


Abstract
Background/Aim: The mammalian, or mechanistic, target of rapamycin (mTOR) pathway has been implicated in several models of human oncogenesis. Research in the role of mTOR in human oncogenesis remains a field of intense activity. In this mini-review, we intend to recount our current understanding of the mTOR pathway, its interactions, and its role in human carcinogenesis in general, and breast cancer in particular. Materials and Methods: We herein outline the discrete components of the two complexes of mTOR, and attempt to define their distinct roles and interactions. Furthermore, we review current developments in the therapeutic targeting of mTOR in human breast cancer. Results: Our understanding of the organisation and interactions of the mTOR pathway continues to evolve. There has been significant incremental, albeit slow, progress in the therapeutic targeting of the mTOR pathway in human breast cancer. Conclusion: Continued progress in the field would require a better understanding of the role of the mTOR pathway in human breast cancer. By summarizing the current literature, this review will provide useful information on the topic.
Rapamycin, also known as sirolimus, is a macrolide produced by Streptomyces hygroscopius. It was initially identified in soil samples taken from the Easter Islands, also known locally as Rapa Nui (1). Its initial application was as an antifungal agent, with activity against Candida species (2).
Subsequently, it was found to have immunosuppressive properties, and is currently used therapeutically in renal transplant patients. Like tacrolimus and cyclosporine, it affects the actions of interlekin-2 (IL-2). Unlike tacrolimus, which reduces IL-2 transcription by T-cells, sirolimus inhibits the proliferative effects of IL-2 on T-cells (3). In mammalian models, rapamycin was known to form a complex with FK506 binding protein 1A, 12 kDa (FKBP12), which interacts with the mammalian, or mechanistic, target of rapamycin (mTOR) subunit of mTORC1 (4).
During the early 1990s, the studies of the effects of rapamycin on yeasts lead to the discovery of the targets of rapamycin (TOR1 and 2) (5). The mammalian, or mechanistic, target of rapamycin (mTOR) was described shortly thereafter. It was found to have homology with yeast TOR1 and 2 (42% and 45% respectively) (4).
mTOR has been characterised as a member of the phosphatidylinositol 3-kinase-related protein kinase (PIKK) family. Other members are the ataxia telangiectasia mutated (ATM), ATM- and Rad3-related (ATR), DNA-PKcs, suppressor with morphological effect on genitalia 1 (SMG-1), and the catalytically inactive transformation/transcription domain-associated protein (TRRAP). PIKKs are a group of evolutionary conserved regulatory enzymes with shared functional motifs. These motifs include a kinase domain (KD), FRAP-ATM-TRRAP (FAT), PIKK-regulatory domain (PRD) and FRAP-ATM-TRRAP-C-terminal (FATC) (6, 7). Furthermore, PIKK enzymes share some commonalities in function and sequence with the better characterised phosphatidylinositol 3 phosphatase family (PI3P) of enzymes (8).
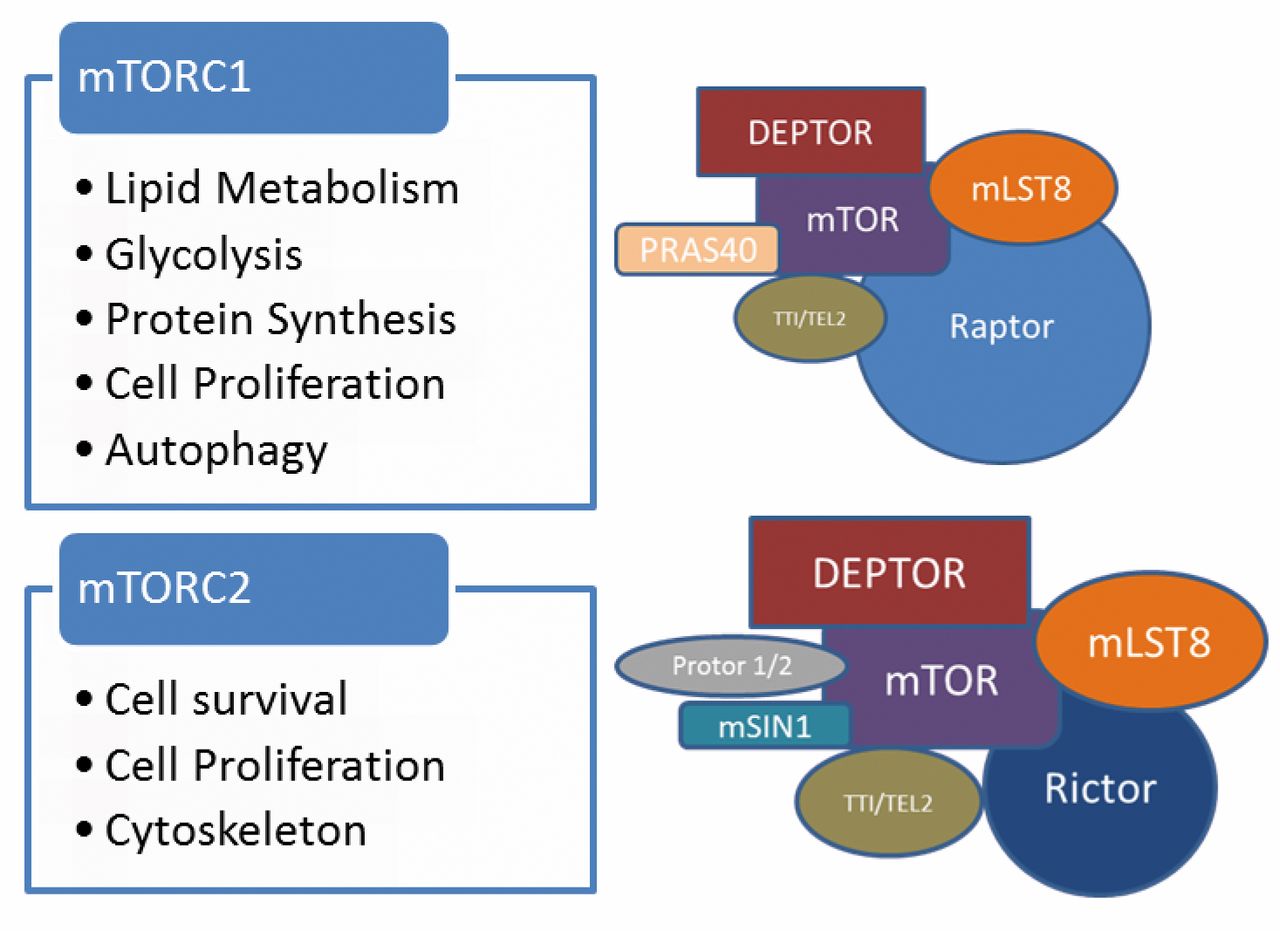
mTOR functions in association with a number of other protein moieties in at least two configurations, which are referred to as complex 1 and 2 (Figure 1).
mTOR Complex 1 (mTORC1)
mTORC1 has been shown to be affected by various stimuli including, but not limited to, hypoxia, nutritional state, stress, growth factors and amino acids. The activity of mTORC1 is inhibited by rapamycin and other rapamycin analogues (rapalogues) (3, 9).
Predominantly, upstream signals are mediated by the interplay of the tuberous sclerosis complex (TSC), and Ras homolog enriched in brain (RHEB). mTORC1 is localised to lysosomal surface by the Ragulator scaffolding complex. In addition, it senses amino-acid levels for mTORC1 (10). TSC contains three proteins, which include TSC 1 and 2 (TSC1 and 2, also known as hamartin and tuberin respectively) and a stabilising component, Tre2-BUB2-cdc16 (TBC) 1 domain family member 7 (TBC1D7) (11, 12). TSC 1 and 2 act as GTPase activating proteins (GAP) for RHEB. RHEB activates mTORC1 by functioning as a GTPase, in a manner similar to the function of the Gα subunit of a G-protein coupled receptor (GPCR) complex (13, 14) (Figure 2).
The majority of upstream stimuli are mediated by a number of effectors, such as protein kinase B (PKB, also known as AKT), extracellular-signal-regulated kinase 1/2 (ERK1/2), and ribosomal S6 kinase (rSK1). The Wingless integration pathway (Wnt) interacts with RHEB, as does the Insulin/PI3K/AKT pathway (15, 16). Alternatively, some upstream components may interact directly with RHEB. This is true with regards to mitogen-activated protein kinase (MAPK), which mediates stimuli from p38 (17). To a certain extent, PKB/AKT also acts directly upon RHEB.
In turn, mTORC1 regulates lipid metabolism, glycolysis, protein synthesis, cell proliferation, and autophagy (18, 19).
In recent studies regarding the regulation of autophagy, death-associated protein 1 (DAP1) has been characterised as co-substrate of mTORC1 along with the autophagy initiator complex, UNC-51-like 1 (ULK1) (20). A recent study by our group has reported inverse associations between DAP1 expression and tumour stage while higher DAP1 expression was associated with improved clinical outcomes (21).
The role of DAP1 in human breast cancer was further explored by developing a DAP1 knocked-down sub-line of breast cancer cells (MCF7). These cells showed an increase in cell proliferation after an initial delay in the course of a five-day growth assay compared to controls. A perusal of the literature suggests a potential involvement of the mTOR pathway downstream of DAP1 as an explanation for these findings. The potential role of DAP1 silencing in mTORC1 pathway recruitment would warrant further investigation (unpublished data).
Other components of mTORC1 are as follows and also shown in Figure 1.
Regulatory-Associated Protein of Mammalian Target of Rapamycin (Raptor)
Hara et al. identified raptor as an mTOR associated protein, providing scaffolding for mTORC1 kinase activity. Furthermore, the results of disruption of raptor and mTOR gene function were found to be phenotypically similar in nematode models (22). It is regarded to be an essential component of mTORC1, needed for stability and function of the complex. Its interaction is needed for function of the TOS motif on mTOR, which is required for phosphorylation (23).
Mammalian Lethal with sec-13 Protein 8 (mLST8, also known as GβL)
Kim et al. characterised mLST8 as an evolutionarily-conserved polypeptide which primarily associates and stabilises the mTOR kinase domain. It promotes mTORC1 kinase activity. In nutritionally deprived conditions, raptor associates with the complex and inhibits this effect. Also, it was shown that GβL was required for rapamycin-mediated inhibition and disruption of mTORC1, as well as for mTORC1 sensitivity to nutritional stimuli (24).
Furthermore, mLST8 has an extensive role in the functioning of mTOR complex 2 (25), which is discussed below.
DEP Domain Containing mTOR-Interacting Protein (DEPTOR)
Peterson et al., in a study of multiple myeloma cell lines, identified DEPTOR as an mTORC1 component interacting with mTOR. It inhibits the kinase activities of mTOR on S6 kinase, SGK, and AKT/PKB (26).
TTI1/TEL2 Complex
The role of the telomere maintenance 2 interacting protein 1 and telomere maintenance 2 (TTI1/TEL2) complex in mTOR was initially observed in yeast models (27). TTI1 was initially identified in mammals by Kaizuka et al., who determined that it contributed to the stability mTORC1 and mTORC2. In addition, it was found to mediate the interactions mTORC1 with telomere maintenance-2 (TEL2, also known as HCLK2) (28). It is suggested that TEL2 may similarly serve in the function of other members of the PIKK family of kinases (29).
Proline-Rich AKT Substrate 40 kDa (PRAS40)
PRAS40 has been characterised as an mTORC1 component which inhibits mTOR phosphorylation of targets by associating with mTOR, specifically by interacting with target of rapamycin signalling (TOS) motifs. Its association with mTOR is reduced by insulin and rapamycin. Its association with TOS motif inhibits mTOR function by controlling substrate binding (30).

Components of the mTOR complexes 1 and 2. Represented here are the mammalian, or mechanistic target of rapamycin (mTOR), regulatory-associated protein of mammalian target of rapamycin (Raptor), mammalian lethal with sec-13 protein 8 (mLST8), DEP domain containing mTOR-interacting protein (DEPTOR), TELO2 interacting protein 1 and telomere maintenance 2 complex (TTI1/TEL2), proline-rich AKT substrate 40 kDa (PRAS40), rapamycin-insensitive companion of mTOR (Rictor), mammalian lethal with sec-13 protein 8 (mLST8), mammalian stress-activated map kinase-interacting protein 1 (mSIN1), and protein observed with Rictor 1 and 2 (protor1/2).

Interaction of the tuberous sclerosis complex (TSC) with Ras homolog enriched in brain (RHEB) and mTOR. The TSC consists of TSC 1 (hamartin), TSC2 (tuberin), and tre2-BUB2-cdc16 (TBC) 1 domain family member 7 (TBC1D7). TSC functions as a GTPase activating protein (GAP) for RHEB. In turn, RHEB acts as a small GTPase with an affinity for mTOR. When retaining a GTP moiety, it activates mTOR. When the GTP is hydrolysed to leave GDP, RHEB, along with associated mTOR, is inactivated.
mTOR Complex 2 (mTORC2)
mTOR also functions as a component of another less well-characterised complex 2 (mTORC2). Our understanding of mTORC2 role in the wider pathway is still evolving. Whilst the study of mTORC1 function was facilitated by the use of rapamycin as an investigation agent, this was not the case for mTORC2. Furthermore, the lethality of rapamycin-insensitive companion of mTOR (Rictor) ablation in most vertebrates precluded study of gene function in null strains (31).
mTORC2 has a role in regulation of cell survival and the cytoskeleton through the stimulation of various kinases, including PKB/AKT. Through PKB/AKT, it is also thought to inhibit mTORC1 (32, 33).
In addition, mTORC2 has recently been implicated in the regulation of cell proliferation by forkhead box-O3a (FOXO-3a) via serum- and glucocorticoidinduced kinase 1 (SGK1) (34).
mTORC2 is not sensitive to rapamycin. However, chronic exposure to rapamycin has been shown to lead to disruption of the mTOR-Rictor complex, and loss of mTORC2 related PKB/AKT phosphorylation. This is presumably due to association of free mTOR with rapamycin, precluding its incorporation into mTORC2 (35). However, the Rictor-mammalian stress-activated MAP kinase-interacting protein 1 (mSIN1) complex has been shown to persist (36).
All the components discussed in relation to mTORC1 are also components of mTORC2, with the notable exception of PRAS40 (18). Subunits distinct to mTORC2, along with the mTORC2-specific role of mLST8 are discussed as follows and shown in Figure 1.
Rapamycin-insensitive Companion of mTOR (Rictor)
Sarbassov et al. characterised Rictor as a 192-kDa evolutionarily-conserved polypeptide which forms the mTORC2 complex with mTOR and GβL, but not with Raptor. It was also noted that this complex was not involved in the phosphorylation of S6 kinase 1, which at that time was taken as a surrogate of mTOR-Raptor function. On the other hand, Rictor-mTOR was shown to have a role in actin organisation and in the phosphorylation of Protein Kinase Cα (37).
Hresko et al. demonstrated in in vitro studies that Rictor-mTORC1 was required for the phosphorylation of AKT/PKB at the Ser-473 locus, which is an important step in the insulin signalling pathway. These studies were conducted with subcellular fractions of 3T3-L1 adipocytes (38).
Guertin et al. demonstrated in murine models that Rictor-mTOR interaction with AKT was specifically required for phosphorylation of forkhead box-O3 (FOXO-3), but not for AKT mediated phosphorylation of TSC-2 or GSK3β (25).
Perumalsamy et al. identified Rictor-mediated PKB/AKT stimulation as a downstream effector of the anti-apoptotic function of the Notch family of receptors (39).
Recent evidence cited SGK1 as a key down-stream affecter of Rictor-mTOR, as detailed below.
Serum- and Glucocorticoid Induced Kinase 1 (SGK1) and Rictor-mTOR
As discussed above, SGK1 is a key regulator of mTORC2-induced cell proliferation (34). Garcia-Martinez et al. identified the serum- and glucocorticoid-induced kinase 1 (SGK1) as an additional effector of Rictor-mTOR function. It was noted that in particular, phosphorylation of SGK1 substrate N-myc downstream regulator gene 1 (NDRG1) was affected by Rictor, mLST8 and mSIN1 knock-down (40).
The role of SGK1 as a Rictor-mTOR substrate was further investigated by Jones et al., who identified a TOR2 deficient strain of Caenorhabditis elegans (round worm). In these strains, mild developmental delay and increased fat storage was noted. Strains with loss of SGK1 homologues were phenotypically similar. These phenotypes were repressed in the case of gain of function mutations in SGK1 (31).
Using highly selective inhibitors, Lu et al. confirmed SGK1 to be an effector of mTORC2, and not of mTORC1. It was also determined that mTORC2 controlled SGK1 mediated activation of epithelial sodium channels (ENaC) in kidney cells (41).
Mammalian Lethal with sec-13 Protein 8 (mLST8, also known as GβL)
As mentioned above, mLST8 is a key component of mTORC1 function (24). In addition, mLST8 has an extensive role in the functioning of mTORC2. Guertin et al., using mLST8-deficient mice, suggested that mLST8 was not critical for mTORC1 signalling, or for its sensitivity to the nutritional state or rapamycin. However, it was shown that mLST8-deleted mice were phenotypically similar to Rictor-deleted mice. Furthermore, it was demonstrated that mLST8 was needed for stable interaction of Rictor with mTOR in mTORC2, was also being essential for mTORC2-mediated phosphorylation of PKB/AKT and subsequent FOXO-3 activation, as well as protein kinase Cα (PKCα) S657 phosphorylation and stability (25).
Mammalian Stress-activated MAP Kinase-interacting Protein 1 (mSIN1)
Wilkinson et al. initially identified stress-activated map kinase-interacting protein 1 (SIN1) in fission yeast and identified homologues in chicken embryos. SIN1 was characterised as an evolutionarily-conserved protein which interacts with members of the stress-activated MAP kinase family of enzymes and plays a role in cellular adaptation to stress (42).
Homologues in humans have been labelled human or mammalian stress-activated map kinase-interacting protein 1 (m- or hSIN1). Yang et al. demonstrated in HeLa cells with hSIN1 knock-down, that hSIN1 was involved in the mTORC2-mediated phosphorylation of PKB/AKT and required for actin organisation and for the stability mTORC2 (43).
Frias et al. enumerated three distinct mSIN1 isoforms (mSIN1.1, mSIN1.2 and mSIN1.5), and demonstrated that only one isomer could exist within an mTORC2 complex. It was also demonstrated in this study that, unlike the other isoforms, mSIN1.5 is insensitive to nutrient starvation and insulin. This is suggestive of at least three configurations of mTORC2 acting upon PKB/AKT in response to distinct sets of upstream signals (36).
Protein Observed with Rictor 1 and 2 (Protor1/2)
Pearce et al. characterised Protor as polypeptides interacting with mTORC2, specifically with Rictor. They are evolutionarily conserved, with homologues identified only in frogs and fish. They exist as one of two isoforms, with three splice variants for Protor 1 (α, β and γ). They all share a conserved sequence, with the exception of Protor 1 γ, which is deficient at the N-terminus. In addition, unlike the other splice variants, Protor 1γ interacts poorly with Rictor (44).
Protor 1 is also known as proline rich protein 5 (PRR5). Woo et al. demonstrated reduced cell proliferation in HeLa cell lines with Protor 1/PRR5 knock-down. Also, whilst PRR5 did not play a direct role in mTORC2-mediated PKB/AKT phosphorylation, PRR5 knock-down did reduce platelet-derived growth factor receptor-β (PDGFRβ) expression, which in turn affected S6K1 and AKT phosphorylation (45).
Pearce et al., in a study on kidney cells in Protor 1 knock-down strains of mice, noted reduced mTORC2-mediated SGK1 phosphorylation and secondary NDRG1 activation, which suggestive of a role of Protor 1 in this function of mTORC2 (46).
mTOR Pathway in Human Breast Cancer Therapeutics
The role of mTOR in cell proliferation and survival has been a major focus of research in human oncogenesis (47). The side-effect profile of rapalogues makes them an attractive therapeutic option. Rapalogues have been clinically proven effective in the treatment of renal cell carcinoma in combination or as a single-agent. However, there has been limited success in their use as single-agents in the case of other cancers (48, 49).
Some efforts have been made to identify potential markers of rapamycin sensitivity, which would theoretically identify the patient population which may benefit from therapeutic targeting of the mTOR pathway (50, 51).
As evident from the preceding discussion, mTOR intersects with several critical and active in oncogenesis cellular pathways. In the context of human breast cancer, cross-talk between oestrogen receptor signalling and the phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT)/mammalian target of rapamycin (mTOR) pathway is believed to be responsible for insensitivity to endocrine interventions. One strategy of therapeutic targeting of the mTOR pathway is co-targeting both signaling pathways to enhance the effectiveness of endocrine therapy (52). Pre-clinical studies have suggested that the mTOR pathway may play a role in the resistance to hormone therapy, trastuzumab and chemotherapy for breast cancer. This concept has been tested in clinical trials for neoadjuvant treatment and for metastatic breast cancer patients.
Everolimus (EVE) was studied under the J2101 trial regarding a potential role in the context of trastuzumab resistance. Specifically, this was a phase II trial comparing the efficacy of EVE combined with trastuzumab and paclitaxel in patients with Her2-overexpressing advanced breast cancer that did not respond to prior trastuzumab and taxane therapy. The preliminary findings of this trial were encouraging. Phase III testing is in progress under BOLERO-1 (53).
Similarly, the recently completed BOLERO-3 trial examines the efficacy of EVE in combination with trastuzumab plus vinorelbine in trastuzumab-resistant disease (54).
In addition, EVE has been the subject of the BOLERO-2 trial. EVE has been validated for use in combination with exemestane (EXE) for the treatment of postmenopausal patients with ER+ and HER2+ tumors that were resistant to hormone therapy (55).
Similarly, the HORIZON trial studied the efficacy of temsirolimus plus letrozole versus letrozole in breast cancer. However, the results thus far are equivocal (56).
Furthermore, the TAMRAD trial looked into the potential use of EVE-plus-tamoxifen vs. tamoxifen-alone in post-menopausal women with HR+/HER2- endocrine therapy resistant breast cancer, with more promising results (57).
In part, owing to the mixed success of rapalogues in the context of breast cancer, there has been an interest in developing mTOR kinase inhibitors (TORKinibs) as dual inhibitors of mTORC1 and mTORC2. It is hypothesised that such an agent would be efficacious by terminating the effects of mTORC2, which would be unaffected by the current generation of rapalogues (58).
However, clinical evidence published by our group regarding the association of Rictor mRNA expression with favourable prognosis in a cohort of 150 breast cancer patients suggest that dual inhibition of both mTOR complexes may in fact be contra-productive (59).
Our knowledge regarding the role of mTOR in human oncogenesis is rapidly evolving. We are on the threshold of effective therapeutic targeting in human neoplastic disease based on this understanding.
Conflicts of Interest
The Authors declare that they have no conflicts of interest.
Acknowledgments
This review was funded by grants from the Breast Cancer Hope Foundation (London, UK).
- Received April 20, 2014.
- Revision received May 25, 2014.
- Accepted June 9, 2014.
- Copyright© 2014, International Institute of Anticancer Research (Dr. John G. Delinasios), All rights reserved
References
In this issue




{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- mTOR Complex 1 (mTORC1)
- Regulatory-Associated Protein of Mammalian Target of Rapamycin (Raptor)
- Mammalian Lethal with sec-13 Protein 8 (mLST8, also known as GβL)
- DEP Domain Containing mTOR-Interacting Protein (DEPTOR)
- TTI1/TEL2 Complex
- Proline-Rich AKT Substrate 40 kDa (PRAS40)
- mTOR Complex 2 (mTORC2)
- Rapamycin-insensitive Companion of mTOR (Rictor)
- Mammalian Lethal with sec-13 Protein 8 (mLST8, also known as GβL)
- Mammalian Stress-activated MAP Kinase-interacting Protein 1 (mSIN1)
- Protein Observed with Rictor 1 and 2 (Protor1/2)
- mTOR Pathway in Human Breast Cancer Therapeutics
- Conflicts of Interest
- Acknowledgments
- References
- Figures & Data
- Info & Metrics
Related Articles
Cited By...
- No citing articles found.