


Abstract
Background: The combination of serum β2-microglubulin and albumin levels is highly prognostic in multiple myeloma (MM), defined as the International Staging System (ISS). Recurrent genomic abnormalities present in myeloma cells also have a strong prognostic power. This study aimed to assess, in a routine diagnostic setting, whether genomic aberrations can be used to identify sub-groups in ISS staging, as this system does not incorporate intrinsic myeloma cell variability at the molecular level. Materials and Methods: A prospective population-based study of 123 patients newly diagnosed with MM with ISS staging were included for karyotyping, interphase nuclei fluorescence in situ hybridization (iFISH) and oligo-based array comparative genomic hybridization (oaCGH) analyses. Results: Clonal abnormalities were identified in 27% of analyses by karyotyping, in 83% by iFISH, and in 99% by oaCGH analysis. ISS staging combined with oaCGH aberrations identified ISS sub-groups. Conclusion: oaCGH analysis is a valuable asset in detecting prognostically relevant genomic abnormalities. The combination of oaCGH data with ISS staging might help define new sub-groups in MM.
Multiple myeloma (MM) is a clonal B-cell neoplasia characterized by malignant plasma cells, which accumulate in the bone marrow, producing a monoclonal immunoglobulin (1). MM is an incurable and heterogeneous disease where survival ranges from a few months to more than 10 years (2).
It is becoming increasingly clear that one of the most important factors related to the clinical aggressiveness of MM is the presence of cytogenetic abnormalities, which can be classified into two main groups: translocations involving immunoglobulin heavy-chain (IGH) locus, and genomic imbalances (3, 4). Patients can have one or more of these abnormalities, and in general, there is an accumulation of additional cytogenetic abnormalities over time (5). Recently, based on chromosomal ploidy, MM is divided into hyperdiploid (≥47 and <75 chromosomes; H-MM) and non-hyperdiploid (NH-MM) groups (6, 7). NH-MM is further sub-divided into three group: hypodiploid (≤44 chromosomes), pseudiploid (45-46 chromosomes) and hypertriploid (≥75 chromosomes). H-MM is defined by multiple chromosomal gains, preferentially of the odd chromosomes 3, 5, 7, 9, 11, 15, 19 and 21, whereas NH-MM has a high frequency of IGH translocations at 14q32. The majority of the IGH translocations involve the following proto-oncogenes as partner genes: i) the cyclin D family consisting of cyclin D1 (CCND1; 11q13), D2 (CCND2; 12p13) and D3 (CCND3; 6p21); ii) Wolf-Hirschhorn syndrome candidate 1 (WHSC1)/fibroblast growth factor receptor 3 (FGFR3) (4p16); and iii) the v-maf musculoaponeurotic fibrosarcoma oncogene homolog (MAF) group consisting of MAF (16q23), MAFB (20q12) and MAFA (8q24) (8, 9). The prognosis for patients of these translocation groups differ, where t(11;14) and t(6;14) are prognostically favorable markers and t(4;14) and t(14;16) are adverse markers. According to ploidy, the NH group in MM has a worse prognosis compared to the H group. In addition, submicroscopic deletions at 17p affecting TP53, loss of the 13q14 region and amplifications of 1q21 region identify subgroups of patients with MM with the highest risk profile (9-11).
Chromosomal abnormalities are currently not included in the diagnostic criteria for MM but they provide important prognostic information by predicting initial response to chemotherapy, remission duration and overall survival (7). At present, cytogenetic evaluation is mandatory for all patients with newly-diagnosed MM and should include metaphase banding analysis together with interphase nuclei fluorescent in situ hybridization (iFISH) of positively identified plasma cells (12, 13). However, detection of ploidy and sub-microscopic imbalances in clonal plasma cells is challenging. Metaphase cytogenetics is hampered by the low ability of the plasma cell to divide, resulting in an abnormal rate of only 20-30%, its limited resolution of about 10-15 Mb, and no assurance that a metaphase even originated from the plasma cell clone (4). FISH on the other hand has a higher resolution and does not require dividing cells but it is imperative that only clonal plasma cells are analyzed, which can be achieved by several measures, including CD138 selection (4). Efforts have been made to develop a FISH panel for identifying H-MM; however, there is no probe combination capable of covering all karyotypic combinations. Since FISH analysis of multiple loci is relatively laborious, and since FISH is a targeted test providing limited views of the genomic landscapes of clonal plasma cells, we aimed to evaluate the diagnostic efficacy of microarray-based whole-genome profiling in MM in a clinical setting.
Although oligo-based array comparative genomic hybridization (oaCGH) analysis is a mature technology and is being used as a first tier test in routine constitutional genetic investigations, it has not yet gained a place in the routine diagnostics of MM or other hematological malignancies. Microarray-based genomic profiling has been applied to enriched plasma cells for detection of copy number alterations (CNAs) (14-18).
The specific aim of the present study was to evaluate the role of oaCGH analysis in detecting genomic imbalances compared to conventional karyotyping by G-banding and myeloma-specific iFISH panel as a feasibility study in a population-based prospective clinical setting of patients newly diagnosed with MM.
The International Staging System (ISS), that combines the use of serum β2-microglobulin (β2-MG) and albumin levels, is a reliable and highly prognostic system in MM (19). However, this system does not directly incorporate the intrinsic variability of the myeloma cell at the genomic level, which is known to be of great prognostic importance (13). It was recently shown that combining iFISH data with ISS staging greatly improves risk assessment (20). Here we combined oaCGH data with ISS staging and investigated the frequencies of aberrations for the different ISS groups. In addition, we determined the rate of chromothripsis and compared it with that reported in a previous study (21).
Materials and Methods
Patient cohort. This prospective single-center study enrolled 123 patients (58 females and 65 males) newly diagnosed with MM in a cohort of Central Denmark Region, according to WHO 2008 criteria (22) in the period from August 2013 to August 2015. The mean age of included patients was 69.9 years (range=39-89 years).
Diagnostic algorithm. The diagnosis of MM is primarily based on evaluation of morphology of bone marrow aspirate and biopsy by pathologists and clinical findings. In addition, diagnosis is supported by the biochemical identification of M-component in urine or plasma, β2-microglobulin, and radiological imaging. For prognosis, cytogenetics analysis is performed on bone marrow aspirate sampled at the same time of sampling for pathology.
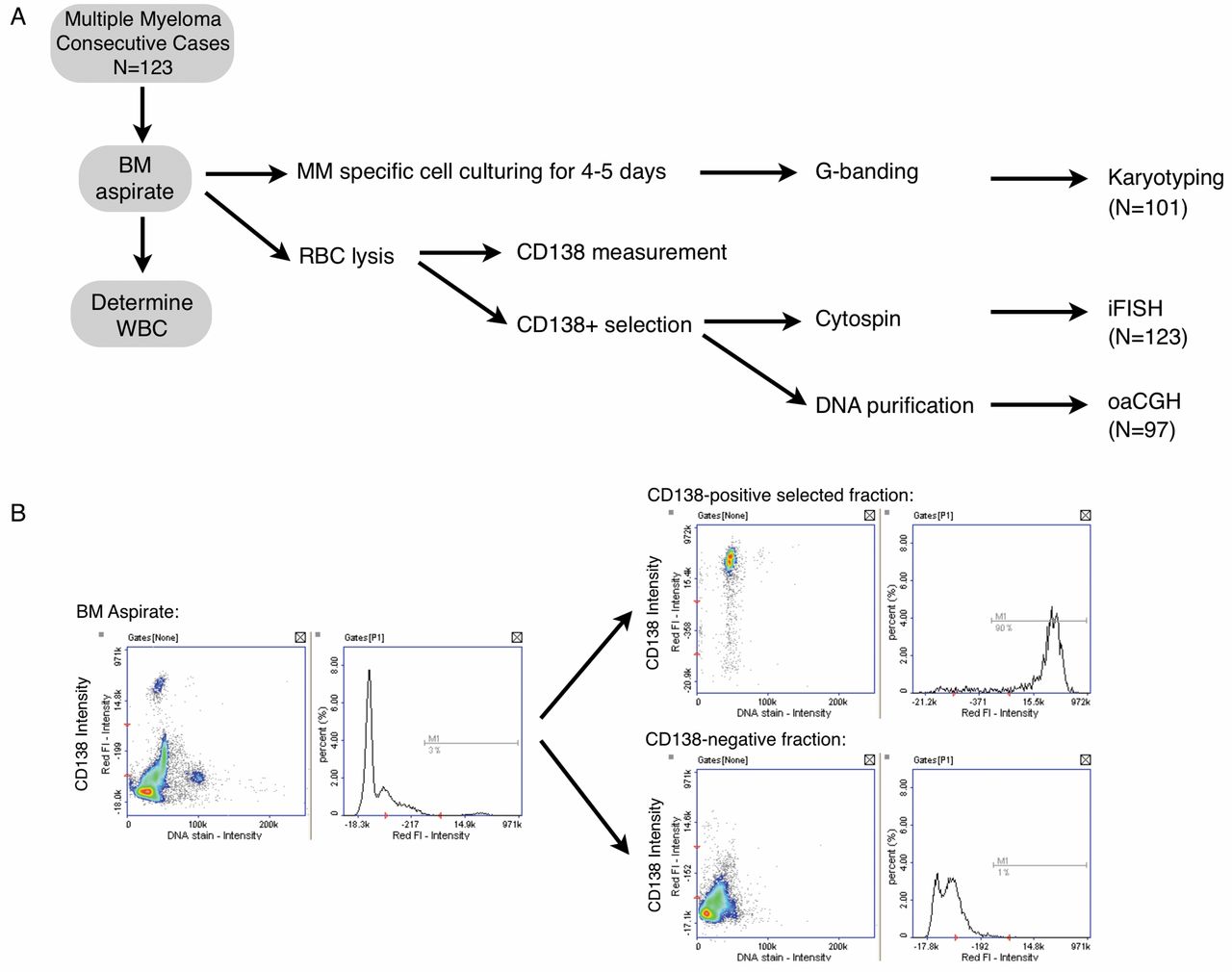
All patients referred to our cancer cytogenetic laboratory for cytogenetic evaluation because of suspicion of MM were subjected to the initial steps of the protocol, i.e. measurement of white blood cells in samples using a Sysmex (se below). Only samples with more than 27 million white blood cells were divided for cell culturing and for red blood cell lysis for later CD138 selection (Figure 1A). Measurement of CD138+ cells in the bone marrow aspirate, CD138+ selection, and preparation of cytospin slides were performed either on the same day of sampling or within 16 h depending upon the time of arrival of the sample at the laboratory. After preparation of up to 12 cytospin slides, the remaining CD138+ cells were subjected to immediate DNA purification for later oaCGH analysis.
Since this was a prospective study, the referral diagnosis covered patients with MM either newly diagnosed or of several years' duration, monoclonal gammopathy of undetermined significance (MGUS), plasmocytoma, and amyloidosis. Out of 331 patients whom were referred for one of these reasons, 123 patients had newly diagnosed MM. All 331 patients had culturing and CD138 selection followed by making of cytospins and DNA purification when the total cell number allowed for it. Only patients newly diagnosed with MM were included in this study and had oaCGH analysis performed within 1-3 weeks after receipt of samples.
Two patients (cases 132 and 186) were referred because of suspected myelodysplastic syndrome (MDS) but immunophenotyping by flow cytometry and morphology revealed MM. The patients were enrolled in the study although it was not possible to purify CD138+ cells from their bone marrow. DNA was purified from the stored bone marrow aspirate without CD138 selection. The cell culturing of these sample was carried out as described below.
Determination of white blood cell counts in bone marrow aspirate. Bone marrow aspirate (1-2 ml) from each patient was sampled into a container with 10 ml RPMI-1640 media before it was send to our laboratory and was received on the same day as sampled. The white blood cell count was measured using the Sysmex KX-21N instrument (Sysmex Denmark, Ballerup, Denmark) according to the manufacturer's instructions.
Cell culturing and karyotyping. A total of 15×106 cells were cultured for 4-5 days in 10 ml RPMI-1640 supplied with 20% fetal bovine serum, phenol red, streptomycin, penicillin and the B-cell mitogens interleukin 4 (IL4) (250 ng) and CD40L (400 ng) as described in (23). The cells were harvested according to our standard methods. The samples from the patients suspected of having MDS were cultured according to our standard methods for myeloid malignancies (24).
Determination of percentage of CD138+ plasma cells. By image cytometry, we determined the percentage of plasma cells in the bone marrow samples. Immunostaining was carried out with mouse anti-human CD138 antibody MI15 labeled with Alexa® 647 (BD Biosciences, Albertslund, Denmark) and cells were counterstained with 4’,6-diamidino-2-phenylindole according to the manufacturers instructions. The labeled cells were analyzed using an NC-3000 image cytometer and NucleoView software (ChemoMetec, Allerød, Denmark) according to the manufacturer's instructions. To determine the degree of purity and efficiency of CD138 selection, the positive and negative fractions were subjected to image cytometry as described above for bone marrow aspirate using the CD138 MI15 monoclonal antibody.

A: Flowchart illustrating the distribution according to karyotyping, iFISH and oaCGH analysis of 123 patients consecutive newly diagnosed with MM. RBC: Red blood cells, WBC: white blood cells. B: CD138 measurement by image cytometry using directly labeled (Alexa® 647) mouse anti-human CD138 antibody MI15 of BM aspirate before and after CD138+ and CD138− selection.
CD138+ selection and cytospin. A volume of bone marrow aspirate equivalent to 50×106 white blood cells was subjected to red blood cell lysis using Lysing buffer (BD Pharm Lyse™; BD Bioscience) according to the manufacturer's instructions. CD138 selection was performed with the EasySep human CD138 selection kit (StemCell Technologies, Grenoble, France) using the purple EasySep magnet up to August 2014; all later samples were subjected to CD138+ selection by the automated RoboSep according to the manufacturer's instructions.
Up to 12 cytospin slides were made per patient using 100 μl per slide at a cell density of 0.2×106 cells/ml and residual CD138+-selected cells were subjected to DNA purification (see below). The cytospin slides were stored dry in the dark at room temperature. When the cytospin slides were older than 2 weeks, a pepsin step was included in the processing as described elsewhere (25), but more than 80% of the slides were examined within 2 weeks.
FISH. Cytospin slides with CD138+-selected cells were used for all iFISH analyses. The following FISH probes were used according to the respective manufacturer's instructions: IGH FISH DNA Probe, split signal (DAKO, Glostrup, Denmark), LSI IGH/CCND1 dual-color dual-fusion probe (Abbott Molecular, Wiesbaden, Germany), XL DLEU/LAMP, XL P53, XL t(4;14), XL IGH/MAF, and XL IGH/MAFB (MetaSystems, Altlussheim, Germany), ON 1q21/SRD (1p36) (Kreatech, Amsterdam, the Netherlands), ELN/D7S486, D7S522 (Abbott), SE3/D3Z1 (Kreatech), SE4/D4Z1 (Kreatech), CBFB split_apart probe (Abbott Molecular), CEP1/D1Z1 (Abbott Molecular) and TGFBR3/RP11-902H1 (Empire Genomics, Buffalo, NY, USA). All cases were screened with the following three FISH probes: XL DLEU/LAMP, TP53, and IGH, and in cases where the IGH result indicated an aberration, IGH translocation-specific probes were used.
At least 100 cells from each of the hybridizations were scored by two independent observers using a DMRXA fluorescence microscope (Leica Microsystems, Ballerup Denmark) equipped with appropriate filters (Chroma Technology, Olching, Germany). The threshold for an abnormal FISH result was set at 10% of cells with aberrant FISH signals.
DNA purification. DNA from the CD138+-selected cells and bone marrow aspirates of the two patients with MDS were purified using Gentra PureGene kit (Qiagen, Copenhagen, Denmark) as described elsewhere (26). We found that at least 8×105 CD138+-selected cells were necessary to obtain sufficient good quality DNA for aCGH analysis. Whenever it was not possible to purify DNA immediately, the CD138+-selected cells were stored frozen after the addition of lysis buffer, which is the initial step in the DNA purification kit. Purified DNA was stored at −20°C.
oaCGH analysis. High-resolution aCGH was performed with the human genome 4×180K Cancer Cytochip (BlueGnome, Cambridge, UK). Digestion, labeling and hybridization were carried out as described in (27) according to the manufacturer's instructions. Briefly, 750 ng high-quality DNA was co-hybridized with normal sex-matched reference DNA (Promega Biotech AB, Nacka, Sweden) for 16 h. After washing, microarrays were scanned using a GenePixPro 4400A multicolor laserscanner (Molecular Devices, Sunnyvale, CA, USA) and feature extraction was performed using BlueFuseMulti software version 3.2 (BlueGnome).
Extracted data were then read into Nexus Copy Number Software version 6.1 (BioDiscovery, Hawthorne, CA, USA). Copy-number abnormalities were defined by using the rank segmentation algorithm with a significant threshold set at 2.4×10−5 with a minimum number of probes per segment set at 3. Maximum contiguous probe spacing was set at 1000 kb. Genomic regions of gain were set at +0.25 and at +1.2 for single-copy gain and high-copy gains, respectively. Genomic regions of loss were set at −0.25 and −1.25 for single-copy loss and bi-allelic loss, respectively. The diploid (2N) value was adjusted based on FISH results when necessary. Regions of gain or loss contained within copy-number variable regions were discarded. All genome-based data reported are based on National Center for Biotechnology Information build 36 (hg18) of the human genome. Bioinformatics analysis was carried out by querying the University California Santa Cruz database (http://genome.ucsc.edu).
Visual estimation of the total chromosome number (modal number) was performed for each of the samples by using the generated ideograms in the overview and chromosome view. Deviation from a normal diploid count of 46 was estimated by assessing the gain or loss of regions bordering the centromere in metacentric and submetacentric chromosomes (i.e. 1-12 and 16-20) and X chromosome, and telomeric of the centromere on the q-arm in the acrocentric chromosomes 13-15, 21 and 22. Metacentric and submetacentric chromosomes with loss or gain of both borders would be counted as a loss or gain of that chromosome, while a continuous segment of loss or gain on the q-arm of the acrocentric chromosomes would be counted as loss or gain of that chromosome. Samples estimated to have ≤44 chromosomes were considered to be hypodiploid, those with 45-46 chromosomes were considered pseudodiploid and grouped together in the NH-MM group; those with ≥47 chromosomes were grouped as H-MM. It is not possible by aCGH in this setup to discern between triploidy, and tetraploidy etc.
Chromothripsis was inferred when a complex pattern of alternating copy-number changes (normal, gain, or loss) along a chromosome or a chromosomal segment was observed according to International System for Human Cytogenetic Nomenclature (28).
Detection of CNA that were present in at least 30% of the tumor was possible by the current oaCGH analysis. Segmented regions shared by 20% or more of the samples were considered recurrent abnormalities and listed as an aggregate.
The oaCGH results were reported within 10 working days after sample receipt for more than 80% of samples. The oaCGH analysis failed in only one case because of low-quality DNA.
Results
A total of 123 consecutive cases newly diagnosed with MM were included in this prospective clinical feasibility study (Table I). With this set-up, we were able to obtain a karyotype result for 101 patients (101/123, 82%), iFISH results for all 123 patients (123/123, 100%) and an oaCGH result for 97 patients (97/123, 79%) (Figure 1A). Using our set-up, we performed measurement of CD138+ using an NC-3000 cytometer, as well as the white blood cell count in the received bone marrow sample to determine the CD138 positivity in the received sample and the purity of CD138 selection (Figure 1B).
The main reason for not being able to obtain a karyotyping result in 22 cases related to the condition of the received bone marrow aspirate. In two of the cases, the bone marrow aspirate contained large coagulates that trapped the cells. In the remaining 20 cases, the sample condition was fine but there were too few white blood cells present in the aspirate.
In 26 out of the 123 included cases, there were too few CD138+ cells for DNA purification to perform oaCGH analysis after making cytospins for iFISH analysis. The main reason for this was a significantly low white blood cell count combined with a low CD138 positivity in the samples (data not shown). In other situations, although the received sample had few white blood cells, this was compensated for in cases with a high CD138 positivity of the cells in the sample. In one case, there were enough cells but the DNA yield and quality was too poor for an oaCGH analysis. To maintain a high rate of successful oaCGH analyses in a clinical setting, it is important that the clinician responsible for bone marrow sample aspiration is aware that a consistently high white blood cell count in the bone marrow aspirate increases the chances of having enough CD138-selected cells for all analyses.
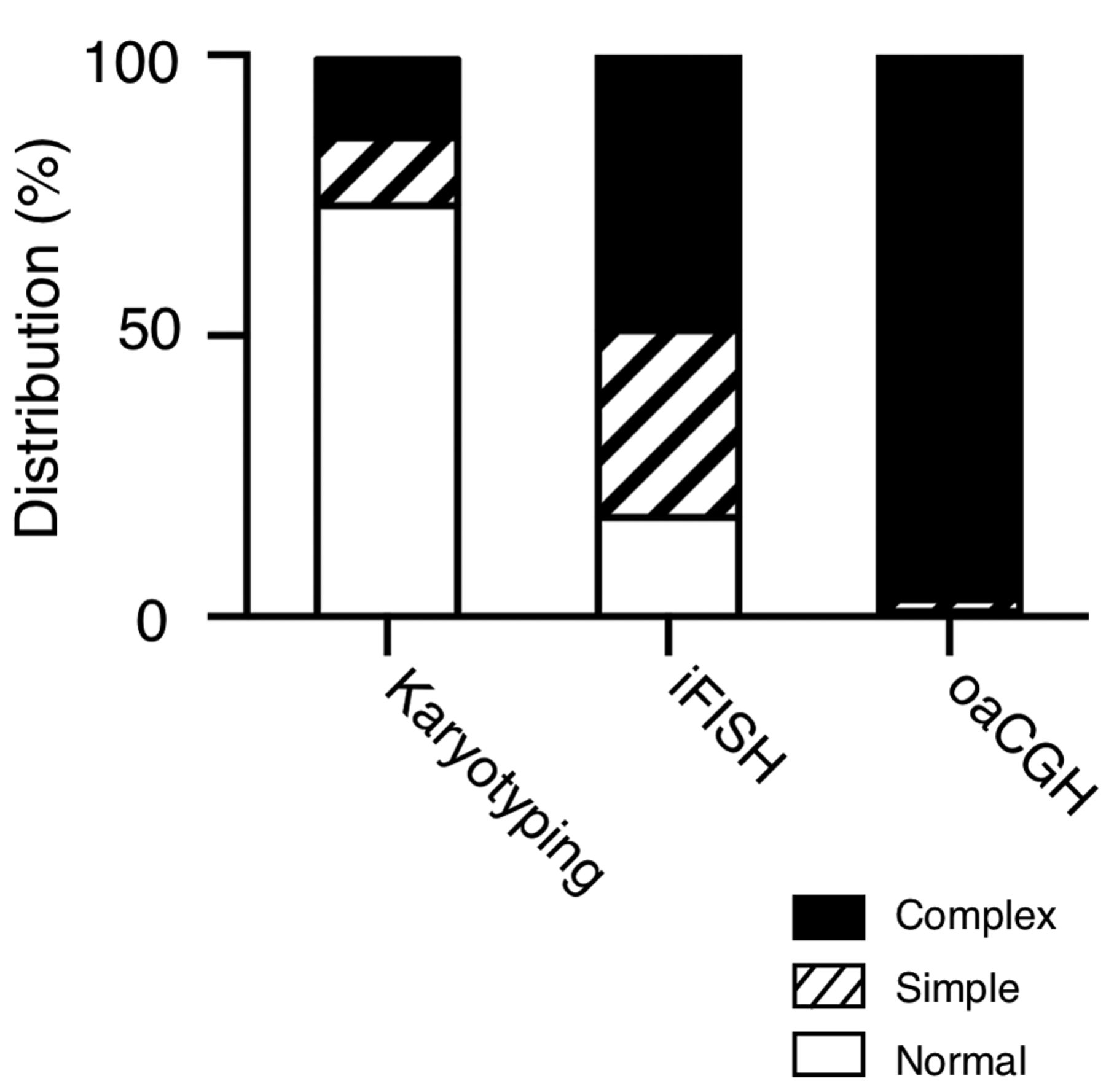
Stacked column diagram showing frequency distribution of results from karyotyping, iFISH and oaCGH analysis.
The distribution of patients with normal results, simple clonal aberrations (defined by one abnormality/case) or complex aberrations determined by each of the examination modalities is shown in Figure 2. The karyotypes were normal in 74 patients (74/101, 74%) and abnormal in 27 patients (27/101, 27%), out of which 12 were simple aberrant, while the remaining 15 were complex. In 102 patients (102/123, 83%), the iFISH result was abnormal and 61 of these cases had more than one aberration. An abnormal oaCGH result was obtained in 96 patients (96/97, 99%), and in 94 of these, the oaCGH result was complex, while two patients only had a single oaCGH aberration. In 21 cases, an abnormal result was obtained with all three types of analyses. In eight cases, aberrations were only detected by oaCGH, and one case only had iFISH abnormalities; there were no cases with only karyotyping abnormalities. Taken together, these findings demonstrate that iFISH and oaCGH analyses are more efficient methods for detecting genomic aberrations compared to karyotyping alone in a clinical setting.
Karyotyping versus oaCGH data. The karyotyping results are listed in Table I. The most common aberration detected by karyotyping was loss of the Y chromosome in 12/74 cases and nine of which were single aberrations. Hypodiploidy with fewer than 45 chromosomes was detected in two cases by karyotyping, cases 106 and 246, with a modal number of 36-41 and 43 chromosomes, respectively. In contrast, in case 106, oaCGH analysis revealed a modal number of 50 including loss of chromosome 13. In case 246, oaCGH analysis revealed a modal number of 45 chromosomes, including loss of chromosomes 4 and 10, and gain of X chromosome, which partially agreed with the karyotyping result. In both cases, iFISH analysis confirmed the oaCGH results. Hyperdiploidy with 47 or more chromosomes was detected in 10 cases by karyotyping. In these cases, the estimated modal number by oaCGH analysis was largely in agreement with the karyotyped modal number. Conversely, hyperdiploidy were inferred in 59 cases by oaCGH analysis and the karyotyping results in most of these cases were normal, suggesting that the abnormal clone had not divided. Unbalanced structural or segmental aberrations were detected by karyotyping in 13 cases, and most of these were of marker chromosomes. In two cases with simple unbalanced structural alterations, oaCGH analysis confirmed the findings: dup(9p) in case 111 and i(17)(q10) in case 221.
iFISH versus oaCGH data. Clonal genomic imbalances were identified by iFISH in 69% (85/123) of the patients at levels ranging from 12% to 99%. iFISH detected 139 CNAs using the IGH, TP53 and 13q screening probes, including aberrations detected by translocation-specific probes applied according to the iFISH result for the IGH locus (Table I). Ninety-seven of these were deletions affecting the following loci 13q, TP53, FGFR3, MAF, and IGH, where mono-allelic loss of the 13q FISH signal was the most frequent. The remaining 42 CNAs were amplifications involving the following loci CCND1, TP53, FGFR3, MAFB, and IGH.
The results of FISH screening for clonal genomic imbalances were in agreement with the array data in 92/97 (95%) of the cases with an oaCGH result. In five cases (cases 147, 258, 336, 376, and 413), iFISH reported loss at 13q or TP53 locus, whereas oaCGH analysis reported a normal result at these loci. This discrepancy was because the number of abnormal cells was between 10 and 22% as determined by iFISH, which is below the detection limit of the oaCGH analysis, which was 30% abnormal cells as defined by locus-specific iFISH analysis.
Alterations in the iFISH pattern using the screening FISH probes DLEU/LAMP, TP53/Cen17 and IGH split-apart probes including translocation-specific probes for IGH/FGFR3, IGH/CCND1, IGH/MAF, and IGH/MAFB may be indicative of chromosomal aneusomy, suggesting ploidy changes. Hyperdiploidy was detected by oaCGH analysis in 59/96 cases with an aberrant oaCGH result but only in 16 of these cases did iFISH analysis suggest hyperdiploidy with the expanded FISH screening probe set panel. Using the basic screening probe set (DLEU/LAMP1, TP53/Cen17 and IGH split-apart probes) this number was reduced to seven cases. Hypodiploidy with 44 or less chromosomes was detected in 13/96 cases by oaCGH analysis. In two of these cases (cases 148 and 326), iFISH analysis showed losses of chromosomes 13, 14 and 17, and in nine cases, loss of chromosome 13 alone using the basic FISH screening probe set. Interestingly, in one case (case 150), iFISH predicted tetraploidy while oaCGH analysis showed diploidy, including a number of segmental alterations. Taken together, these results demonstrate that iFISH analysis is of limited value in predicting ploidy changes using the complete FISH screening probe set. The ploidy changes detected by oaCGH analysis was confirmed by additional FISH probes not part of the FISH screening probe set.
G-Banding, interphase nuclei fluorescent in situ hybridization (iFISH) and oligo-based array comparative genomic hybridization (oaCGH) results of patients with newly diagnosed multiple myeloma including scoring by International Staging System (ISS).
As expected, oaCGH analysis did not detect translocations involving the IGH locus, whereas iFISH detected rearrangements in 64 out of the included 123 patients (52%) with the IGH split-apart probe. Of the 64 cases with an IGH rearrangement, 23 cases involved CCND1 (11q23), 16 FGFR3 (4p16), three MAF (16q23), one MAFB (20q), and 21 cases were without an identified partner chromosome.
oaCGH aberration characteristics. We next examined the characteristics of aberrations that were detected by oaCGH analysis in the present cohort (Figure 3A, Tables I and II). A total of 1,278 imbalances were detected by oaCGH analysis in 96 patients with an aberrant oaCGH analysis, giving an approximate average of 13 (range=1-29) aberrations per affected patient. The majority of patients (61%, 59/97) had a hyperdiploid karyotype, 13/97 (13%) had a hypodiploid karyotype with 44 or less chromosomes, and 25/97 (25%) were pseudodiploid with 45-46 chromosomes. The hyperdiploid karyotypes resulted from gains of any or combinations of chromosomes 3, 5, 7, 9, 11, 15, 19 and 21 in 92% of the samples, while monosomy 13 (43%) and loss of X chromosome (26%) were the most frequent whole-chromosome losses. Gain of chromosome 15 was the most frequent and seen in 55% of the patients, followed by gain of chromosomes 9 and 19 (53%), 5 (47%), 7 and 11 (42%), 3 (40%), and 21 (12%). High copy number gains were most frequently observed for chromosomes 15, 19, 9 and 11, in declining order, in various combinations in the hyperdiploid karyotype group in 31% (18/59) of the patients.
Segmental losses (425/1278) were more frequent than whole-chromosome losses (91/1278) and segmental gains (300/1278) (Table II). The genomic location of losses detected in >20% of the patients after significant peak analysis are summarized in Table III. A significant common minimal region of deletion was found in the NH-MM group at chromosome 1 region 1p22.2-p22.1 (91,597,326-92,050,583) and contained the TGFBR3/TBR3 genes. The BAC probe RP11-902H1 covers the TGFBR3 gene and it was used together with a centromeric chromosome 1 probe confirming the deletion in aCGH-defined positive cases (data not shown). In cases where aCGH analysis did not identify a deletion in 1p22.2-p22.1, the iFISH result showed a normal pattern.
Characteristics of oligo-based array comparative genomic hybridization aberrations in total cohort and in sub-groups of multiple myeloma, hyperdiploid (H-MM) and non-hyperdiploid (NH-MM).
Segmental gains (300/1278) were less frequent than whole-chromosome gains (434/1278). The genomic location of gains recorded in >20% of the patients after significant peak analysis are listed in Table IV.
The distribution of oaCGH aberrations were examined with respect to commonly used risk stratification systems in MM. It is common to divide the MM tumors into H-MM and NH-MM groups where H-MM generally confers a more favorable prognosis compared to NH-MM (6, 7). When the present cohort was classified according to aneuploidy groups, as determined by oaCGH analysis, 59 cases (61%) were H-MM and 38 cases (39%) cases were NH-MM (Table II). The main differences between the two groups, apart from the apparent whole-chromosome gains, were higher frequencies of losses at 1p, 12p and 14q, and that gains at chromosomes 11 and 19 are more frequent at the long and short chromosome arms, respectively (Figure 3B).
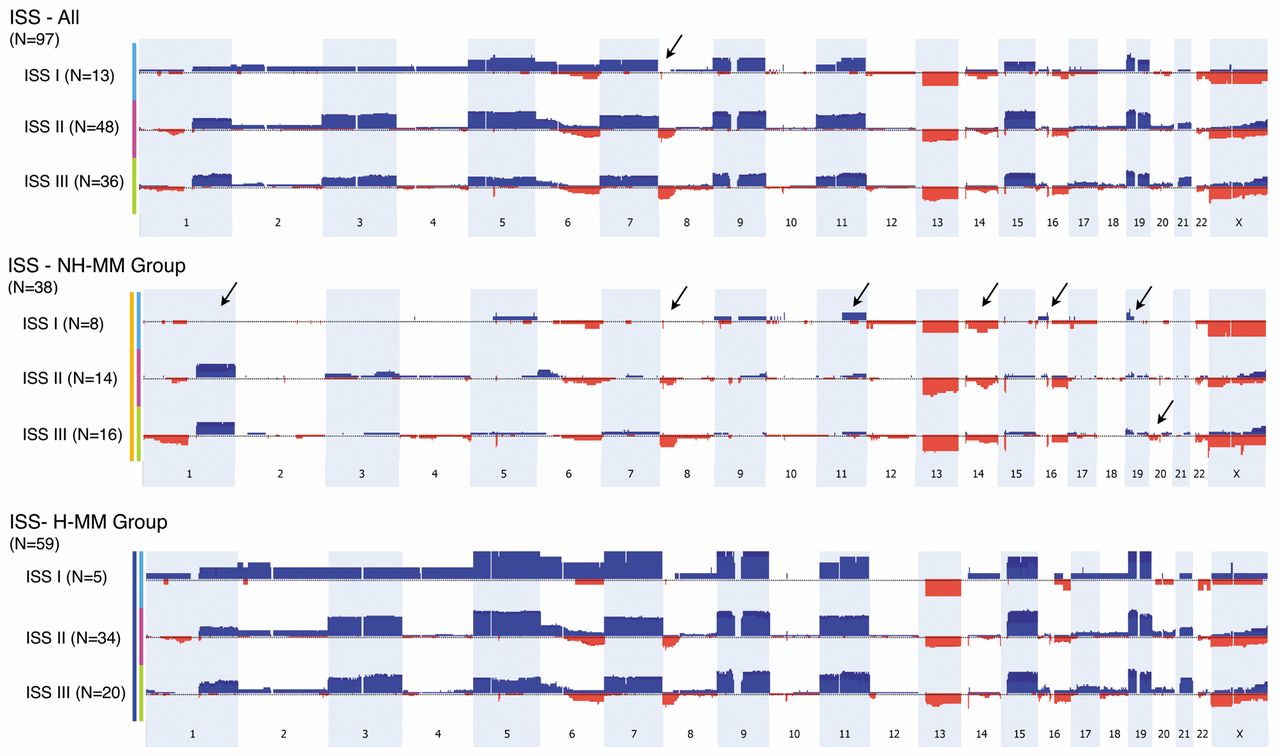
The ISS is a simple and reproducible prognostication system for predicting overall survival, regardless of age, treatment type, and geographic location (3). The system is widely used and is based on serum albumin and β2-microglubulin levels. It is applicable to both young and elderly patients treated with conventional therapy (19) or treated with novel agent-based therapies (29). ISS-I generally confers a more favorable prognosis compared to ISS-II and ISS-III (19). When the present cohort with an oaCGH result was divided according to ISS staging, 13, 47, and 37 cases belonged to the ISS-I, ISS-II and ISS-III groups, respectively (Table V). Apart from differences in the mean levels of β2-microglubulin between the three groups, the most striking difference is the significantly higher number of cases with complex karyotypes in the ISS-III group.
It has been suggested that genomic complexity defined as three or more oaCGH aberrations of 5 Mb or more in size may serve as an independent risk factor for disease progression in chronic lymphatic leukemia (30), and in acute myeloid leukemia (AML), five or more oaCGH aberrations may correlate with prognosis (31). There were no significant differences between the three ISS staging groups for these parameters (Table V).
Comparison of the oaCGH profiles in the three ISS staging groups revealed a couple of striking differences. The size of the commonly deleted region of 8p was found to be significantly smaller in the ISS-I group (0.64 Mb, genomic position 7,214,673-7,858,439) compared to deletion of most of 8p (43.7 Mb, genomic position 0-43,660,173) in the other two groups (Figure 4). Interestingly when the ISS system is combined with the ploidy groups, H-MM and NH-MM, the most striking differences are that no cases in the ISS-I/NH-MM group had 1q amplifications and that the 8p deletion was significantly smaller in ISS-I compared to the other groups (Figure 4). In addition, there were differences related to 11q, 14q, 16p, 19p and 20p between the groups. In selected cases, iFISH with 1q21/1p36 probes were used and confirmed the oaCGH results (Table I).
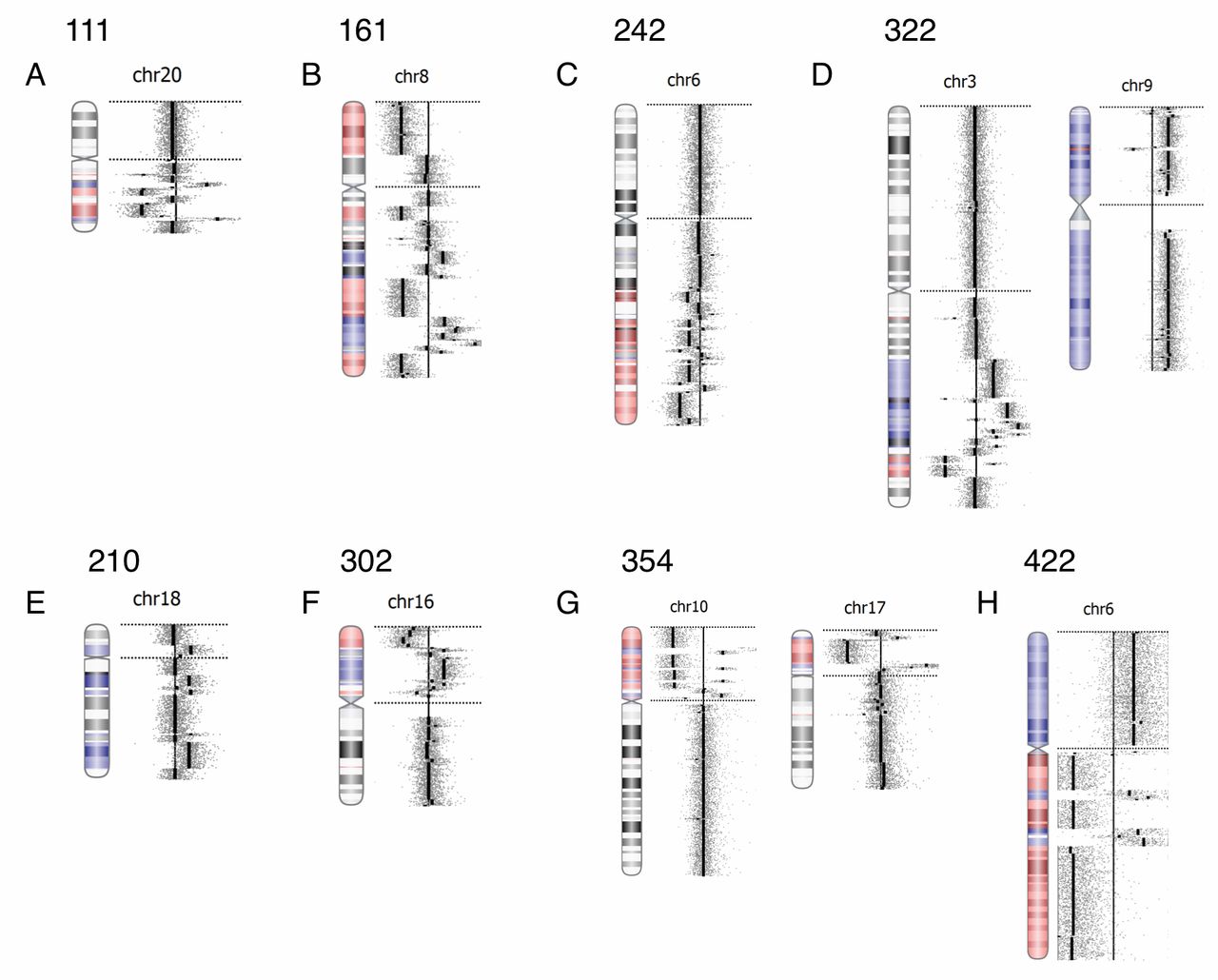
Chromothripsis. The copy number profiles derived from the 180K oligo-based Cancer Cytochip microarrays indicated a complex genomic rearrangement compatible with the hallmarks of chromothripsis in 22% of the patients newly diagnosed with MM (22 out of 96 cases with an abnormal oaCGH result). A total of 29 chromosomes were affected by chromothripsis in this study cohort, and representative chromothriptic chromosomes are shown in Figure 5. The most commonly affected chromosome was chromosome 16, being involved in five out of 22 cases. The second most commonly affected chromosomes were chromosomes 1, 6, 8 and 20, each involved in three out of the 22 cases. Five cases had two chromosomes affected by chromothripsis, and in one case, three chromosomes were affected. The vast majority of cases were segmental chromosomal aberrations, with an average size of 59.1 Mb (range=2.35-183.31 Mb) (Table II). Fifteen out of the 22 cases with chromothriptic chromosomes belonged to the hyperdiploid prognostic group and 12 of these cases belonged to the ISS-III group.
Genomic regions of common losses.
Genomic regions of common gains.
Discussion
In the present prospective study of 123 patients consecutive newly diagnosed with MM, we showed that the 4×180K oaCGH array in conjunction with iFISH for the loci IGH, TP53, and DLEU1 detected clinically relevant genomic risk prognosticators in a clinical setting of MM diagnosis in a cost-effective manner.
It is imperative that the screening for genomic aberrations in MM is performed on positively identified plasma cells because of the frequent low percentage of plasma cells in diagnostic samples (13). Immunomagnetic sorting of CD138+ cells from bone marrow of patients with MM has the advantage that the sorting can be automated (32) and that the positively isolated cells can be used to make cytospin slides for iFISH studies, and high-quality DNA for oaCGH analysis can be isolated from the surplus of these isolated cells. In a clinical setting, we found that we were unable to obtain enough CD138+ cells for oaCGH analysis in 26 out of 123 patients, giving a success rate of 79%. The main reason for this was a combination of a low number of white blood cells, as well as a low percentage of CD138+ cells in the diagnostic bone marrow samples. Additionally, because karyotyping is currently the method of choice for whole-genome scanning in MM at diagnosis, approximately 10×106 cells were used for this purpose. Consequently, if karyotyping were omitted, the oaCGH success rate would increase to above 95%. In this study, we had a limit of at least 8×105 cells to be used for DNA isolation to obtain high-quality DNA and to avoid whole-genome amplification.
A karyotyping result was obtained in 82% (101/123) of cases but the abnormality rate was as low as 27% (27/101), which is in agreement with another study (33). This highlights the limited value of karyotyping in MM, although it can still be useful for detecting balanced translocations that are not screened for by iFISH and to distinguish cases with proliferative disease (20).
Comparison of aberrations detected by karyotyping, and interphase nuclei fluorescent in situ hybridization (iFISH) in cases with an oligo-based array comparative genomic hybridization (oaCGH) result with respect to International Staging System (ISS).
oaCGH analysis is limited by its inability to detect balanced gene rearrangements and to identify multiple clones and low level clonal aberrations. In this study, oaCGH analysis failed to detect low-level aberrations present in fewer than 30% of cells. The clinical relevance of small clones is presently unclear. It has been shown that for 17p deletions, the poor prognostic impact was greatest for clone sizes above 60%, and above 30% for 1q gain (16, 34, 35). Clearly, further studies are required to confirm these findings and define cut-off levels for other loci. We detected subclones with locus-specific iFISH probes and karyotyping in two and three cases, respectively. These were low-level clones and therefore of unknown clinical relevance.
The inability of oaCGH analysis to detect balanced translocations is a major drawback. However, there are a significant number of apparently balanced translocations, which in fact are unbalanced due to sub-microscopic alterations (17). In our study, seven cases had CNAs at 11q13 in the CCND1 locus detected by oaCGH analysis, suggestive of a translocations involving this locus, which were confirmed by iFISH. Conversely, oaCGH analysis detected no CNAs at the CCND1 locus in three cases, while iFISH showed translocation there. In addition, we found that iFISH showed translocations at the FGFR3, MAF, or MAFB loci in several cases, while oaCGH analysis showed no CNAs. Taken together, even though oaCGH analysis may be suggestive of apparently balanced translocations, iFISH is indispensable for their definitive identification. A variant of aCGH analysis which allows for detection of recurrent balanced translocations, tCGH, may in the future, prove useful in making a complete aCGH approach in MM (36).
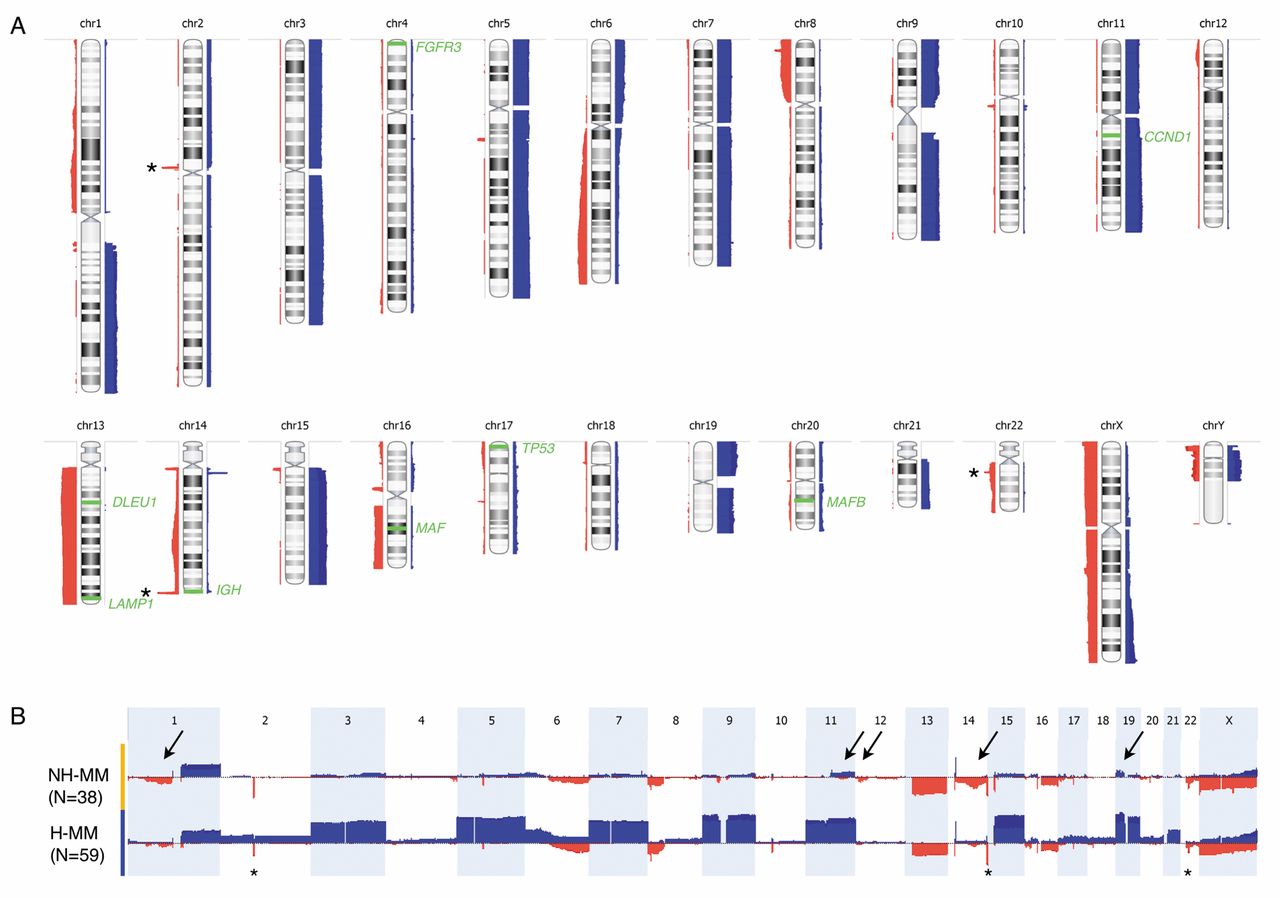
A: Summary frequency plot of genomic copy number changes identified in CD138+ selected cells from BM of 97 patients with newly diagnosed multiple myeloma. Red bars to the left of each chromosome ideogram indicate genomic losses, light blue bars to the right indicate genomic gains, and high copy number gains are indicated by dark blue. The heights of the bars indicate the frequency of genomic imbalances in the relevant regions. The asterisk's indicate sub-microscopic losses at 2p11, 14q32 and 22q11, representing rearrangements at the immunoglobulin genes Ig-light chain-kappa, Ig-heavy chain and Ig light chain-lambda, respectively. Losses are predominantly segmental (1p, 6q, 8p, 12p, 14q, 16p11, 16q, and 17p, and 22q) except for the whole-chromosome losses of chromosome 13 and X. Gains mostly involve whole chromosome arms (1q, 6p and 19p) or whole chromosomes (3, 5, 7, 9, 11, 15, 19, and 21). Loci examined by iFISH are indicated by green bars and text. B: Frequency plot of aCGH abnormalities detected in the cohort grouped according to ploidy category, NH-MM (top, yellow bar) and H-MM (bottom panel, blue bar). Chromosomes 1 to X are represented from left to right. Gains are depicted as upward blue bars and losses are depicted by downward red bars. The amplitude represents the frequency (%) of each copy-number abnormality. Arrows point to differences in oaCGH abnormalities between the groups. N: Number of patients in each group. FGFR3: fibroblast growth factor receptor 3; CCND1: cyclin D1; MAF: v-maf musculoaponeurotic fibrosarcoma oncogene homolog.
Comparison of the oaCGH results with the iFISH assays showed a high degree of concordance except for low-level clonal aberrations, as discussed above. Imbalances involving the IGH, IGLK and IGLL loci are usually not reported because they are considered to present copy number variations. However, their presence in many MM cases indicates the presence of a monoclonal B-cell population (37).
We found that assignment of ploidy groups by oaCGH analysis often depends upon the iFISH results in agreement with other studies (17). In more than 20 cases, the diploid (2N) value had to be adjusted in the Nexus software based on FISH results to avoid misinterpretation of aneuploid chromosomes and thereby ploidy assignment. We therefore suggest that iFISH consistently be performed using the following loci: IGH, TP53 and DLEU1/LAMP1 in order to be combined with the oaCGH results. Other studies have suggested that oaCGH combined with IGH/FGFR3 iFISH could be sufficient as first-line testing for risk assessment in MM (15).
MM is an incurable and very heterogeneous disease group. Some patients have severe disease with rapid disease progression and short survival, while others are less severely affected (2). Among a large number of predictive parameters, a major advance was achieved by ISS. According to this system, patients are classified into three groups with very different overall survival. An important limitation to this system is that it does not directly incorporate intrinsic plasma cell variability at the genomic level. Recurrent genetic aberrations in malignant plasma cells have been shown to have a strong prognostic power, among which the most important are t(4;14)/IGH-FGFR3, del(17p)/TP53 and 1q gains (8, 38). In a recent study, it was shown that risk assessment is considerably improved by considering ISS staging in combination with iFISH results (20). Prognostic information is also provided by chromosome ploidy group (39). The H-MM group, with more than 47 chromosomes, is characterized by gain of odd-numbered chromosomes, low incidence of IGH-involved translocations, monosomy 13, and a good prognosis. The NH-MM group, encompassing hypodiploid, pseudodiploid and hypotetraploid clones, is characterized by a high incidence of IGH-involved translocations, monosomy 13, and a poor prognosis. In the present study, we combined the oaCGH results with ISS and ploidy (Figure 4), which has not been done previously to our knowledge. From this we show that 1q gain was absent in the NH-MM/ISS-I group and that the 8p deletion was also considerably smaller in this group. Although our prospective study design did not allow for evaluation of overall survival, these findings suggest combining ISS staging with genomic profiling might improve risk assessment in MM. Interestingly, we showed that complex karyotypes were more frequent in the ISS-III group, known to be associated with a shorter overall survival. Genomic complexity defined as three or more oaCGH aberrations of 5 Mb or more in size has been suggested as an independent risk factor for disease progression in chronic lymphatic leukemia (30) and in AML that five or more oaCGH aberrations may correlate with prognosis (31). We examined these aberration types with respect to ISS but found no significant differences between the three groups except for a tendency for more frequent and larger aberrations in ISS staging groups II and III compared to group I. Studies on larger cohorts and long follow-up are necessary for further clarification.

Frequency plot of aCGH abnormalities detected in the cohort grouped according to ISS stage I (light blue bar), II (red bar) and III (green bar) (top panel), and the ISS staging according to ploidy category, NH-MM (middle panel, yellow bar) and H-MM (bottom panel, blue bar). Chromosomes 1 to X are represented from left to right. Only copy-number abnormalities greater than 1.0 Mb are included and copy-number variations are excluded. Arrows point to major differences in oaCGH abnormalities between the groups.
A common loss of 1.55 Mb mapped at chr1:90,52-92,05 within 1p22.2-p22.1 was shared by 21% of the samples in this study. There was no correlation to ploidy or ISS grouping. One of the genes in this region is TGFBR3 that has only been described in other array-based studies. The TGFβ signaling pathway plays an important role in regulating normal hematopoiesis by inhibiting proliferation and stimulating differentiation when appropriate (40). It was recently demonstrated that loss of TGFBR3 expression is a frequent alteration in the TGFβ signaling pathway in MM (41). Importantly, restoring TGFBR3 expression reduced cell growth, proliferation, heterotropic adhesion, and migration in myeloma cells, whereas silencing of endogenous TGFBR3 expression increased cell growth, heterotropic adhesion, and migration (41).

Chromothripsis in patients newly diagnosed with MM. Chromotriptic chromosomes from eight patients (panels A-H) are shown with copy-number profiles on the right handside for the corresponding chromosomal ideograms with indicated copy-number changes. Case numbers are given at the top of each copy-number profile. Amplifications are indicated by blue and losses are indicated by red.
A commonly deleted region in 12p13 has been described in other microarray studies in approximately 16% of patients with MM, although the precise genomic position is debated (15, 16). In the present prospective study of only patients newly diagnosed with MM, we found that 11% of the patients had deletion in 12p13. Further studies are warranted to clarify the clinical or diagnostic significance of this deletion.
Chromothripsis is an emerging genomic risk marker in hematological and other cancer types. The term describes complex patterns of alternating copy number changes (normal, gain or loss) along the length of a chromosome or chromosomal segment because of chromosome shattering and restitching (42). The detection of these complex rearrangements requires high-resolution techniques such as next–generation sequencing or microarray analysis and can therefore not be visualized by conventional cytogenetics (43). The number of CNAs can vary greatly from several hundreds to a few per chromosome. Magrangeas et al. were the first to describe chromothripsis in patients newly diagnosed with MM (21). By microarray analysis, they found chromothripsis in 1.3% of samples in a study of 764 patients. More recently, Berry et al. (18) and Rack et al. (17) analyzed 20 and 112 patients, respectively, with monoclonal plasma cell disorders and did not find evidence of chromothripsis in their cohorts. We found chromothripsis in 22 out of 96 cases with newly diagnosed MM with an aberrant oaCGH result affecting 29 chromosomes (Figure 5 and Table I). Magrangeas et al. showed that nine chromosomes exhibited chromothripsis in 10 patients, affecting chromosomes 1q, 3q and 16q, recurrently, and chromosomes 2, 17, 8q, 10q, 18q and 12q on a single basis. In our study, chromosome 16 was most frequently chromothriptic, being involved in five cases; chromosomes 1, 6, 8 and 20 were chromothriptic in three cases; and chromosomes 19 and 22 were involved in two cases. These observations indicate that chromosomes 8 and 16 are more frequently chromothriptic and suggest that certain chromosomes may either be more prone to chromothripsis or that it appears so after possible selective growth advantage in vivo. The reasons for the observed differences between the various studies remain elusive and must await larger prospective studies. The study by Magrangeas et al. indicated that chromothripsis was an adverse marker, and they suggested that patients with chromothripsis might be biologically different (21). Our study design did not allow for such a comparison as we had only limited follow-up data.
The exact mechanisms driving chromosome shattering is unknown, while aberrant mitosis producing micronuclei and premature chromosome compaction have been implicated (44). The mechanisms driving the stitching process are also unknown, although DNA repair mechanisms such as non-homologous end-joining, fork-stalling and template switching, and microhomology-mediated break-induced replication are involved (45).
Conclusion
We showed that oaCGH analysis using the 4×180K oaCGH array platform combined with iFISH for the loci IGH, TP53, and DLEU1 in CD138+-selected cells from patients newly diagnosed with MM greatly enhances the rate of detection of genomic abnormalities as compared to G-banding and iFISH alone in a clinical setting. In addition, we found that it is possible to obtain sufficient material for these analyses with a success rate of approximately 80%, that could be increased to >95% if sample material for G-banding was instead used for aCGH analysis. With the growing amount of data from previous aCGH studies and from our results, we suggest that future cytogenetic diagnosis of genomic aberrations in MM is carried out primarily by oaCGH analysis combined with iFISH for IGH, TP53 and DLEU1/LAMP1. It will be necessary to evaluate the prognostic value of recurrent oaCGH findings, using larger cohorts, combined with other predictive parameters such as, for example, ISS staging and ploidy grouping. Our results suggest that oaCGH aberrations may define ISS subgroups and that a combination of ISS staging and genomic profiling might improve risk assessment. The major drawback of aCGH analysis is its sensitivity, which is about 20-30% depending on aberration size, whereas iFISH can detect abnormal clones below 10%. However, the clinical relevance of these low-level clones needs to be determined.
Acknowledgements
The biotechnologists Kirsten V. Madsen, Bente Madsen, Pia Kristensen, Majbritt D. Jensen and Mette Rasmussen are greatly thanked for their excellent technical assistance with karyotyping and iFISH analyses. In addition, Bente Madsen and Pia Kristensen are greatly thanked for their excellent technical assistance in performing the oaCGH analyses. The clinicians at the Department of Hematology, Aarhus University Hospital and in particular consultant Niels Frost Andersen, MD, are greatly thanked for referring patients and providing clinical details. The Danish Cancer Society supported the study.
Footnotes
Conflicts of Interests
The Author has no conflicts of interest to declare.
- Received September 28, 2015.
- Revision received November 27, 2015.
- Accepted December 2, 2015.
- Copyright© 2016, International Institute of Anticancer Research (Dr. John G. Delinasios), All rights reserved




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}