


Abstract
Background/Aim: Multiple myeloma is a B-cell neoplasm, which can spread within the marrow of the bones forming many small tumors. In advanced disease, multiple myeloma can spread to the blood as plasma cell leukemia. In some cases, a localized tumor known as plasmacytoma is found within a single bone. Despite the approval of several agents such as melphalan, corticosteroids, proteasome inhibitors, thalidomide-based immuno-modulatory agents, histone deacetylase inhibitors, a nuclear export inhibitor and monoclonal antibodies daratuzumab and elatuzumab, the disease presently remains uncurable. Materials and Methods: In order to define new targets and treatment modalities we searched the literature for microRNAs, which increase or inhibit in vivo efficacy in multiple-myeloma-related xenograft models. Results and Conclusion: We identified six up-regulated and twelve down-regulated miRs, which deserve further preclinical validation.
- Antisense-oligonucleotides
- microRNA delivery
- microRNA mimetics
- locked nucleic acids
- treatment resistance
- multiple myeloma
- review
Multiple myeloma (MM) is a B-cell malignancy characterized by an excess of monoclonal plasma cells in the bone marrow (BM) (1). In 2019, the incidence of MM in the US was 32,000 new cases and 13,000 deaths (2). Regarding pathogenesis, it has been shown that monoclonal gammopathy of undetermined significance (MGUS), a premalignant state of the disease, progresses to MM which is associated with osteolytic bone lesions in most patients (3). MM homes to the BM by binding to adhesion and extracellular matrix proteins (4). Considerable variability in MM evolution has been observed in patients with MM due to evolution of different dominant subclones (5). In the majority of patients translocations which deregulate oncogenes such as cyclin D1 and D3, fibroblast growth factor receptor 3 (FGFR3), nuclear protein multiple myeloma SET (MMSET) or transcription factors such as c-MYC and c-MAF have been identified (6). Despite approval of several agents for treatment, the disease is presently incurable (7, 8). Among the approved agents are chemotherapeutics such as melphalan, corticosteroids dexamethasone (dex) and prednisone, immuno-modulatory agents such as thalidomide, lenalidomide and pomalidomide, proteasome inhibitors bortezomib, carfilzomib and ixazomib, histone deacetylase (HDAC) inhibitors such as panobinostat and nuclear export inhibitor selinexor (7, 8). In addition, two monoclonal antibodies (mabs) have been approved: daratuzumab, directed against cluster of differentiation 38 (CD38) and elatuzumab targeting SLAM family member 7 (SLAMF7) (9, 10). For palliative treatment of osteolytic bone disease, bisphosphonates, such as pamidronate and mab denosumab have been approved (11). Immuno-therapy approaches based on bispecific antibodies and chimeric antigen receptor (CAR) T-cells with focus on B cell maturation antigen (BCMA) as a target are ongoing (12, 13). Nevertheless, due to incurabilty of the disease with presently available agents, identification of new targets and treatment modalities are urgently requested. In this review, we focused on the role of microRNAs (miRs) in the context of the pathogenesis of MM with in vivo efficacy in MM-related models.
Micro RNAs and Their Role in Oncology
miRs are small noncoding RNAs in the range of 22 nucleotides (nts) in length. Their function is post-transcriptional mRNA silencing. This can be achieved by cleavage of the corresponding mRNA, destabilization of the target mRNA through shortening of the polyA tail and/or attenuation of its translation (14-16). At least 1,000 miR-related genes transcribed by RNA polymerase II have been identified in humans (17). The biogenesis of miRs involves generation of pri- and subsequently of pre-miRs (15, 18). miRs are transcribed as RNAs forming short hairpins resulting in generation of pri-miRs. These are recognized by nuclear protein DiGeorge Syndrome critical region (DGCR8) which associates with ribonuclease DROSHA to form the MICROPROCESSOR complex (15, 18). The processing products are referred to as pre-miRs and are exported from the nucleus by exportin 5, a member of the karyopherin family. In the cytoplasm, DICER, an RNAse III enzyme, cleaves the pre-miRs yielding the miR/miR* duplex of about 22 nts in length (15, 18). One strand is incorporated into the RNA-induced silencing complex (RISC) where miR and corresponding mRNA interact. Nts 2-7 of the miR, referred to as seed region, must be perfectly complimentary to the mRNA (15, 18). Multiple miRs can target the same mRNA and a single miR can target several different mRNAs (19). Therefore, miRs have the potential of reprograming malignant cells to benign cells (20).
The importance of miRs in oncology was shown in patients with chronic lymphatic leukemia (CLL) which is caused by loss of miRs 15a/16-1 functioning as tumor suppressors (TSs) by targeting B-cell lymphoma 2 (BCL2) (21, 22). Further work has identified miRs as regulators of oncogenes and TSs (23), tumor immunity (24) and metastasis (25-28). The role of miRs in MM is summarized in several reviews (29-31). In this review, we focus on miRs with in vivo efficacy in MM-related models in order to identify new targets and treatment modalities for treatment of MM.
Up-regulated miRs With Efficacy in Myeloma-related In Vivo Models
miR-17-5p affects iron transport. miR-17-5p (Figure 1) promotes cell proliferation and inhibits apoptosis in ARP1 and OCI MM cells (32). miR-17-5p transfected ARP1 cells exhibit increased tumor growth (TG) in comparison to control cells after subcutaneous implantation into immune-deficient mice (32). Ferroportin 1 (FPN1), a transmembrane protein, which transports Fe from the cytoplasm to the outside of the cell, was identified as a direct target of miR-17-5p (32, 33). Decreased FPN1 has been shown to promote MM cell growth and osteoclast differentiation (34). Furthermore, decreased FPN1 correlates with bad prognosis in MM (34). FPN1 is regulated by hepsidin, a hormone produced in the liver. Hepsidin binds to FPN1 and limits its iron-efflux capacity (33).
miR-20a inhibits a zinc finger transcription factor. miR-20a (Figure 1), a member of the miR-17-92 cluster, is elevated in the plasma of MM patients (35). miR-20a promotes proliferation of NCI-H929 and U266 cells (36). Intratumorally injected miR-20a mimetics into palpable tumors established by subcutaneous implantation of OPM2 MM cells promote TG in immune-deficient mice (36). Early growth response protein 2 (EGR2), a transcription factor with three Zn fingers, was identified as a direct target of miR-20a (36). EGR2 acts as a TS and inhibits tumor cell proliferation as well as metastasis and stimulates apoptosis (37, 38).

Up-regulated microRNAs promoting growth of multiple myeloma xenografts in preclinical in vivo models. The miRs are displayed according to their numerical designation. The corresponding targets are displayed. BAK: Pro-apoptotic protein BAK; BAX: BCL2-associated X protein; BTG2: BTG family member 2; EGR2: early growth response protein 2; FPN1: ferroportin 1; PHLPP2: PH domain and leucine rich repeat protein phosphatase; PI3K: phosphoinosite 3-kinase; PTEN: phosphatase and tensin homolog; PUMA: p53 up-regulator of apoptosis; CDKN1B: cyclin-dependent kinase inhibitor 1B; CDCN1C: cyclin-dependent kinase inhibitor 1C; RHOB: RAS homolog gene family, member B.
miRs affecting signaling and cell cycle
mir-27 inhibits proliferation-related pathways. miR-27 (Figure 1) is up-regulated in MM patients in comparison to BM derived from normal donors (39). Increased expression of miR-27 in MM patients predicts poor prognosis (39). miR-27 promotes proliferation, survival and motility of U266 and H929 MM cells (39). H929 cells treated with a miR-29 inhibitor display reduced TG after subcutaneous injection into the flanks of nude mice (39). Sprouty homolog 2 (SPRY2) has been identified as a direct target of miR-27 (39). SPRY2 acts as an inhibitor of the mitogen-activated protein kinase (MAPK) pathway in MM cells (40). SPRY2 also inhibits transforming growth factor β receptor (TFGβR), signal transducer SMAD2 and epidermal growth factor receptor (EGFR) signaling (41, 42).
miR-125b promotes AKT activation. Levels of miR-125b (Figure 1) are increased in plasma samples from MM patients compared to healthy controls (43). miR-125b stimulates proliferation and migration of MM cells and inhibition of miR-125b reduces the levels of phosphorylated signal transducer serine-threonine kinase AKT (43). Expression of a miR-125b inhibitor in MM cells suppresses TG in a xenograft model (43). Pleckstrin homology domain leucine-rich repeat protein phosphatase (PHLPP2) was identified as a direct target of miR-125b (43). PHLPP2 acts as a negative regulator of the phosphoinosite 3-kinase (PI3K) pathway (44).
miRs promoting growth of MM cells by simultaneous inhibition of several cell-cycle-, apoptosis-regulatory genes and tumor suppressors.
miR-21. miR-21 (Figure 1) expression correlates with disease progression in MM and inhibition of miR-21 affects growth, survival and clonogenicity of KMS-26, U-266 and OPM-2 MM cells (45). Enforced expression of miR-21 mimetics enhances proliferation of MM1S cells (45). miR-21 inhibitors abrogate the supporting activity of human BM stromal cells (45). Growth of palpable OPM-2 MM cells is inhibited by intra-tumoral injection of antisense oligonucleotides (ASO) directed against miR-21 (45). Phosphatase and tensin homolog (PTEN), ras homolog gene family member B (RHO B) and B-cell translocation gene 2 (BTG-2) were identified as targets of miR-21 (46). PTEN acts as a TS and has phosphatase-dependent and - independent (scaffold) activities such as maintainance of genomic stability, modulation of cell survival, migration, proliferation and metabolism (47, 48). RHO B is a member of the RHO family of small GTPases which regulate actin stressfibers, cytoskeletal actin organisation and vesicle transport in cancer cells (49). BTG-2 regulates pre-B-cell differentiation through protein arginine N-methyltransferase 1 (PRMT1) mediated methylation of cyclin-dependent kinase 4 (CDK4) leading to cell-cycle arrest and limitation of pre-B-cell expansion (50). Furthermore, BTG-2 acts as a TS and is involved in adaptation to cellular stress (50).
miRs-221/222. miRs-221/222 (Figure 1) exhibit increased expression levels in BM of MM patients compared to BM from normal donors (51). In vitro, ASO and locked nucleic acid (LNA) 13 mer (LNA-i-miR-221) mediate significant anti-proliferative effects in OPM2 and NCI-H929 MM cells (51, 52). Palpable OPM2 xenografts in immuno-deficient mice treated intratumorally with unformulated or lipid-emulsion particles of ASO directed against miRs 221/222 give rise to inhibited TG (51). Also, LNA-i-miR-221 induces anti-tumoral acitivity in sucutaneously implanted OPM2 xenografts after i.p. or i.v. injection (52). Only MM cells with t(4;14) translocations are growth-inhibited by LNA-i-miR-221 in vitro and in vivo (52). Up-regulation of canonic miR-221/222 targets such as PTEN, cyclin-dependent kinase inhibitors 1B and 1C (CDKN1B, CDKN1C), p53 up-regulated modulator of apoptosis (PUMA) and impairment of AKT activation was observed after administration of LNA-i-miR-221 in vitro and in vivo (52). CDKN1B acts as a cell cycle inhibitor and TS and its inhibition promotes tumor cell proliferation (53, 54). CDKN1C functions as a TS and inhibits several G1 cyclin/CDK complexes resulting in inhibition of cell proliferation (55). PUMA has a pro-apoptotic function through interaction with anti-apoptotic BCL2 family members (56). The therapeutic potential of targeting the cell cycle and apoptotic pathways in MM are summarized in (57, 58).
Down-regulated miRs Mediating In Vivo Efficacy in Myeloma-related Models
miRs affecting angiogenesis. miRs-15a, -16. These are underexpressed in primary MM cells, established MM cell lines and in advanced stage MM patients in comparison to normal B-cells (Figure 2) (59). Vascular endothelial growth factor-A (VEGF-A) has been identified as a direct target of miRs-15a and -16 (59). RPMI-8226 cells transfected with pre-miR-15a or pre-miR-16a suppress pro-angiogenic activity of these cells, such as endothelial cell (EC) tube formation (59). Conditioned media from miR-15a or miR-16a-transfected RPMI-8226 MM cells inhibit proliferation and chemotactic motility of bone marrow endothelial cells (BMEC) (59). RPMI-8266 cells transfected with lentivirus expressed miR-15a exhibit significant inhibition of TG after subcutaneous implantation into immune-deficient mice in comparison to control cells (59). Also, systemic delivery (i.v.) of synthetic miRs-15a/16 conjugated with neutral lipids inhibits growth of green fluorescent protein (GFP)-labeled RPMI-8266 cells in immuno-deficient mice as shown by reduced fluorescence in comparison to the control cell line (59). Interestingly, miRs-15a and -16 are deleted in CLL and are causative for this disease (21, 22). VEGF-A, the target of miRs-15a and -16 induces synthesis of interleukin 6 (IL6) in the endothelium of the micro-vasculature and in BM and acts a growth factor for MM cells (60). Increased BM-related angiogenesis in MM has been shown to be due to aberrant expression of angiogenic factors by MM cells (61). In line with these findings is the observation that approved agents for treatment of MM, such as bortezomid, thalidomide and its derivatives mediate anti-angiogenic activity (62-64).

Down-regulated miRs inhibiting growth of multiple-myeloma-related xenografts in preclinical in vivo models. The miRs are displayed according to their numerical designation. The corresponding targets are displayed. AURKA: Aurora kinase A; BCL2: B-cell lymphoma 2; BCL9: B-cell CLL/lymphoma 9; CDK6: cyclin-dependent kinase 6; c-MET: transmembrane tyeosine kinase c-MET; DDR1: discoidin domain receptor family, member 1; DNMT3A/3B: DNA methyltransferase 3A/3B; FGF1: fibroblast growth factor 1; FOXP1: forkhead box protein P1; HDAC4: histone deacetylase 4; HIF-1α: hypoxia inducible factor 1alpha; IL8: interleukin 8; MCL1: induced myeloid leukemia inducible factor 1alpha; NOTCH1: tyrosine kinase receptor notch 1; NFĸB: nuclear factor ĸB; PTEN: phosphatase and tensin homolog; PSMβ5: proteasome subunit β type 5; USP5: uniquitin specific peptidase 25; VEGF-A: vascular endothelial growth factor-A.
miR-199-5p. miR-199-5p (Figure 2) decreases cell proliferation and increases apoptosis in MM cells under hypoxic conditions (65). miR-199-5p targets hypoxia-inducible factor-1α (HIF-1α) which interferes with the MM/endothelial cell (EC) loop promoting the basic events of the angiogeneic response (65). OPM2 MM cells transfected with synthetic miR-199-5p show reduced expression of angiogenic factors such as VEGF-A, interleukin 8 (IL8) and basic fibroblast growth factor (bFGF) and their conditioned media reduce EC migration (65). Enforced expression of miR-199-5p in MM cells represses discoidin domain receptor 1 (DDR1), which is involved in promoting cancer cell proliferation, drug resistance, angiogenesis and metastasis (65-67). DDR1 is a predicted target of miR-199-5p (68). Growth of subcutaneously injected, palpable NCI-H929 MM cells was significantly inhibited after intratumoral injection of miR-199-5p formulated as neutral emulsion particles (65). It was also shown that miR-199-5p induces adhesion of MM cells to hypoxic BM stromal cells (BMSCs) (65). HIF-1α, the major target of miR-199-5p, plays a pivotal role in adaptation of tumor cells to nutrient-deprived conditions by up-regulation of transcription of several hypoxia-inducible oncogenic factors and acts as a master switch for angiogenesis (69, 70). Furthermore, HIF-1α activation contributes to the pathogenesis of MM by stimulating angiogenesis via up-regulation of IL8 and VEGF-A (71). In MM, impairment of cell-adhesion and promotion of invasion and metastasis is caused by binding of HIF-1α to the promoter of zinc finger transcription factor SNAI1 leading to decreased expression of E-cadherin (72, 73).
miRs affecting proteasomal degradation
miR-125a. miR-125a (Figure 2) expression is low in MM cell lines and tissues in comparison to matching control cells and tissues (74). Overexpression of miR-125a in NCI-H929 and U266 MM cells decreases cell viability and colony-forming ability by promotion of apoptosis (74). Overexpression of miR-125a in a mouse xenograft model inhibits TG (74). Ubiquitin-specific peptidase 5 (USP5) was identified as a direct target of miR-125a (74). USP5 cleaves linear and branched ubiquitin polymers and therefore inhibits the proteosomal pathway in MM cells (75, 76). Inhibition of USP5 has been shown to result to degradation of transcription factor c-MAF and apoptosis MM cells (77).
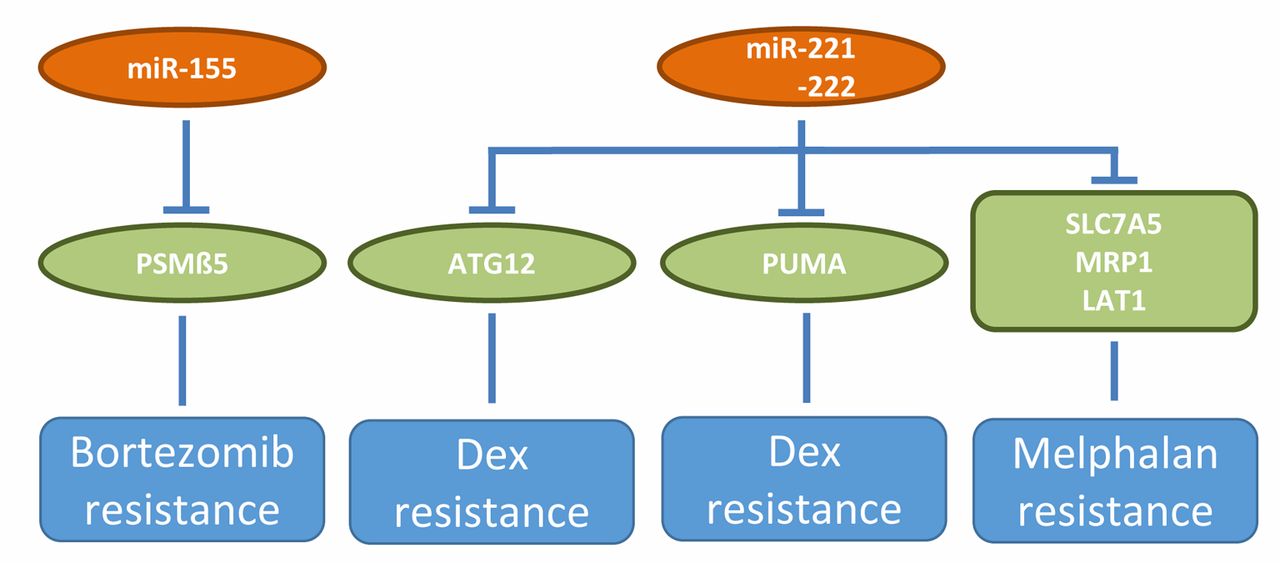
miRs involved in resistance to bortezomib, dexamethasone and melphalan in preclinical in vivo models of multiple myeloma. Up-regulation of miR-155 mediates bortezomib resistance, whereas down-regulation of miR-221/222 is involved in dexamethasone and melphalan resistance. ATG12: Autophagy-related 12; Dex: dexamethasone; LAT1: large neutral amino transporter 1; MRP1: multidrug resistance associated protein 1; PSMβ5: proteasome subunit β type 5; PUMA: p53 up-regulated modulator of apoptosis; SCL7A5: solute carrier family 7, member 5.
miR-155. miR-155 (Figure 2) is down-regulated in MM patient-derived plasma cells compared to healthy plasma cells (78). Synthetic miR-155 mimics trigger anti-proliferative and pro-apoptotic effects in RPMI 8226 and OPM2 MM cells (78). OPM2 cells transduced with miR-155 exhibit moderate inhibiton of TG after subcutaneous implantation into nude mice, however combination with bortezomib greatly enhances in vivo activity (78). Proteosomal subunit PSMβ5 has been identified as a direct target of miR-155 (78, 79) and therefore miR-155 is an inhibitor of proteasome activity. PSMβ5 is the target of bortezomib and bortezomib resistance in MM is associated with PSMβ5 overexpression and polyploidy (80).
miRs with an impact on cell cycle and apoptosis
miR-26a. miR-26a (Figure 2) mimics suppress growth of LP1 and U266 MM cells (81). This is achieved by induction of cell-cycle arrest at G0/G1 phase (81). In vivo, a miR-26a mimetic, injected into subcutaneously established, palpable LP1 xenografts, causes inhibition of TG (81). Cyclin-dependent kinase 6 (CDK6) was identified as a direct target of miR-26a (81). miR-26a mimics inhibit protein levels of transcription factor E2F-1, p53 and p21 (81). CDK6 phosphorylates TS retinoblastoma (Rb), resulting in release of its binding partner E2F-1 which activates DNA replication (82). CDK4/6 inhibitors palbociclib and ribociclib in combination with aromatase inhibitor letrozole have been approved by the FDA for treatment of hormone receptor-negative, advanced-stage breast cancer (83). Palbociclib has been successfully applied in MM therapy, further research has to define the subgroup of patients who receive greatest therapeutic benefit (84).
miRs-137 and -197. Basal expression of miR-137 and miR-197 (Figure 2) is significantly higher in normal plasma cells compared to those derived from MM patients and cell lines (85). miRs-137 and -139 are negative regulators of cell viability, colony formation and migration of MM1S and My5 MM cells (85). In miR-137-lentivirus transduced MM1S cells significant inhibition of tumor formation and increased survival in immuno-deficient mice was observed in comparison to the non-transduced cell line (85). Synthetic miR-137, -193 formulated as non-lipid emusion particles injected into palpable MM 1S xenografts were able to induce tumor regressions (85). Anti-apoptotic protein myeloid cell leukemia-1 (MCL-1) was identified as a direct target of miR-137 (85). MCL-1 plays a complex role in the pathophysiology of MM (86). AZD 5991, a macrocyclic inhibitor of MCL-1 is presently in clinical trials in patients with hematological malignancies (87). Aurora kinase A (AURKA) was identified as another direct target of miR-137 (88). miR-137 overexpression together with bortezomib treatment significantly inhibited TG in a MM xenograft model (88). AURKA is required for the correct duplication and separation of the chromosomes during mitosis, but also exhibits non-mitosis-related functions (89). AURKA inhibitors are presently evaluated in clinical studies in patients with MM (90).
miRs involved in transcription and signaling. miR-30-5p. The members of the miR-30-5p family of miRs (30a,b,c,d,e) exhibit low levels of expression in MM cells compared to normal plasma cells (91). Overexpression of miR-30c reduces proliferation, colony formation, migration and cancer stem cell (CSC) generation in H929 and OPM1 MM cells (91). In vivo efficacy of miR-30c was evaluated in three in vivo models (91). TG of subcutaneously implanted H929 cells transduced with miR-30c was inhibited in comparison to the non-transfected cell line. Tail-vein injection of miR-30c transduced H929 cells resulted in decreased tumor burden in intestine, spine and skull in comparison to the control cell line. Intraperitoneal delivery of miR-30c using lipid nanoparticles decreased tumor burden and increased survival compared to non-transfected H929 cells (91). B-cell CLL/lymphoma 9 protein (BCL9) was identified as a direct target of the miR-30c (91, 92) (Figure 2). BCL9 functions as an essential co-activator of WNT/β-catenin transcriptional activity promoting proliferation of several types of cancers (92-94). Aberrant WNT signaling mediating proliferation has been observed in MM cells (95, 96). Identification, preclinical and clinical development of inhibitors of the WNT signaling pathway is pursued by several companies and institutions (97).
miR-144-3p. Expression of miR-144-3p (Figure 2) in MM cell lines is significantly lower than in plasma samples from healthy individuals (98). MM1S MM cells, transfected with a miR-144-3p mimic, show inhibited cell growth and increased apoptosis (98). MM1S cells transduced with an expression vector for miR-144-3p give rise to decreased tumor size in comparison to the control cell line after subcutaneous implantation into immune-deficient mice (98). Transmembrane tyrosine kinase c-MET was identifed as a direct target of miR-144-3p (98). c-MET is the receptor of hepatocyte growth factor (HGF) and is involved in invasion, migration, proliferation and angiogenesis (99, 100). Dysregulation of c-MET is a hallmark of disease in MM patients (101). Several c-MET inhibitors have been identified and are under preclinical and clinical evaluation (102-105).
miRs involved in epigenetic regulation
miR-145-3p. miR-145-3p (Figure 2) down-regulation is associated with disease progression in MM (106). miR-145-3p exerts a TS function in MM by inducing autophagic cell death (106). miR-145 mimics potentiate anti-MM activity of bortezomib in vitro and in vivo (106). HDAC4 was identified as a direct target of miR-145-3p (106). Suppression of HDAC4 results in up-regulation of pro-apoptotic protein BCL2-like protein 11 (BCL2L11) and causes inactivation of mammalian target of rapamycin complex 1 (mTORC1) (106). HDAC4 is involved in histone deacetylation, chromatin condensation and transcriptional repression (107). An emerging role of histone acetylation in autophagy has been observed (108). It is well documented that HDAC inhibition can induce autophagy in tumor cells (109). However, the physiological consequences of autophagy in cancer are context-dependent (110).
miRs inhibiting several distinct targets including epigenetics-related programmes. It has been shown that miR-29b (Figure 2) can function as a TS by targeting epigenetic regulation, modulation of cell proliferation, apoptosis, differentiation, invasion and metastasis and regulation of diverse signaling pathways in tumors (111). The function of miR-29b in cancer is context-dependent, acting as a tumor promoter or as a TS (111). A plethora of miR-29b-related targets have been identified such as key tumor suppressor PTEN, signaling molecules protein kinase B2 (AKT2) and cyclin D2 (CCND2), apoptosis-related molecules BCL2 and MCL1, prometastatic regulators such as VEGF-A, inhibitors of metastasis such as transforming growth factor β (TGFβ), integrins α6, β1 and WNT inhibitory factor-1 (WIF-1), an inhibitor of WNT/βcatenin signaling, modulators of epithelial mesenchymal transition (EMT) such as E-cadherin, transcription factors TWIST and SNAIL and extracellular matrix (ECM) remodeling molecules such as collagens (111). Context-dependent functions of specific miRs also have been observed in neuronal development (112). Herein we focus on miR-29b targets which are MM-related. miR-29b targets HDAC4 in several MM cell lines (113). HDAC4 mRNA expression inversely correlates with miR-29b expression in MM samples (113). Silencing of HDAC4 up-regulates miR-29b and phenocopies miR-29b effects in MM cells (113). Multiple myeloma growth in vivo was inhibited by targeting HDAC4 (113) and DNA methyltransferases DNMT3A and DNMT3B (115). Neutral-lipid emulsion formulated miR-29b reduces tumor volume by 50% after intratumoral injection (113), whereas intraperitoneally administered pan-HDAC inhibitor SAHA increases TG inhibition (113). Mechanistic interpretations of the findings have been discussed above (106-109). In addition, it has been shown that disruption of the HDAC4/Rel associated protein B complex blocks MM growth (114). Synthetic miR-29b mimics inhibit cell-cycle progression in MM cells (115). An inverse correlation between expression levels of miR-29b and DNMT3 has been noted in MM cell lines and primary MM samples (115). Synthetic miR-29b mimetics affect DNA methylation after injection as non-lipid emulsion particles into SKMM1 xenografts induced tumor growth (TG) inhibition in comparison to corresponding controls (115). Also, tail vein injection of miR-29b mimetics formulated with non-lipid emulsion particles gave rise to TG inhibition of SKMM1 xenografts (115). miR-29b mimetics were also evaulated in severe combined immuno-deficient (SCID)-synthetic hu mice (116). In this model MM adhere to BMSC in a biopolymeric scaffold in immuno-compromised mice, recapitulating growth of MM cells. Growth of BM dependent cell line INA-6 in a human BM microenvironment was inhibited by miR-29b (116). It has been shown that DNA methylation and other epigenetic events are involved in MM initiation, progression and high individual variability (117-119). Also, DNA methylation has been shown to be involved in inactivation of TS genes in MM (120). H929 and U266 MM cells transfected with miR-29b mimetics display reduced proliferation and increased apoptosis (121). In vivo, H929 cells expressing lentivirus transduced miR-29b show reduced TG after subcutaneous implantation into nude mice in comparison to the non-transduced cell line (121). In this system, Forkhead box protein 1 (FOXP1), a transcription factor, was identified as a direct target of miR-29b (121). FOXP1 functions as a repressor of TS genes (122). FOXP1 is involved in B-cell maturation and differentiation and is expressed in neoplastic plasma cells, but not in their normal counterparts (123, 124).
miR-34a modulates a plethora of cancer hallmark-related targets. Up-regulation of miR-34a (Figure 2) in cancer cells is associated with cell-cycle arrest, apoptosis, senescence and TS function (125, 126). Proliferation-related targets are cyclin D1, CDK4, CDK6, angiogenesis mediator VEGF, apoptosis-related BCL2 and NAD-dependent deacetylase Sirtuin (127, 128). A comprehensive list of mir-34a-related targets can be found in (127, 128). However, the targets as well as anti-tumoral or pro-tumoral function of miR-34a are context-dependent (127, 128). miR-34a (MRX-34) was the first miR evaluated in clinical studies as substitution therapy. However, clinical studies had to be closed due to multiple immune-related severe adverse effects (127, 128). Herein, we focus on miR-34a in the context of MM. Substitution of miR-34a was shown to correlate with in vivo efficacy in several in several preclinical in MM-related vivo models (129-131). Stable expression of miR-34a inhibits proliferation of p53-mutant SKMM1 cells (129). TG is inhibited in SKMM1 cells after lentiviral transduction of miR-34a in immuno-deficient mice (129). Intratumoral delivery of formulated miR-34a results in inhibition of growth RPMI-8226 MM xenografts (129). Also, systemic delivery of formulated miR-34a inhibits MM xenografts in mice (129). In the SCID-synth-hu mice model (116, 129) carrying a 3-dimensional polymeric scaffold reconstituted with human BMSCs and then engrafted with primary MM cells, it was shown that miR-34a mimetics overcome the BM microenvironment-dependent protective effect in vivo displaying reduced tumor infiltration, increased caspase 3 and reduction of Ki67 expression. Further improvement of in vivo efficacy after systemic delivery in MM-related in vivo models was achieved by encapsulation of miR-34a into chitosan/polylactide glycolic acid nanoparticles (130). Combined expression of miR-34a and second mitochondria-derived activator of caspase (SMAC) with a lenti-viral system in RPMI-8226 xenografts resulted in synergistic anti-tumor effects (131, 132). Both, miR-34a and SMAC induce mitochondria-initiated apoptosis through release of cytochrome c (131). SMAC is known to bind to inhibitors of apoptosis (IAPs), releasing caspases to activate apoptosis (132). In the preclinical in vivo models for MM as outlined above, it was shown that miR-34a targets validated genes such as BCL2, CDK6, Cyclin D, SIRT1 and VEGF (129-131). No clinical-related parameters are available for miR-34a in MM.
Chemo-resistance. miRs-221/222 (Figure 3) regulate expression of drug transporters solute carrier family 7 member 5/ large neutral amino transporter (SLC7A5/LAT1) and multi-drug resistance protein 1 (ABCC1/MRP1) and through these pathways induce resistance to melphalan (133, 134). In vivo, a 13 mer locked nucleic acid (LNA-i-miR-221) injected intraperitoneally, significantly inhibited growth of subcutaneously implanted, melphalan-resistant U266/LR7 MM xenografts (133). Making use of isogenic dexamethasone (dex)-sensitive and -resistant MM cell lines MM1S and MM1R, pro-apoptotic protein PUMA was identified as a mediator of dex resistance (135). Enforced expression of miR-221/222 in MM1S induces dex resistance in vivo (135). ASO directed against miR-221 abrogates dex resistance of MM1R cells in vivo and increases survival of MM1R xenografts in vivo (135). In another system it was shown that inhibitors of miR-221/222 increased the expression of its target autophagy-related 12 (ATG12) and functionally induced autophagy and cell death in MM cells (135, 136). miR-155 (Figure 3) targets proteosomal protein PSMβ5, the target of bortezomib, thus reducing proteasome activity (78, 79). As outlined previously, OPM2 cells transduced with miR-155 exhibit moderate inhibiton of TG after subcutaneous implantation into nude mice, however combination with bortezomib greatly enhances in vivo activity (78). Substitution therapy with miR-155 therefore may antagonize bortezomib resistance in a clinical setting.
Synopsis
We identified six up-regulated miRs promoting and 12 down-regulated miRs inhibiting in vivo efficacy in MM-related xenografts. Up-regulated miRs are candidates for inhibition of activity, down-regulated miRs are candidates for substitution therapy, as outlined in greater detail in the next chapter. Down-regulation of the target of miR-17-5p, FPN1, correlates with bad prognosis in MM patients (34) and expression of miR-21 increases during disease progression (45, 137). miR-27 and miR-221/222 expression is increased in BM of MM patients and miR-27 expression correlates with bad prognosis (39, 51). miRs-20a and -125b are increased in plasma of MM patients in comparison to healthy probands (35, 43). All the described up-regulated miRs are associated with clinical parameters supporting their further preclinical validation.
Twelve down-regulated miRs are involved in inhibition of TG in preclinical in vivo models of MM. Their functional reconstitution as a mode of therapeutic intervention with MM growth in patients will be outlined in greater detail in the next chapter. miR-34 is de-validated as a target for treatment of cancer because clinical studies were terminated due to strong side-effects (127, 128). However, the de-validation holds true only for the specific formulation used in clinical studies. HIF-1α, one of the targets of miR-199-5p is involved in the pathogenesis of MM (71). Reduced expression of miRs 15a/16 (59), miR-29b (111-114,138) and miR-145-3p (106) are closely related to progression of MM. For miR-30-5p (91), miRs-125a (74), miR-144-3p (98), miRs-137/197 (85), miR-155 (78) reduced expression in tumor cells compared to corresponding normal cells was shown. With the exception of miR-34 in the specific formulation used in clinical studies, the remaining down-regulated eleven miRs deserve further preclinical validation as targets for inhibition of MM in patients.
Therapeutic Intervention
As outlined, the therapeutic options of miR-related agents for treatment of MM are inhibition of miRs or their functional reconstitution both in combination with other agents (139-141). miR inhibitors are single-stranded RNAs such as ASO or LNA, 12-25 nts complementary to the corresponding mRNA (142). miR sponges are designed to contain multiple miR-binding sites that compete with the natural mRNA target for binding to the corresponding miR (142). Low-molecular weight inhibitors can interfere with the transcription of the corresponding miRs or secondary structures, however, there are concerns about the specificity of such as compounds (139, 141). Functional reconstitution of miRs can be performed with miR-mimetics or by forced expression of the corresponding miR after tranduction of tumor cells with expression vectors for the miR under study. miR mimetics are chemically synthesized double-stranded RNAs which mimic endogenous miRNAs after transfection into cells (139-141).
It should be kept in mind that MM spreads in the BM, creating small tumors, therefore the disease is referred to as MM (142-146). In some cases one localized tumor forms in a single bone as a plasmacytoma (142-145). In rare cases the disease is found as extracellular myeloma in soft tissues or other organs. In advanced disease, the disease can be located in the blood system as plasma cell leukemia (142-144). For plasmacytoma, intratumoral administration, for MM and plasma cell leukemia systemic administration of miR-related therapeutic agents might be indicated. In the preclinical in vivo experiments outlined in this review, intratumoral injection is the favoured mode of adminstration followed by systemic administration of the corresponding miR-related agents in plasmacytoma. With the exception of plasmacytoma, preclinical validation of miR-related agents should, therefore, focus on systemic administration.
However, various issues have to optimized in the preclinical valdation process, case by case. Critical issues are: Optimization of pharmaco-kinetic and pharmacodynamic properties due to removal by the reticulo-endothelial system, renal excretion, entry into the tumor cells and efficiency of intracellular endosomal escape. Further critical issues are hybridization-dependent and -independent side-effects, immunogenicity, haematological and hepato-toxicity and cytokine-release syndrome (147-154). These important aspects are not discussed in this review.
Therapeutic Landscape
As already outlined, clinical studies in cancer patients with MRX-34 as reconstitution therapy with a miR-34 mimetic have been terminated due to unacceptable side-effects (155). However, clinical studies with a LNA anti-miR-155, COBOMERSEN, revealed a favorable side-effect profile and are presently continued in patients with several types of leukemias and lymphomas (155). However, in patients with MM, miR-155 is down-regulated and therefore treatment with a miR-155 inhibitor is not a therapeutic option. Further setbacks were noticed in clinical studies with miR-related agents in other indications (156). Clinical studies in patients with hepatitis C infection and kidney disease had to be terminated due to severe side-effects (156). Presently, second-generation miRs are clinically evaluated in these indications (156). However, recently Onpattro, also known as Patisiran, a small interfering RNA (siRNA) targeting transerythrin mRNA was approved by the FDA and the European Medicines Agency (EMA) for patients with familial amyloid neuropathy (157, 158). The emerging importance of si-RNA-based therapeutics is emphasized by a recent deal between REGENERON and ALNYLAM covering 39 targets for diseases of the eye, central nervous system and liver (159). However, it should be kept in mind that siRNAs are specific for a single target, whereas miRs hit several targets with impact on several pathwys and signaling networks (160). Therefore, in oncology, miR-related agents might have a conceptual advantage over miR-related agents unless toxicity issues de-validate the corresponding miR-based agent.
Footnotes
Authors' Contributions
AN and UHW jointly designed and wrote the manuscript.
This article is freely accessible online.
Conflicts of Interest
AN is and UHW was an employee of Roche.
- Received February 17, 2020.
- Revision received March 9, 2020.
- Accepted March 11, 2020.
- Copyright© 2020, International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved




{kind=link}
{kind=link}
{kind=link}