


Abstract
Background: Intratumoral heterogeneity (ITH) is a major cause underlying therapeutic difficulty of cancer. Although an understanding of ITH is critically important in order to develop novel therapeutic strategies, experimental models that enable the examination of ITH in a time series are lacking. Materials and Methods: We developed an experimental approach based on patient-derived xenograft (PDX) mice and a multiregional sequencing approach (MRA). The multiple regions of primary colorectal cancer (CRC) and serially transplanted PDX tumors were analyzed via whole-exome sequencing and bioinformatic analyses. Results: Our PDX-MRA of CRC indicated the spatiotemporal genetic transition of ITH. It was found that the subclonal architecture of CRC dynamically changes during serial transplantation. Furthermore, our data suggest that environmental selective pressures drive the development of minor pre-existing subclones in PDX-MRA. Conclusion: PDX-MRA is a useful tool for understanding the spatiotemporal dynamics of ITH.
Cancer consists of multiple subclones with different genetic backgrounds (1). This intratumoral heterogeneity (ITH) is a major cause of the therapeutic difficulty because the presence of multiple subclones boosts adaptation to pharmacological interventions (2, 3). Thus, the understanding of how ITH changes over space and time, or the ‘clonal dynamics’ of cancer cells, is essential for developing new therapeutic strategies.
Recent studies that have employed a multiregional sequencing approach (MRA), in which DNA samples from spatially separated multiple regions within a single tumor are analyzed via next-generation sequencing, have identified ITH for various types of malignancies (4-6). For example, our MRA previously revealed ITH of colorectal cancer (CRC) and colorectal precancerous lesions of CRC (7, 8). However, one limitation of the MRA is that it obtains only a snapshot of ITH at the time of surgical resection, although the evolutionary history can be derived from the snapshot through phylogenetic analysis. In order to analyze the clonal dynamics of cancer cells more directly, an alternative in vivo experimental approach is needed.
Patient-derived xenograft (PDX) is an animal model of cancer in which tumor tissues derived from patients are transplanted into immune-deficient mice. PDX is highlighted as a preclinical model to investigate drug response and develop personalized medicine because tumor cells in PDX are considered to represent the genomic features of primary tumor cells in contrast to conventional cell lines cultured in vitro (9, 10). We hypothesized that the combination of MRA and PDX might serve as a novel approach to examine the clonal dynamics of cancer cells.
In this study, we combined MRA and PDX to directly examine the clonal dynamics of CRC. MRA of primary and PDX tumors was conducted via whole-exome sequencing (WES), which was followed by estimation of the subclonal architecture, as well as analysis of single-nucleotide variants and copy number alterations (CNAs). Our PDX-MRA indicated the spatiotemporal transition of genetic ITH and the population dynamics of subclones under environmental selective pressures. We propose PDX-MRA as a novel tool that provides a better understanding of ITH.
Materials and Methods
Ethics statement. Surgically resected samples of primary CRC were obtained from a patient who provided written informed consent according to a protocol reviewed and approved by the Institutional Review Boards and Ethics Committees of Kyushu University (Protocol Number: 659-00). All experiments with human samples were conducted according to the principles expressed in the Declaration of Helsinki. This study was conducted in strict accordance with the recommendations indicated in the Guide for the Care and Use of Laboratory Animals, 8th Edition (11). All animal surgeries were performed under isoflurane anesthesia, and all efforts were made to minimize suffering. The design of the animal experiments was reviewed and approved by the Committee for Animal Experimentation of Kyushu University (Protocol Number: A29-140-0).
Sample collection, preparation and establishment of PDX-bearing mice. A 68-year-old female patient with CRC in the ascending colon who underwent surgical resection of the primary tumor at Kyushu University Beppu Hospital provided written-informed consent and was enrolled in this study. The primary tumor was diagnosed as moderately to poorly differentiated adenocarcinoma and classified as T3N0M0, stage IIA. Samples were obtained from the surgically resected primary CRC tissues and matched normal colon tissue. Resected primary CRC tissues were immediately placed in RPMI-1640 medium supplemented with 1% Pen-strep and cut into pieces of 1 mm. The tumor samples were transplanted using injectors with 18 G needles subcutaneously to immune-deficient NOD.CB17-Prkdcscid/J (NOD/SCID) mice (CLEA Japan, Tokyo, Japan). The mice were observed weekly for tumor growth. Tumor sizes were measured weekly using a Vernier caliper and calculated using the following formula: tumor volume=length × width2 ×0.5. At a size of 1,000-2,000 mm3, the xenografted tumor was removed and split into four, and each tumor sample was serially re-transplanted to NOD/SCID mice in the same manner. The primary and PDX tumor samples and the normal colon sample were immediately snap-frozen using liquid nitrogen and stored at −80°C until DNA extraction and histopathological analysis.
Histopathological analysis of primary and PDX tumor tissues. The frozen tumor tissue samples were cut in a cryostat at −20°C and stained with hematoxylin-eosin (HE). The morphology of the stained tissue samples was independently reviewed by an experienced pathologist in the Kyushu University Beppu Hospital Department of Clinical Laboratory Medicine.
DNA extraction, library preparation and WES. The extraction of genomic DNA from fresh frozen tissues was conducted using an AllPrep DNA/RNA Mini Kit (Qiagen, Chatsworth, CA, USA). Genomic DNA samples extracted from CRC tissues, normal colon tissues and PDX tumor tissues were subjected to library preparation using a SureSelect Human All Exon V5 kit (Agilent technologies, Santa Clara, CA, USA). The captured libraries were subjected to WES using HiSeq 2500 (Illumina, San Diego, CA, USA) with the paired-end 100-bp sequence read option according to the manufacturer's protocols.
Separating human- and mouse-specific sequencing reads. In order to remove contaminating mouse-specific reads originating from the DNA of mouse stroma, sequencing reads were separated by species of origin using Xenome version 1.0.1 (12). Human-specific reads and the reads that could be aligned to both human and mouse genomes were subjected to alignment and downstream analysis.
Alignment and detection of mutations. The sequence data were processed through an in-house pipeline Genomon 2.5.0 (http://genomon.hgc.jp). Briefly, the sequencing reads were aligned to the NCBI Human Reference Genome Build 37 hg19 with BWA version 0.7.8 using default parameters (http://bio-bwa.sourceforge.net/). PCR duplicate reads were removed with Picard (http://www.picard.sourceforge.net). Mutation calling was performed using the EBcall algorithm (13) with the following parameters: (i) mapping quality score ≥30, (ii) base quality score ≥15, (iii) both the tumor and normal depths ≥8, (iv) variant reads in tumors ≥4, (v) variant allele frequency in tumor samples ≥0.05, (vi) variant allele frequency in normal samples <0.1, (vii) minus logarithm of p-value of Fisher's exact test ≥1.3, and (viii) minus logarithm of the p-value of EBcall ≥5. Mutations shared by all samples and other mutations were defined as ubiquitous mutations and heterogeneous mutations, respectively. Heterogeneous mutations were further divided into shared mutations, which were shared by multiple samples, and private mutations, which uniquely existed in a single sample.
Estimation of somatic CNAs. In order to estimate the somatic CNAs of primary CRC cells and PDX CRC cells, CNVkit version 0.9.0 (14) was used on aligned WES data derived from the normal colon sample, and primary and PDX CRC samples. The CNAs were inferred applying the standard procedure with default parameters (https://cnvkit.readthedocs.io/en/stable/pipeline.html).
Estimating the composition of tumor subclones. The composition of subclones in each primary and PDX CRC sample was calculated using Clomial version 1.16.0 (15). The numbers of subclones were determined as five, including one noncancerous subclone, and the best genotyping model of the subclones was inferred by simulating 1,000 times with the following parameters: C=5 and binomTryNum=1,000.
Data availability. All WES data have been deposited in the Japanese Genotype-phenotype Archive with the following accession number: JGAS00000000154 (https://ddbj.nig.ac.jp/jga/viewer/view/study/JGAS00000000154).
Results
Establishment of PDX-MRA. In order to track the clonal dynamics of CRC cells, we established PDX-bearing mice from a CRC tumor. As shown in Figure 1A, four spatially separated samples were collected from a surgically resected CRC in the ascending colon and one control sample from the normal colon mucosa. One of the four primary tumor samples was divided into two samples, followed by subcutaneous implantation of each sample into two NOD/SCID mice (first-generation tumor, PDX G1). The developed tumors were resected and serially re-transplanted into other mice (second-generation tumor, PDX G2). Subsequently, the primary tumor samples, the paired normal sample and the PDX tumor samples were subjected to multiregion WES. In total, one normal colon sample, four primary tumor samples (samples A to D) were obtained and 22 PDX tumor samples (samples E1 to N) developed in 10 mice.

Establishment of patient-derived xenograft (PDX) mice for use in the multiregional sequencing approach. A: The overall experimental design of the PDX-MRA study. B: A heatmap presenting growth curves of PDX tumors. C: Representative images of hematoxylin-eosin staining of primary colorectal cancer (CRC) tumor (left), first-generation PDX tumor (G1) tumor (middle) and second-generation PDX tumor (G2) tumor (right). Arrows indicate glandular structures. Original magnification: ×200.
The phenotypic characteristics of CRC cells under the PDX environment. We observed that the growth speed of PDX tumors increased during the serial passages (Figure 1B). The mean time to harvest shortened from 147 days for PDX G1 to 56 days for PDX G2, which is consistent with a previous report of the PDX models of squamous cell carcinoma and adenoid cystic carcinoma (16). These data suggest that CRC cells had adapted to the PDX environment through passaging. The histopathological characteristics of the PDX tumors were also compared with the primary tumors. In contrast to the growth patterns, the PDX tumors mostly retained the original moderately to poorly differentiated morphology of the primary tumor (Figure 1C).
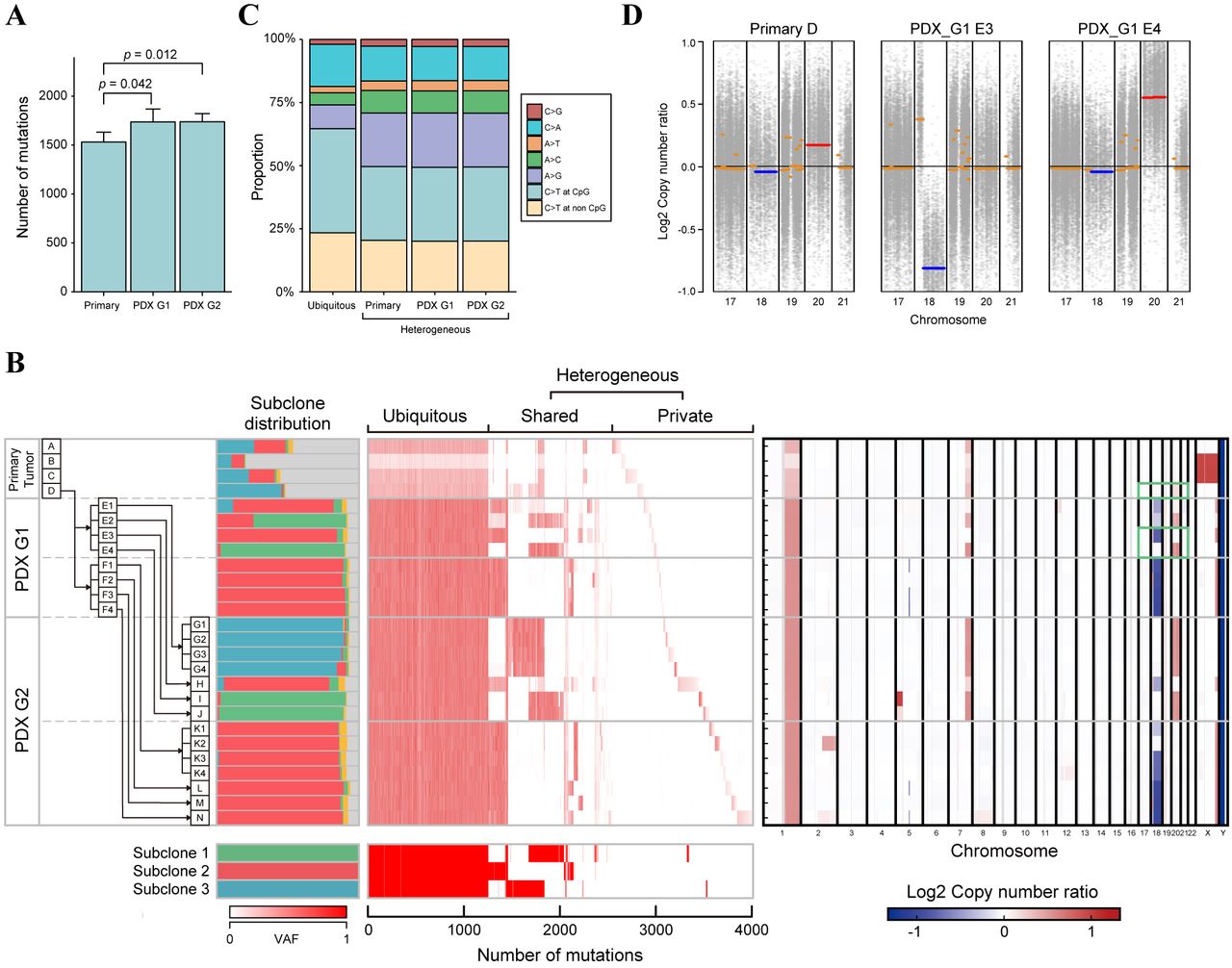
The genetic landscape of the CRC cells over space and time. The overall genomic alterations of the CRC cells in PDX-MRA were assessed by conducting WES on DNA from 22 PDX tumor samples, four primary tumor samples and one matched normal colon sample. The WES of these samples, which had a median sequencing depth of 134.30 (range=93.61-181.88), detected 4,012 mutations in total, with a median of 1,686.5 (range=1,402-1,981) mutations for each sample. From these observations, each sample was estimated to have a median mutation rate of 33.73 mutations per megabase (range=28.04-39.62), which categorized the primary tumor as a hypermutated CRC (17). Although the number of mutations was significantly increased in the PDX tumor samples compared with the primary tumor samples, there was no significant difference between PDX G1 and G2 (Figure 2A). The difference between the primary and PDX samples likely reflects that the proportion of tumor subclones in the PDX samples was larger than that in the primary samples, which was demonstrated by our clone composition analysis as described below.
Based on multiregional mutation profiles, mutations were categorized as ubiquitous or heterogeneous mutations; ubiquitous mutations are maintained in all samples in contrast to heterogeneous mutations. The heterogeneous mutations were further subcategorized into shared mutations, which were observed in some of the samples, and private mutations, which were observed in a single sample (Figure 2B). The ubiquitous mutations accounted for only 30% of the total mutations, which suggests that the genomic features of PDX tumors were altered from those of the primary tumors during passage. Our analysis of mutational types demonstrated that ubiquitous mutations tended to have a higher proportion of C to T transitions at CpG sites and a lower proportion of A to G transitions than heterogeneous mutations (Figure 2C), consistent with our previous analysis (7).

The genomic landscape and clonal dynamics of colorectal cancer indicated by patient-derived xenograft (PDX) mice in the multiregional sequencing approach (MRA). A: Bar plots representing the number of mutations in the primary and in PDX tumors. p-Values were calculated using the Wilcoxon rank-sum test. B: Multiregion genomic profiles of primary and PDX tumors. Left: The history of passaging in PDX-MRA and the subclonal structure inferred by Clomial. Major tumor subclones are depicted as green, red and blue, while minor tumor subclones and noncancerous subclones are depicted as yellow and gray, respectively. Middle: Variant allele frequencies (VAFs) of all mutations are presented as a heat map. Bottom: Mutation patterns of subclones inferred by Clomial. Right: The copy number alteration (CNA) profile of CRC in PDX-MRA inferred by CNV kit. Heat map represents the presence of CNAs (red, amplification; blue, deletion). Green boxes represent the chromosomal regions indicated in Figure 2D. C: Stacked bar plots representing mutational signatures calculated from ubiquitous mutations and heterogeneous mutations in PDX-MRA. The mutational signatures of heterogeneous mutations were calculated for primary, first-generation PDX (G1) and second-generation PDX (G2) tumors. D: Copy number plots representing chromosomal amplification (red) and deletion (blue) in the primary tumor and PDX tumor.
The transition of subclonal architecture of CRC in PDX-MRA. We subsequently estimated the composition of subclones that constituted each tumor sample by utilizing Clomial, which is a computational method to infer genomic subclones using the profiles of somatic mutations (15). Primary and PDX tumor samples were found to comprise three dominant subclones with distinct mutational profiles (Figure 2B). The proportions of tumor subclones were significantly higher in the PDX tumors than in the primary tumors (Mann-Whitney U-test, p<0.001), which reflects the removal of mouse-specific reads during the processing of the WES data. Notably, the composition of subclones substantially varied across tumors. Although subclone 3 was dominant in the primary tumor (sample D), other minor subclones, such as subclones 1 and 2 expanded to dominate the PDX G1 tumors. Interestingly, we observed that these subclones were replaced by subclone 3 again during passaging in several PDX tumors (i.e. E, G, H and I in Figure 2B). Moreover, some PDX tumors retained stable subclonal distributions in which subclone 2 was dominant in multiple mice (i.e. F, K, L, M and N in Figure 2B). These data suggest that the subclonal architecture of PDX tumors dynamically changes from that of their origin.
Dynamics of CNAs in PDX-MRA. Finally, CNAs from WES data were estimated using CNVkit, which is a previously described computational inference algorithm (14). Using these data, the spatiotemporal transition of CNAs in PDX-MRA was analyzed. There were few CNAs in the primary tumor, which is consistent with the characteristics of hypermutated CRC (17). In contrast, PDX tumors acquired several CNAs, such as the deletion of chromosome 18q and amplification of chromosome 20 (Figure 2B). These CNAs were also less frequently observed in the primary tumor, which suggests that environmental selective pressures had driven the development of pre-existing subclones that harbored specific CNAs (Figure 2D). Furthermore, these CNAs have been reported to be positively correlated with metastasis in patients with CRC and have been established as driver events for CRC progression (18, 19). These data suggest that the CNAs that contribute to the malignancy of CRC were subject to selective sweep in the PDX environment.
Discussion
In this study, we presented PDX-MRA as a novel experimental approach that enables the determination of the transition of ITH over space and time. We demonstrated that multiple distinct genomic subclones are present in the primary CRC tumor and PDX tumors. Moreover, our analysis indicated the dynamic expansion of minor subclones in PDX-MRA, which is consistent with previous genomic studies of PDX-bearing mice (20, 21). These observations indicate that environmental selective pressures drive the development of minor pre-existing subclones in the initial establishment of PDX. Our analysis of CNAs also supports the evidence of clonal selection in PDX-MRA; several CNAs, which are reportedly correlated with the malignancy of CRC, existed as minor genomic alterations in the primary tumors, but had become dominant in the PDX tumors. Consistent with these results, it has been reported that PDX-specific CNAs are the result of positive biological selection of pre-existing minor subclones (22).
These findings are in contrast to our previous study of advanced CRC in which subclones were generated by neutral evolution (7). Compared to a real human tumor, a PDX tumor presumably grows in an environment with limited resources; thus, PDX-MRA might not completely reproduce the clonal dynamics of the original tumor. Moreover, the PDX environment in immunodeficient mice lacks the interactions between cancer and immune cells. Although it is unclear whether the lack of tumor–immune cell interactions was implicated in shaping the ITH in PDX-MRA, it should be noted that the tumor–immune microenvironment reportedly has a substantial role in the clonal evolution of human cancer (23, 24). Despite these limitations, PDX-MRA has an advantage that therapy-driven clonal evolution with the use of chemotherapeutic agents can be tracked, which should be addressed by future studies. Collectively, we believe that PDX-MRA would serve as a useful tool for understanding the clonal dynamics of cancer cells in order to develop novel therapeutic strategies.
Acknowledgements
This work used the supercomputing resources provided by the Human Genome Center, The Institute of Medical Science, The University of Tokyo (http://sc.hgc.jp/shirokane.html).
The Authors thank K. Oda, M. Kasagi, S. Sakuma, N. Mishima and T. Kawano for their technical assistance. This work was supported, in part, by the following grants and foundations: the Japan Society for the Promotion of Science Grant-in-Aid for Science Research (Grant Numbers 15H05707, 15H05912, 16H01576, 16K10543 and 16K19197); the OITA Cancer Research Foundation; the Daiwa Securities Health Foundation; Priority Issue on Post-K computer (Grant Numbers hp170227 and hp160219); Eli Lilly Japan K.K. Grant; and the Japanese Foundation for Multidisciplinary Treatment of Cancer.
Footnotes
This article is freely accessible online.
Conflicts of Interest
The Authors have no conflict of interest.
- Received October 15, 2018.
- Revision received October 31, 2018.
- Accepted November 2, 2018.
- Copyright© 2019, International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved




{kind=link}
{kind=link}