


Abstract
Antibody-based molecules can be delivered into cells either by intracellular expression or through cellular uptake. We describe technologies for identification and expression of intracellular antibodies for target validation, intracellular immunization and tumor therapy, such as intracellular antibody capture technology, suicide or silencing technology, antigen-antibody interaction dependent apoptosis and their application for inhibition of oncogenic intracellular proteins and induction of apoptosis. These strategies have to be viewed in the context that inhibition of protein-protein interactions by small molecules is often limited due to their large interaction surface. We summarize antibodies with the ability to penetrate cells and strategies to induce uptake of antibodies after modification with protein transduction domains. Interference in oncogenic pathways is described for moieties based on antibody 3E10, which translocates into the nucleus after extracellular administration. Finally, we discuss examples of tumor immunotherapy and vaccination against intracellular antigens, and possible interactions mediating their mode of action.
- Autoimmune disease-related antibodies
- cancer immunotherapy against intracellular targets
- intracellular immunization
- inhibition of oncogenic pathways
- mechanisms of antibody uptake
- protein knock-down
- target validation
- protein transduction domain
- review
Based on the hallmarks of cancer (1-3), deregulated pathways as well as overexpressed and mutation-activated molecules have been identified as targets for potential therapeutic intervention. Secreted or cell surface-expressed target molecules are accessible to antibody-based entities, immunotoxins, or agents based on novel antigen-binding scaffolds (4-6). On the other hand, in general, intracellular targets are inhibited by small molecules. Some potentially important targets which function through protein-protein interaction (PPI), however, are usually classified as ‘undruggable’ due to the large protein surface to be covered by a small molecule inhibitor in order to interfere with such a PPI. Targeting of PPIs is an important strategy for the generation of anticancer drugs since PPIs are basic units in oncogenic signaling networks promoting uncontrolled proliferation and sustained cell survival (7, 8). Amplified c-MYC leads to binding to MYC-associated factor X (Max) or mothers against decapentaplegic (Mad), thereby inducing transcription of growth-promoting genes and inducers of cell cycle progression such as cyclin D (9). Induction of tumorigenesis by human papilloma viruses (HPV) is based on neutralization of the tumor-suppressor functions of the phosphorylated form of retinoblastoma protein (pRb) and p53 by binding of HPV proteins E7 or E6, respectively (10). Dysregulated epigenetic mechanisms are important contributors to the generation and progression of cancer. Altered histone methylation and acetylation based on the recognition of binding proteins such as methyl-lysine or acetyl-lysine-binding bromodomains are important contributors to tumorigenesis (11, 12). The interface of many protein-protein complexes is typically hydrophobic and relatively flat, often lacking deep grooves where small molecules can dock (13, 14). Intracellular delivery of antibodies or antibody-related molecules which in theory should be able to target large surface PPIs, could be a method of choice to tackle this problem.
Basic options for the intracellular delivery of antibodies are either based on the transfer of the genetic information required to synthesize the antibody within the target cell itself [plasmid- or virus-based (adeno-, adeno-associated virus, retroviruses)] or on direct delivery of the antibody-based molecules themselves alone or in the context of dendrimers, liposomes, nanoparticles or fusion of antibody-related moieties with protein transduction domains (Figure 1) (15). In the following, we review achievements in more detail.
Systems for Intracellular Expression of Antibodies
Numerous technologies have been described for the generation of antibodies directed against defined antigens. These include hybridoma technology, DNA-based immunization, phage display libraries and transgenic animals (16-18). In the following we describe technologies for expression of antibodies inside bacterial, yeast, or mammalian cells. In eukaryotic cells, the correct folding and proper disulfide bond formation of antibodies takes place in the endoplasmic reticulum (ER) and is supported by ER-associated chaperones such as binding protein (BiP) and protein disulfide isomerase (PDI) (19-21). In addition, retention of newly-synthesized antibodies in the ER can be achieved by adding an ER-retention signal, such as the KDEL peptide motif, to the carboxy-terminus of the antibody. A strategy for intracellular production of antibodies in Escherichia coli is based on the twin arginine translocation (TAT) system which mediates the transport of proteins folded in the cytoplasm through the cytoplasmic membrane into the periplasm of bacteria. Here, a TAT-specific signal peptide and β-lactamase are fused to a single-chain variable fragment antibody (scFv) at the N- and C-terminus, respectively, thus conferring ampicillin resistance to cells in the case of scFv fusion protein correctly transported from the cytoplasm (22).
Selection of intracellular antibodies has also been achieved by intracellular antibody capture technology (IACT). This technology is based on a two-hybrid screen in yeast relying on scFv or intracellular domain antibody (iDab) antigen interaction. For example, the antigen is expressed as a DNA-binding domain-target antigen fusion protein and the corresponding antibody as an activation domain antibody moiety fusion protein. Their interaction results in activation of a reporter gene conferring resistance to ampicillin or tetracycline (Figure 2A). In a variation of this method, in the case of antibody moiety antigen interaction, production of the tetracycline repressor will be stopped and the imidazoleglycerol-phosphate dehydratase (HIS3) gene will be expressed, allowing selection of antibody moiety expressing yeast cells based on the growth in the absence of histidine (23-25).
Another variation on the theme was the development of suicide or silencing intracellular technology (SIT), based on the inducible degradation of intracellular antibodies equipped with proteasome-targeting sequences and thus converted into suicide antibodies (26). Cellular proteolysis is predominantly carried-out through the ubiquitin/proteasome pathway, leading to the degradation of a target upon ubiquitinylation in a multistep process. SIT relies on recruitment of target scFv cellular substrate complexes to the specific binding pocket of one of the members of the E3-E2 ubiquitinylation complex, for example the F-Box protein, resulting in degradation of the cellular substrates by the proteasome (Figure 2B). Inhibitor κBα (IκBα) protein undergoes stimulus-mediated degradation induced by tumor mecrosis factor-α (TNFα) and was used as a bridging molecule in the context of a fusion protein between the target protein recognized by the antibody and the cellular degradation machinery, resulting in selective protein knock-down. Proof-of-concept experiments were performed with β-galactosidase and τ protein (26).
Another powerful method is the induction of cell death by antigen antibody interaction-dependent apoptosis (AIDA) (27). The underlying principle is autoproteolysis and activation of pro-caspases through dimerization of antibody moiety procaspase fusion proteins after interaction with the corresponding antibody-related epitopes (Figure 2C). Corresponding epitopes can be located on a single protein, two different proteins, or on different parts of a cancer-related fusion protein generated by chromosome translocation.
Intracellular Monoclonal Antibodies (mAbs) Directed Against Oncogenic Kinases
An scFv antibody directed against epidermal growth factor receptor (EGFR) and inhibiting the binding of EGF was directed to the secretory pathway after intracellular expression in NIH/3T3 fibroblasts. EGF-induced activation of the receptor was shown to be reduced and anchorage-independent growth of the cells was inhibited (28). Intra-molecular disulfide bridges are important for antibody stability. They are formed during expression in the secretory pathway, whereas scFv expressed in the reducing microenvironment of the cytosol is often inactive. Therefore, in order to isolate mAbs directed against the intracellular domains of EGFR, a combination of biopanning of a combinatorial library and subsequent expression in yeast was used to isolate the corresponding mAbs. Their co-localization with EGFR at the plasma membrane was shown (29). Other proof-of-concept experiments for the expression of intracellular antigens have focused on human epidermal growth factor receptor-2 (HER2) as a target. Intracellular antibody directed against HER2 down-regulated HER2 on the surface of SKOV3 ovarian carcinoma cells and inhibited proliferation of HER2-overexpressing cells (30). Functional inactivation of activated HER2 and reversion of the transformed phenotype was demonstrated in scFv-transfected NIH/3T3 cells engineered for synthesis in the lumen of the ER and prevention of secretion (31). Studies of an intracellular scFv in T47D breast cancer cells, which express all four members of the HER family, revealed suppression of cell surface expression of HER2 and impairment of Neu differentiation factor (NDF) and EGF signaling (32). Transmembrane receptor kinase receptor d’origine nantais (RON) and its ligand macrophage stimulating protein (MSP) have a significant impact on the development and progression of human cancer (33). Making use of glutathione S-transferase RON intracellular domain fusion protein as an antigen, a phage display derived scFv was isolated and shown to interact in vivo with the intracellular domain of RON in mammalian cells (34). The potential of intracellular antibodies as tools for target validation was demonstrated with neutralizing antibodies directed against endothelial and epithelial kinase (ETK), an intracellular kinase involved in the SRC-induced cell transformation process (35). When single-domain light-chain variable regions derived from phage libraries were expressed intracellularly, inhibition of kinase activity and clonogenic cell growth of mouse NIH 3T3 cells overexpressing vSrc (NSR) was observed.

Basic options for intracellular delivery of antibodies. A mammalian cell is represented by the light blue oval and some of its organelles are highlighted. The options for delivery are outlined in ovals at the periphery of the cell. ER, Endoplasmic reticulum; G, Golgi apparatus; M, mitochondria; N, nucleus. The Ingenuity Pathway Analysis Path Designer tool supplier was used to prepare this figure.
An important potential of intracellular antibodies is their capability to interfere with PPIs mediating stimulation of oncogenic pathways. As a prototype of inhibiting these kinds of interactions, intracellular antibodies to rat sarcoma (RAS) have been investigated in detail (36). scFv antibodies based on intracellular capture frameworks and binding to RAS with high affinity were shown to impair oncogenic transformation (37). These findings were extended to single-domain intracellular antibodies directed against RAS (38). It was shown that variable heavy-chain domain (VH) and light-chain domain (VL) regions of anti-RAS Fv can be intrinsically stable and independent of intra-domain disulfide bonds (39). The neutralizing RAS antibody 413-259 (40) was investigated in detail after microinjection of expression plasmids for the whole antibody or scFv fragments targeted to the cytosol into Xenopus laevis oocytes. Co-localization with RAS and block of meiotic maturation were observed. In mammalian cells it was noticed that anti-RAS scFv fragments sequestered the antigen in aggregated structures referred to as aggresomes, leading to inhibition of RAS function (41, 42).
Oncogenic fusion proteins based on chromosome translocations have been identified as major oncogenic drivers in many subtypes of cancer. Neutralization of oncogenic function with intracellular antibodies is another potential of this class of molecules. Making use of IACT, panels of antibodies were identified which bind intracellularly to the breakpoint cluster region protein (BCR) and Abelson murine leukemia viral oncogene homolog 1(ABL), parts of the BCR-ABL fusion protein which is involved in the pathogenesis of human chronic meloid leukemia (43). It has been shown that the SRC-homology domain 2 (SH2) of the BCR protein is essential for the transforming properties of the BCR-ABL fusion protein. Therefore, blocking of this domain with an intracellular mAb may neutralize its oncogenic function.
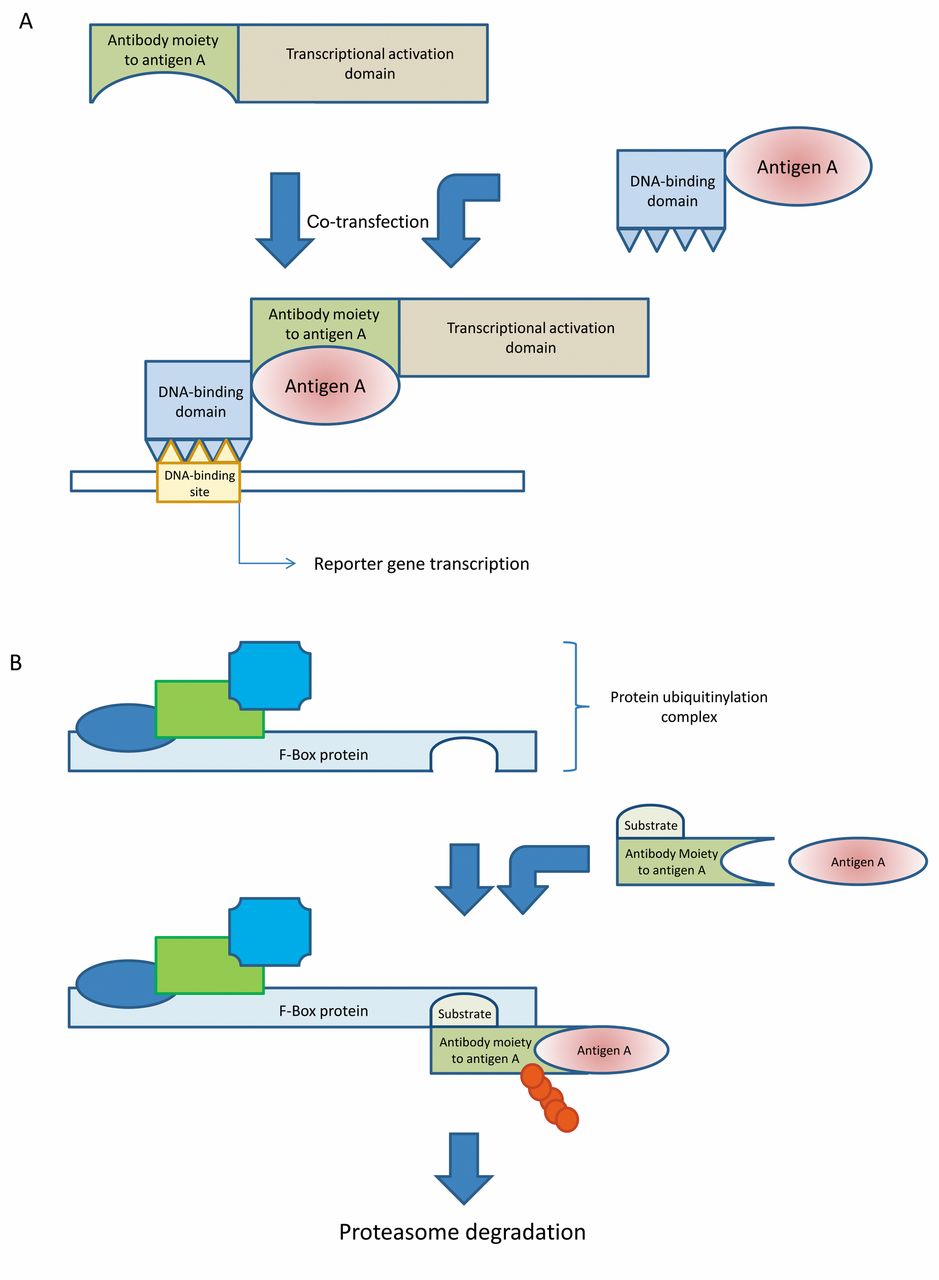
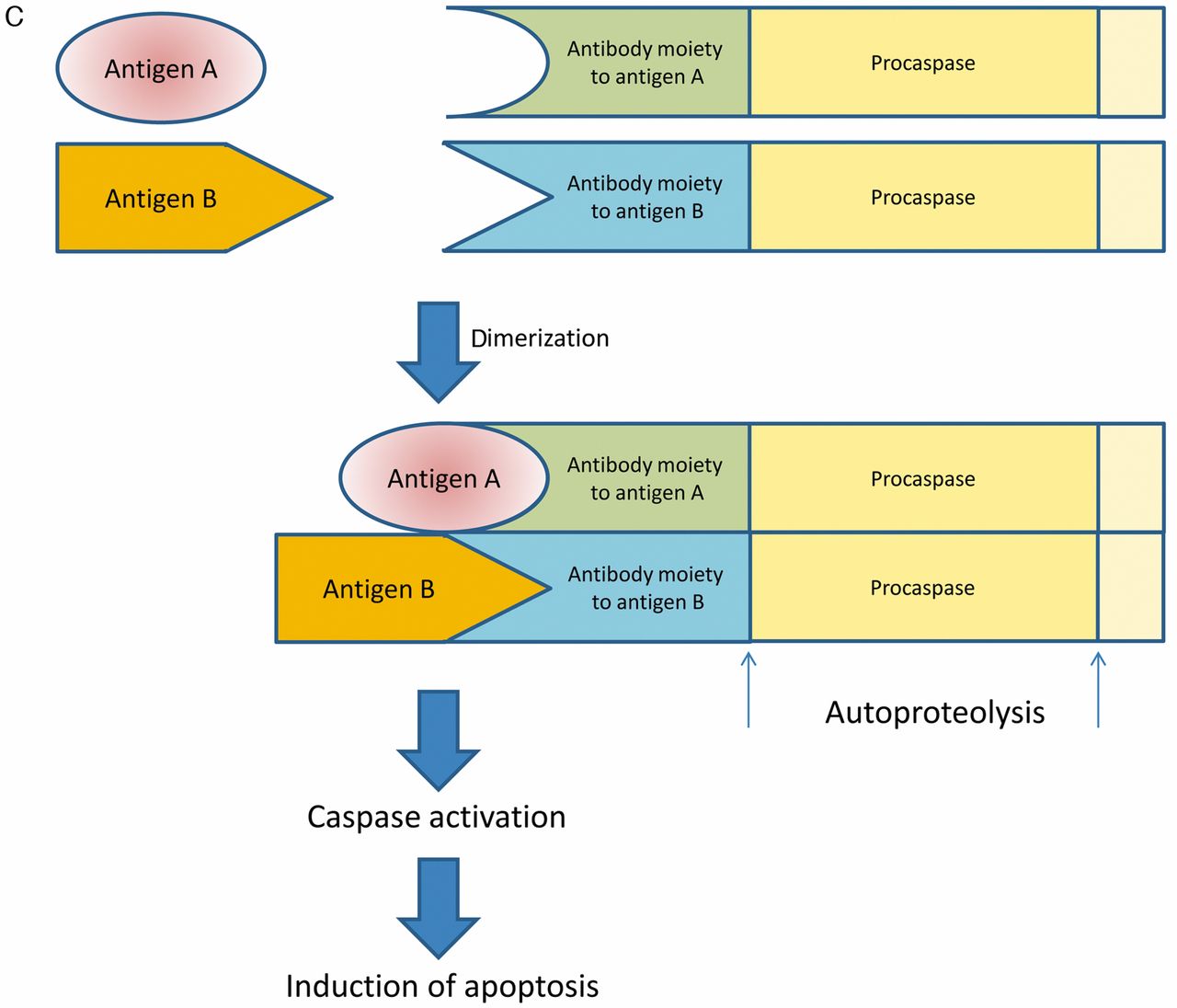
Schematic outline of intracellular antibody capture technology (IACT), suicide (or silencing) intracellular technology (SIT) and antigen-antibody interaction-dependent apoptosis (AIDA). A: ICAT technology. Antigen antibody interaction results in activation of transcription of a reporter which may be a selection marker. B: SIT technology. After interaction of F-box protein with a fusion protein of F-box protein substrate and an antibody moiety directed against antigen A, which is complexed with antigen A, the complex is ubiquitinylated (red bubbles) and subsequently degraded by the proteasome. C: AIDA technology. After dimerization of fusion proteins of antibody moieties directed against antigens A and B and procaspase, autoproteolysis of procaspase leads to caspase activation and subsequently to induction of apoptosis.
A method was devised in which intracellular interaction between an antigen and the corresponding antibody induced killing of tumor cells (44). It is based on the finding that caspase 3, an inducer of cell death by apoptosis, can undergo autoactivation when two or more scFv-caspase 3 fusion proteins bind to the epitopes of the same or different antigens that are close together. This was shown for anti-β galactosidase scFvR4 caspase 3 fusion protein and anti-HIV integrase scFv IN33 caspase 3 fusion proteins. Making use of this method, cells expressing a defined target antigen were selectively killed.
Cell-penetrating Antibodies
Cell penetrating antibodies have been described, mostly in the context of autoimmune diseases such as systemic lupus erythematosis. Most of these autoantibodies were found to be directed against nuclear antigens.
In a landmark study, it was shown that antibodies directed against nuclear ribonucleoprotein penetrated live human mononuclear cells through fragment crystallizable (Fc) receptors in patients with mixed connective tissue disease. Intra-nuclear immunoglobulin was observed in skin biopsies of these patients by direct immunofluorescence (45). More recently, anti-P-mAb 9B6-4, recognizing ribosomal phosphoproteins P0, P1 and P2 (46), was described. This antibody penetrates Jurkat cells, astrocytes and lung cancer cells, probably via interaction with P0 on the cell surface. Although it was shown that the antibody penetrated more than 90% of Jurkat cells, only 20% were killed by apoptosis, indicating that sensitivity may vary with cell-cycle phase. Similarly, antibodies to DNA were observed in patients with systemic lupus erythematosis. In a corresponding mouse model, murine monoclonal antibodies to DNA were able to penetrate cells, bind to nuclei and to induce glomerular proliferation and proteinuria in vivo (47). In addition, penetration of patient-derived auto-antibodies into living epithelial Colo16 tumor cells was demonstrated (48). Specifically, 5/36 (14%) of the antinuclear antibody positive sera from patients scored positive for cell penetration. Antibodies were internalized by an Fc- and complement-independent mechanism which was inhibited by cytochalasin B blocking receptor-mediated endocytosis (48). In addition, intranuclear immunoglobulin G (IgG) was detected in keratinocytes of a patient with penetrating IgG (48), and antibodies to heat shock protein 27(HSP27) were identified in patients with ovarian cancer (49). An HSP27-specific mAb able to internalize into neuronal and glial cells, activate caspases and to induce apoptosis was described. Internalization was Fc receptor-independent, and it is not known whether interaction with cell surface HSP27 was responsible for internalization (50). The mAb binds to actin and mediates depolymerization and proteolytic cleavage of actin in a dose-dependent manner.
It might be argued that antibodies reactive with intracellular antigens might contribute to the peripheral deletion of autoreactive clones (51). In summary, quite likely the mechanisms of uptake and intracellular transport are not uniform. Specific or cross-reactive antigens on the cell surface have not been identified in all cases, and uptake can be Fc-receptor-dependent or -independent. Some antibodies are also internalized via clathrin-associated vesicles and later released or transit may occur with facilitators or chaperones (52).
Cellular Uptake of Modified Antibodies
The concept of generating cell-permeable antibodies (transbodies) through conjugation or genetic fusion of protein transduction domains (PTD) with antibody-derived moieties was recently put forward (53).
For example, neutralization of oncogenic drivers after transfer of modified, cell-permeable antibodies would be an exciting prospect of this technology. In fact, a fusion protein consisting of glutathione-S-transferase, an scFv antibody directed against murine thyoma viral oncogene homolog (Akt1), and a membrane translocating sequence (MTS) from Kaposi fibroblast growth factor accumulated intracellularly in 293T, BT-474 and polymoma virus middle T-antigen (PyVmT)-expressing cells, and inhibited Akt1 as well as Akt2 and Akt3 phosphorylation. The fusion protein reduced tumor volume and mediated inhibition of angiogenesis in PyVmT-expressing tumors implanted into mouse dorsal window chambers (54). Similarly, cyclin D1 is often overexpressed in cancer. A polyarginated antibody to cyclin D1 inhibited cell-cycle progression in NIH3T3, HT29 and SW480 cells after internalization (55). MYC is a transcription factor deregulated in many types of cancer (56). Thus, an sc-Fv directed against c-MYC was fused with the internalization domain int of antennapedia for intracellular delivery. Anti-proliferative activity in HCT-116 cells was noted after internalization of the fusion protein (57).
However, one has to bear in mind that antibodies such as these described above do not have a targeting component specific for tumor cells, therefore they are probably also internalized by other non-transformed cells. Consequently, combination of these binders with a targeting moiety directed against cell surface-exposed tumor cell antigens, resulting in bi-specific fusion proteins, might be mandatory for therapeutic applications in patients with cancer.
Antibody 3E10-based Internalizing Moieties
A cell-penetrating antibody, 3E10, was isolated from a mouse model of systemic lupus erythematosis. A 3E10-derived scFv penetrates cells and nuclei through equilibrative nucleoside transporter 2 (ENT2), a nucleoside transporter which is ubiquitously expressed in human cells, including malignant cells (58-60).
In the context of studying lupus autoantibodies to deliver proteins for protection of normal cells from therapeutic ionizing radiation, it was discovered that one of the antibodies, 3E10, was able to sensitize tumors to radiation treatment (61). The mode of action is based on binding of 3E10 to single-stranded DNA, thus interfering with DNA repair, making cells more susceptible to DNA damaging agents such as doxorubicin or radiation. mAb 3E10 as a single agent is toxic to cancer cells with deficiencies in DNA repair pathways, such as those harboring mutations of breast cancer susceptibility type 2 antigen (BRCA2) (61). Since previous phase I clinical studies in patients with systemic lupus erythromatosis have demonstrated the safety of 3E10, studies in patients are expected soon.
3E10 also has the potential as a delivery vehicle (62). 3E10 and its single-chain variants proved effective in delivery of cargo proteins to cell nuclei in cell culture and in animals. Protein complexes consisting of 3E10 Fab fragments bound to alkaline phosphatase-conjugated goat anti-mouse κ chains were transported into the nucleus of COS-7 and CHO cells. Cellular penetration of 3E10 was markedly enhanced by a single mutation in the VH region. A fusion protein between scFv 3E10 and green fluorescent protein (GFP) penetrated COS-7 cells and localized in the cell nucleus (62). PAb421, an antibody which binds to the C-terminus of p53, was shown to restore the wild-type function of several p53 mutants (63). A bi-specific single chain antibody composed of scFv of 3E10 and scFv PAb421 was evaluated in SW480 colon cancer cells, which carry a mutation at position 273 of p53 and are responsive to treatment with mAb PAb421. Indeed, the bispecific single-chain antibody was toxic for SW480 cells but not for COS-7 cells, in which the presence of SV40 large T-antigen inhibits the binding of pAb421 to p53. Mutant bispecific antibody was not cytotoxic to SW480 cells (64). However, only data for SW480 cells but not for additional cells with other p53 mutations were shown.
Protein transduction with wild-type p53 as a 3E10 p53 fusion protein selectively kills tumor cells with functionally inactivated p53 (65). In an experimental mouse model of colon cancer metastasis to the liver, splenic injection of the 3E10Fv-p53 fusion protein inhibited metastasis of CT26.CL25 cancer cells significantly after their injection into the portal vein (66).
Forkhead box p3 (FOXP3), a forkhead family nuclear transcription factor, is deleted or mutated in the majority of human breast cancer tissues, providing a rationale for replacement therapy (67). Thus, 3E10 Fv-FOXP3 fusion protein inhibited proliferation and induced apoptosis in a dose-dependent manner in breast and ovarian cancer cell lines such as MDA-MB 231, MCF-7, SKOV-3 and OVCAR-3. 3E10 Fv-FOXP3 killed CT26 syngeneic colon cancer cells in vitro, and inhibited the development of liver metastases in vivo (68).
Mouse double minute homolog (MDM2), an E3-type ubiquitin ligase, down-regulates the function of p53 by repressing its transcriptional activity. Inhibition of MDM2 can, thus, induce senescence of tumor cells (69, 70). mAb 3G5 binds to MDM2 and thus blocks binding of MDM2 to p53. A bispecific 3E10 3G5 antibody created by genetic linkage of two scFvs retained both cell-penetrating properties and MDM2-binding activities, which resulted in increased tumor p53 levels and growth inhibition of MDM2-addicted tumors (71).
mAb-based Immunotherapy Against Intracellular Targets
Proof-of-concept experiments for mAb-based immuno-therapy directed against intracellular cancer-related targets have been performed in several systems for members of the phosphatase of regenerating liver (PRL) family. Dysregulation of protein tyrosine phosphatases resulting in aberrant tyrosine phosphorylation is implicated in cancer formation and progression (72). PRLs have been characterized as farnesylated proteins and were shown to be associated with the inner leaflet of the plasma membrane (73). Global gene expression profiling has implicated PRL3 in metastasis of colorectal cancer to the liver as the only gene to be highly expressed in all 18 liver metastases investigated (74). There is evidence that PRL1 and PRL2 are also involved in metastasis (75). Expression of PRL3 was detected in developing heart, blood vessels and erythrocytes, but not in their corresponding mature tissues (76). PRL3 overexpression promotes cell migration, invasion and metastasis (77) and is found in many types of cancer such as acute myeloid leukemia (AML) and lung cancer (31% squamous cell carcinoma, 26% adenocarcinoma) (77). From the mode of action point of view, activation of the human v-akt thyoma viral oncogene homolog (AKT) pathway by PRL3 and its tight regulation by poly C-binding protein (PCBP1), which suppresses the translation of PRL3, seem to have a significant impact on PRL3 function in cancer (78). Activation of AKT signaling by PRL3 is based on down-regulation of phosphatase and tensin homolog expression and activation of phospho-inositol-3-kinase signaling to promote epithelial mesenchymal transition (79). Involvement of PRL3 in endothelial recruitment and new blood vessel formation underlines the role of PRL3 as a multi-tasking phosphatase (76). Loss of transforming growth factor-β signaling, which often occurs during progression of colorectal cancer, was shown to activate PRL3 by releasing product of sma gene (SMA) and MAD-dependent inhibition of PRL3 transcription (79).
An important proof-of-concept experiment demonstrated that mAbs directed against intracellular PRL phosphatases inhibited cancer metastases in mice (80).
Chinese Hamster Ovary (CHO) cells expressing EGFP PRL1 or EGFP PRL3 fusion proteins, or enhanced GFP were injected into nude mice. Mice treated with PRL1- or PRL3-specific mAbs showed 90% inhibition of lung metastases. PRL1-specific mAbs inhibited metastasis of PRL1-, but not PRL3-expressing cells, while PRL3 specific mAbs blocked tumor formation of PRL3-, but not PRL1 expressing cells. In addition, metastasis formation by A2780 human ovarian cancer cells, which express endogenous PRL3, was blocked by PRL3 antibodies, whereas the PRL3 antibodies had no impact on metastasis of CT26 mouse colon cancer cells which do not express the PRL3 protein (80). In vitro, 10% of non-permeabilized MCF-7 breast cancer cells took up mouse anti-GS28 directed against a Golgi marker, and rabbit anti-p53, with enhancement of uptake to 70% of cells after serum starvation. No uptake was noticed in non-permeabilized mammary epithelial cells (MCF-10A). Uptake of antibody was inhibited by endosome inhibitor NH4Cl.

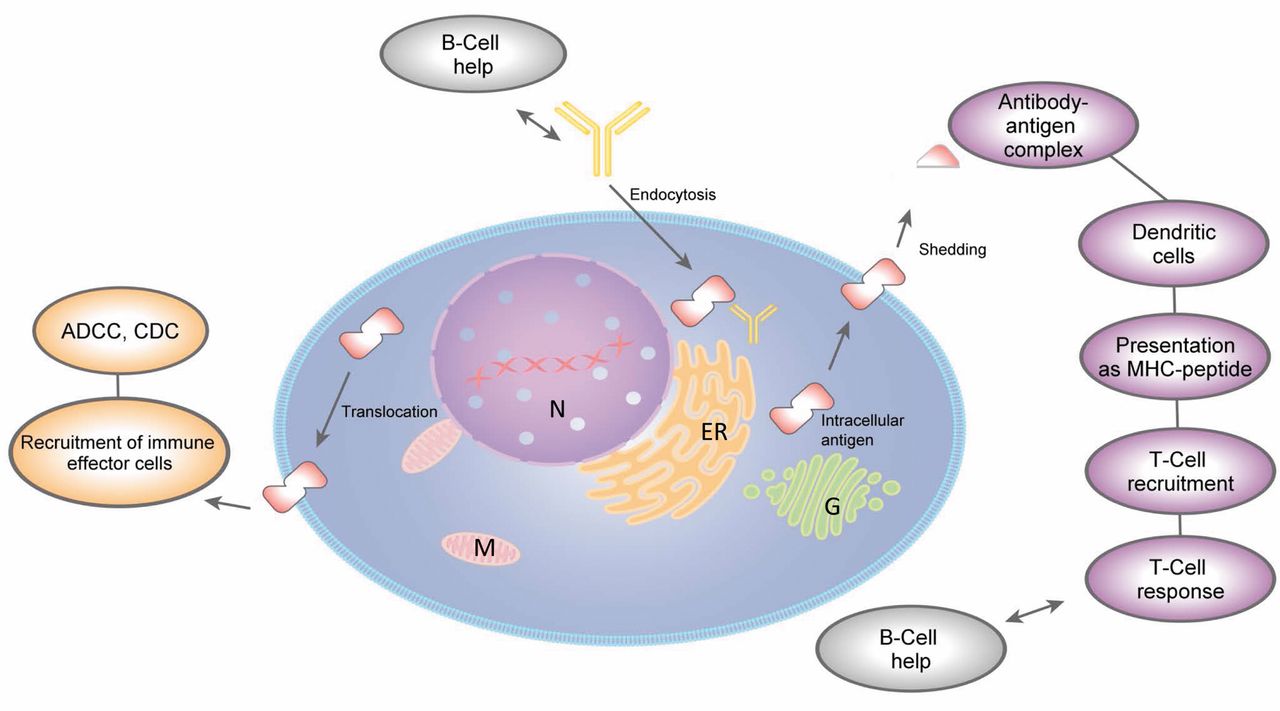
Antibodies directed against intracellular tumor antigens: possible mechanisms for generation of an anti-tumoral response. The following mode of-action scenarios are outlined: intracellular interaction of antigen with antibody, recruitment of immune effector cells to antigens translocated to the plasma membrane; generation of a T-cell response against the tumor antigen based on its translocation to the plasma membrane; shedding and formation of antigen antibody complexes. ER, Endoplasmic reticulum; G, Golgi apparatus; M, mitochondria; N, nucleus; MHC, major histocompability complex; ADCC, antibody-dependent cytotoxicity; CDC, complement-dependent cytotoxicity. Ingenuity Pathway Analysis Path Designer tool (IPA) was used to prepare this figure.
A chimeric anti-PRL3 human IgG1 antibody was evaluated with respect to inhibition of lung metastasis formation after tail vein injection of B16 melanoma cells with high and low expression of endogenous PRL3, respectively (81). Significant inhibition of metastasis was seen only in the high PRL3-expressing cell line in nude mice. These findings were extended to the PRL3 high-expressing human colorectal HCT-116 and human ovarian A2780 cell lines, whereas PRL-3-negative human lung cancer cell line NCI-H460 was unaffected. Depletion experiments indicated the functional involvement of B-cells by comparison of the antibody-mediated efficacy between nude mice lacking mature T-cells and severe combined immunodeficiency (SCID) mice lacking both mature B- and T-cells (81). Dependence of anti-tumor activity on B-cells has also been shown for a death receptor DR5-specific mAb in mouse breast and colon adenocarcinoma models (82). PRL3 localized to the cell surface was demonstrated in the cell lines under investigation. This finding was supported by the observation that calreticulin, another intracellular protein, is expressed on the cell membrane when cells are exposed to chemotherapeutic agents or radiation (83). As an extension to a more relevant model for clinical application, in vivo experiments were performed in immune-competent C57BL/6 mice (84). Here, an IgG1 antibody against PRL3 effectively retarded growth of metastatic tumors that expressed endogenous PRL3, and the efficacy of treatment highly correlated with the expression of endogenous PRL3. B-cells enhanced internalization of these antibodies into cancer cells in vivo. As an extension of these experiments, an IgG2a-antibody directed against enhanced GFP was evaluated in melanoma cells overexpressing enhanced GFP and corresponding control cell lines. Results indicated that only enhanced GFP-expressing cell lines responded to the mAb therapy. In MMTV-PymT transgenic mice, middle T-antigen (mT)-specific rat IgG2b antibody prevented progression of mT-expressing mammary tumors. Antigen-induced antibodies (by vaccination) in C57BL/6 mice prevented formation of PRL3- or enhanced GFP-expressing tumors, and the formation of mammary gland mT tumors was retarded in female MMTV-PymT transgenic mice vaccinated with mT antigen. Thus, in cancer genetically predisposed, vaccination with an appropriate oncogenic protein was able to prime the immune system against the oncogenic protein. Unfortunately, data covering treatment of established, solid syngeneic or xenograft tumors are not yet available. In addition, the mode of action of mAbs directed against intracellular antigens has not yet been properly dissected and might be an amalgamate of several mechanisms (Figure 3). B-Cell-facilitated internalization of mAbs might initiate a cell death-inducing program with a yet unresolved mode of action. Tumor antigens could move to the cell surface and be recognized by antibodies directed against the specific antigen and might thus induce antibody-dependent cellular cytotoxicity by recruitment of natural killer cells via the Fc moiety of the antibody. Tumor antigens might be shed to the tumor microenvironment or into the circulation and form complexes with the corresponding mAbs. These complexes could then be taken up by dendritic cells which process the corresponding antigen and present the resulting tumor antigen-derived peptides to natural killer cells which mediate tumor cell killing after activation. It is claimed that T-cells do not play a role in the antitumor activity of intracellular tumor antigen mAbs in nude mice. It remains to be tested, however, whether these conclusions are also valid for immune-competent mice (85, 86).
Conclusion
Intracellular expression of antibodies is an important method for target validation of PPIs and is complementary to RNA-based approaches which result in knock-down of the corresponding proteins (87-91). Recombinant expression of tumor-specific antibodies via plasmid-based or viral delivery is dependent on the identification of selective tumor antigens and hampered by the immunogenicity of viral delivery vectors. Overall, improvements in delivery of constructs for the expression of antibody-based molecules in human tumors is progressing slowly and will require significant breakthroughs to achieve successful clinical applications. The demonstration that some autoimmune disease-related antibodies can be taken-up by mammalian cells is a major achievement in the field, allowing molecules based on these antibodies to interfere with intracellular PPIs through delivery of antagonistic peptides, proteins or antibody-based moieties. The efficacy of antibodies directed against intracellular antigens as shown for selected antigens with respect to inhibition of metastasis is probably based on a combination of mechanisms which need to be resolved in more detail, but may open an avenue for new achievements. The characteristics of intracellular antigens for which this technology is applicable also need to be worked out. Finally, the demonstration of treatment efficacy for established, solid syngeneic or xenograft tumors is a pending issue.
- Received October 8, 2013.
- Revision received November 7, 2013.
- Accepted November 8, 2013.
- Copyright© 2013, International Institute of Anticancer Research (Dr. John G. Delinasios), All rights reserved
References
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- Systems for Intracellular Expression of Antibodies
- Intracellular Monoclonal Antibodies (mAbs) Directed Against Oncogenic Kinases
- Cell-penetrating Antibodies
- Cellular Uptake of Modified Antibodies
- Antibody 3E10-based Internalizing Moieties
- mAb-based Immunotherapy Against Intracellular Targets
- Conclusion
- References
- Figures & Data
- Info & Metrics