;9)(q31;q34) and a der(9)t(4;9) (q31;q34), and a translocation between chromosomes 1 (p34~35) and the der(9)t(4;9)(q31;q34). The breakpoint on the der(9) occurred just upstream (proximal) of the ABL1 gene (Figure 3B and C). Thus, part of 9q34 together with 4q31-qter material is translocated to the der(1). Further, the segment from 1pter to 1p34~35 is translocated onto 9q34 from the der(9)t(4;9)(q31;q34) (Figure 3C) which is why we observed two green signals by interphase FISH; a part of the “green” region of the probe hybridized to der(1) whereas the other part hybridized to der(9) (Figure 3C). If we therefore reassess the G-banding karyotype in light of the FISH data, we arrive at the following karyotype: 46,XY,der(1)(4qter->4q31::9q34->9q34::1p34~35->1qter),der(4)t(4;9) (q31;q34),der(9)t(1;9)(p34~35;q34)[7]/46,XY[4].

FISH analyses of a pediatric soft tissue perineurioma. A) FISH with the ABL1 break apart probe on a normal nucleus and a nucleus with aberrant hybridization pattern suggesting rearrangements of both ABL1 and the region proximal to ABL1. B) A diagram of the ZytoLight ABL1 break apart probe. Vertical arrows indicate the rearranged regions. C) Diagram showing two hypothetical cytogenetic events that would explain the observed FISH results: Left, translocation between chromosomes 4 and 9 generating the derivative chromosomes der(4)t(4;9)(q31;q34) and der(9)t(4;9)(q31;q34); right, translocation between chromosomes 1 (p34~35) and der(9)t(4;9)(q31;q34) which gives rise to der(1)(4qter->4q31::9q34->9q34::1p34~35->1qter) and der(9)t(1;9)(p34~35;q34). The chromosomes are not in scale.
PCR/cycle (Sanger) sequencing with the primer combination M13For-GAB1-1676F1 and M13Rev-ABL1-167R1 revealed a fusion of GAB1 exon 6 (nucleotide 1898 in the reference sequence NM_002039.4) with ABL1 exon 2 (nucleotide 83 in the reference sequence NM_005157.4) (Figure 4A). Based on the fusion point detected and the reference sequences NM_002039.4 and NM_005157.4, the GAB1-ABL1 fusion transcript is in frame and coding for a 1632 amino acid residues (aa) protein in which the first 528 aa come from GAB1 while the other 1104 aa are from ABL1 (Figure 4B).
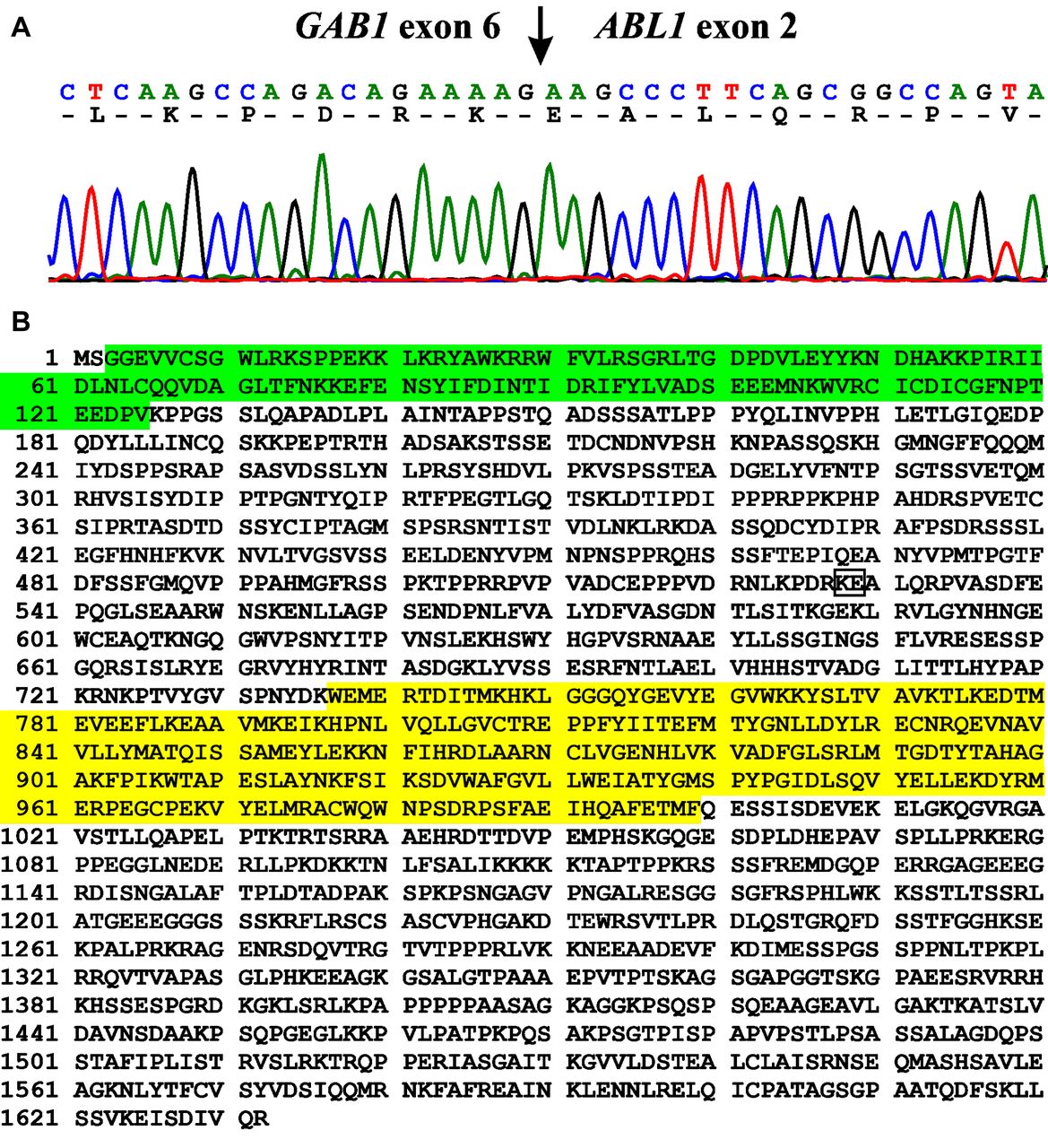
Results of Sanger sequencing. A) Partial sequence chromatogram of the cDNA amplified fragment showing the junction position of the GAB1 and ABL1 genes (arrow). The exon numbers were based on the sequences with accession numbers NM_002039.4 for GAB1 and NM_005157.4 for ABL1. B) The putative 1632 amino acid residues GAB1-ABL1 protein based on the sequences with accession number NP_002030.2 for GAB1 and NP_005148.2 for ABL1. The pleckstrin homology (PH) domain of GAB1 is in green. The catalytic domain of the protein tyrosine kinase of ABL1 is in yellow. The GAB1-ABL1 junction is in box.
Discussion
We herein present a soft tissue perineurioma with a der(4)t(4;9)(q31;q34) which gave rise to a GAB1-ABL1 fusion gene. A search of the literature revealed 3 additional cases with similar genetic and clinicopathologic characteristics (Table I, cases 9, 11, and 12).
Clinicopathological data on published intraneural, sclerosing, and soft tissue perineuriomas with abnormal karyotypes. The solitary fibrous tumor with der(4)t(4;9)(q31.1;q34) and the soft tissue angiofibroma with GAB1-ABL1 and associated (assumed) t(4;9) are included.
The first tumor was found in the right shoulder of a 9-year-old girl (Table I, case 11). It was diagnosed as a benign solitary fibrous tumor (SFT). Among various chromosome aberrations, also a der(4)t(4;9)(q31.1;q34) was found (37). SFT may mimic other tumors, among them soft tissue perineurioma (21, 23, 33, 38, 39). However, SFTs are characterized by the pathognomonic NAB2-STAT6 fusion gene resulting from an intrachromosomal inversion involving 12q13.3, which leads to nuclear expression of STAT6 (38-44). Thus, “One should be cautious to call an “SFT-look-alike” tumor an SFT without a positive STAT6 immunostain result” (44).
The second tumor was found in the forearm of a 14-year-old girl (Table I, case 9). It was diagnosed as a soft tissue perineurioma and had a t(2;9;4)(p23;q34;q31) chromosome translocation as the sole cytogenetic aberration (45). Additional experiments with array-comparative genome hybridization and FISH showed that the three-way translocation resulted in interstitial deletions in 2p and 9q as well as rearrangement of ABL1. The 5‘part of ABL1 was deleted whereas the 3‘part was moved to the der(4) (45). The authors concluded that “the translocation between the distal end of the 9q deletion, which is located at 9q34.12, and 4q31 could very likely be significant because 3‘ABL1 is involved” (45). Because the tumor we examined displayed a similar cytogenetic pattern to that of the above-mentioned tumors, i.e., the initial karyotype showed a three-way translocation with breakpoints in chromosome bands 4q31 and 9q34, we performed FISH with an ABL1 break apart probe. This showed splitting of ABL1 (Figure 3A). We considered the possibility that an ABL1-fusion might be located on the der(4), and therefore searched the relevant literature for ABL1 fusions in mesenchymal tumors. This gave information on a third tumor which was located in the foot of a 7-year-old boy, diagnosed as a soft tissue angiofibroma and carrying a GAB1-ABL1 fusion gene (46) (Table I, case 12). However, soft tissue angiofibromas are characterized by the pathognomonic t(5;8)(p15;q13) chromosome translocation or variants thereof resulting in an AHHR-NCOA2 fusion gene (46-50). Moreover, in a case of soft tissue angiofibroma (51), a t(7;8;14)(q11;q13;q31) translocation was found resulting in a GTF2I-NCOA2 fusion. This further emphasizes the role of NCOA2-rearrangements in the development of soft tissue angiofibroma (51). Because there are histologic and immunohistochemical similarities between soft tissue angiofibroma and soft tissue perineurioma (21, 23, 48, 49, 52-55) and because GAB1 maps on chromosome band 4q31, we investigated the present tumor to see if a GAB1-ABL1 fusion gene had been generated. Using RT-PCR/cycle (Sanger) sequencing, we detected an in-frame GAB1-ABL1 fusion transcript with the fusion point identical to the one previously described (46). Because both the GAB1 and ABL1 genes on 4q31 and 9q34, respectively, are transcribed from centromere to telomere, the GAB1-ABL1 fusion gene is predicted to be formed on der(4). Thus, we conclude that the tumor with der(4)t(4;9)(q31.1;q34) (37) and the one with t(2;9;4) (p23;q34;q31) (45) both carried a GAB1-ABL1 fusion gene. In fact, a simple, balanced t(4;9)(q31;q34) chromosome translocation could generate a GAB1-ABL1 fusion.
The GAB1-ABL1 fusion gene would code for a chimeric protein in which the first 28 aa of ABL1 are replaced by the first 528 aa of GAB1. It retains the pleckstrin homology (PH) domain of GAB1 and all the functional domains of ABL1, including the kinase domain (2, 9, 56-58). Thus, GAB1-ABL1 is predicted to be a chimeric tyrosine kinase with similar functions to the chimeric kinases BCR-ABL1, ETV6-ABL1, NUP214-ABL1, ZMIZ1-ABL1, and EML1-ABL1 which were found in hematologic malignancies (9).
Only a limited number of perineuriomas have been studied genetically. Karyotypic data exist on two intraneural tumors, three sclerosing tumors, and, including the present case, five soft tissue perineuriomas (Table I) (45, 59-63). Molecular genetic studies, including FISH, are also very few (28, 60, 64-66).
In intraneural perineurioma, a deletion in chromosome band 22q11 was reported in the first tumor examined whereas structural aberations of 2q11 and 3q12 were seen in the second (59, 61) (Table I, cases 1 and 2). Whole-exome sequencing and copy number variation analysis of another 16 intraneural perineuriomas detected mutation in the TRAF7 gene (which maps on 16p13) in 10 (60%) and larger deletions of chromosomes 10, 11, and 22 in two tumors (65).
In the three reported sclerosing perineuriomas with abnormal karyotypes, alteration of chromosome 10 was seen (60, 61) (Table I, cases 3, 4, and 5). One tumor had two chromosome translocations involving 10q24 (Table I, case 3), a second had loss of chromosomes 10 and 22 (Table I, case 4), and the third had a deletion involving 10q24 and loss of chromosome 10 (Table I, case 5) (60, 61, 67). Molecular studies of sclerosing tumors suggested a tumorigenic role of NF2 (on 22q12.2) abnormalities (60, 64). Lasota et al. (64) found point mutations in NF2 coding sequences in three of five sclerosing perineuriomas while Sciot et al. (60) found a cryptic deletion in NF2, in addition to a deletion involving 10q24, in another sclerosing perineurioma (Table I, case 5).
In soft tissue perineuriomas other than the above-mentioned tumors, three more cases have been reported with abnormal karyotypes (Table 1, cases 6, 7, and 8): a tumor of the thigh in a 26-year-old woman showing loss of chromosome 13 (Table I, case 6), an intrabdominal tumor in a 13-year-old girl with a t(8;9)(q13;q22) as the sole cytogenetic aberration (Table I, case 7), and a tumor of the foot in a 43-year-old man whose tumor cells had an add(2)(q33) and t(4;10)(q25;q24) (Table I, case 8) (61-63). Whole exome sequencing and copy number variation analysis of 14 soft tissue perineuriomas showed deletions of 22q12 encompassing NF2 in 6 tumors, whereas 4 tumors had deletion of 17q11 encompassing NF1. No point mutations were detected in NF1, NF2, or TRAF7 (66).
The existing data, those previously published together with what we describe here, therefore indicate three different pathogenetic pathways in perineuriomas. The first pathway involves rearrangements of chromosome 22/NF2 gene or chromosome 17/NF1 gene, the second involves chromosome band 10q24, whereas the third involves chromosome translocations in which chromosome bands 4q31 and 9q34 are recombined to generate a GAB1-ABL1 fusion gene.
Acknowledgements
The Authors thank Professor Jason Hornick, Department of Pathology, Brigham and Women's Hospital, Boston USA for help with the diagnosis. This work was supported by grants from Radiumhospitalets Legater.
Footnotes
Authors' Contributions
IP designed and supervised the research, performed molecular genetic experiments and bioinformatics analysis, and wrote the article. LG performed cytogenetic analysis and evaluated the FISH data. KA performed molecular genetic experiments, FISH analyses, and evaluated the data. ST performed pathological examination. ML-I performed pathological examination. IL performed pathological examination. FM evaluated the cytogenetic and FISH data. SH assisted with experimental design and writing of the article. All Authors read and approved the final manuscript.
This article is freely accessible online.
Conflicts of Interest
The Authors declare that they have no potential conflicts of interest in regard to this study.
- Received April 21, 2020.
- Revision received May 22, 2020.
- Accepted June 1, 2020.
- Copyright© 2020, International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved
References
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.