


Abstract
In addition to chemotherapy, targeted therapies have been approved for treatment of locally advanced and metastatic gastric cancer. The therapeutic benefit is significant but more durable responses and improvement of survival should be achieved. Therefore, the identification of new targets and new approaches for clinical treatment are of paramount importance. In this review, we searched the literature for down-regulated microRNAs which interfere with druggable targets and exhibit efficacy in preclinical in vivo efficacy models. As druggable targets, we selected transmembrane receptors, secreted factors and enzymes. We identified 38 microRNAs corresponding to the criteria as outlined. A total of 13 miRs target transmembrane receptors, nine inhibit secreted proteins and 16 attenuate enzymes. These microRNAs are targets for reconstitution therapy of gastric cancer. Further target validation experiments are mandatory for all of the identified microRNAs.
- Apoptosis
- invasion
- proliferation
- microRNA mimetics
- reconstitution therapy
- transmembrane receptors
- secreted factors and enzymes
- target validation
- xenograft models
- review
Gastric cancer (GC) is the third-leading cause of cancer worldwide and is the fourth most common cancer with an annual death toll of 700 000 worldwide (1). From a molecular point of view the following subtypes have been characterized: Epstein–Barr virus, microsatellite instability, genomically stable and chromosomal instability subtypes, all correlate with differential prognosis (2). Of all GCs, 90% are adenocarcinomas which arise from the glandular epithelium (2). The only curative treatment is surgery. Neo-adjuvant and adjuvant treatment are integrated with chemotherapy and radiation, nevertheless the 5-year survival rate for patients with locally advanced disease is less than 30% and in the metastatic setting, the median survival is in the range of 1 year (3-5). Preferential organs of metastasis are the liver (48%), peritoneum (32%), lung (15%) and bone (12%) (6). New treatment modalities have been introduced, such as trastuzumab in the subclass of patients with human epidermal growth factor receptor 2 (HER2)-positive tumors, and ramucirumab as second-line treatment or in combination with paclitaxel (3-5). More recently, immune-checkpoint inhibitory monoclonal antibodies (mAbs) against programmed cell death protein 1 (PD1), such as nivolumab and pembrolizumab, have been approved for patients with heavily pre-treated GC (3-6). Promising clinical studies are ongoing in claudin 18.2-positive GCs and in those with fibroblast growth factor receptor 2 (FGFR2) amplification (3-6). Nevertheless, there is an urgent need to identify new targets and treatment modalities which lead to durable responses and improved survival. Many of the recently identified targets, e.g., those involved in epigenetic modification or tumor suppressors, are undruggable or difficult to interfere with (7-9). In this review, we focus on microRNAs (miRs) which are down-regulated in GC and interfere with controllable targets (transmembrane receptors, secreted factors and enzymes) with demonstrated efficacy in GC-related preclinical in vivo models. The identified targets need to be validated for treatment of GC and concomitantly the identified miRs can be evaluated as tools for reconstitution therapy.
miRs in Cancer
miRs are synthesized in the nucleus as pri-miRs containing a hairpin loop, processed to pre-miR hairpin structures, and are finally exported to the cytoplasm (10, 11). Subsequently, pre-miRs are cleaved by endoribonuclease DICER to produce a 22-nucleotide miR duplex with 5’phosphate and a two-nucleotide overhang at each end (10, 11). One strand of the 22-nucleotide duplex is maintained (guide strand), the other strand (passenger strand) is degraded (12, 13). Binding of the guide strand to the 3’-untranslated region of the corresponding mRNA leads to degradation or translational repression of the target mRNA (12, 13). In contrast to small interfering RNAs which target a single mRNA species, a single miR species may repress up to 100 mRNAs and vice versa each mRNA can be inhibited by up to 100 different miRs. This indicates the potential of miRs to modulate several pathways and cellular networks (14). In oncology, miRs can affect the whole cascade of oncogenic events from tumor initiation, tumor progression and metastasis, as well as angiogenesis, to interactions with the immune system and the tumor microenvironment (15-17). Several miRs may act as oncogenes or tumor suppressors in a context-dependent way, depending on the cell-type in which they are expressed (18). In a proof-of-concept (POC) experiment, genetic deletion of the miR-15/16 cluster in mice recapitulated the features of chronic lymphocytic leukemia, supporting their role as tumor suppressors (19). An oncogenic role was identified for miR-221 which induced hepatocellular carcinoma in transgenic mice after liver-specific expression and subsequently many other examples have followed (20). We recently summarized the role of miRs in metastasis of breast, ovarian, prostate, non-small-cell lung carcinoma and pancreatic cancer (21-25). In this review, we focus on miRs down-regulated in GC which have controllable targets and which mediate efficacy in preclinical GC-related in vivo models after reconstitution therapy.
Transmembrane Receptor Tyrosine Kinases
miR-7 and miR-133. miR-7 (Figure 1A) was shown to be down-regulated in GC cell lines and its expression was inversely correlated with metastasis (26, 27). miR-7 suppressed migration of 9811-P GC cells in Matrigel-based transwell assays and tail vein injection of GC9811-P cells stably expressing miR-7 gave rise to reduced numbers of metastatic nodules in the liver of nude mice (26). miR-133a (Figure 1) was shown to inhibit proliferation of GC cell lines, their colony formation, migration, invasion, epithelial–mesenchymal transition (EMT), inducing apoptosis and repressing tumorigenicity of GC cell lines SGC-7901 and MGC-803 in nude mice (27). Both miRs target insulin-like growth-factor receptor 1 (IGF-1R), a transmembrane tyrosine kinase receptor which is overexpressed in many tumor types and acts as an oncogene by conferring anti-apoptotic, pro-survival and transforming properties (28). Expression of IGF-1R was shown to be positively correlated with poor prognosis in patients with GC (29). Clinical studies targeting IGF-1R with mAbs or small molecules are currently underway in several types of cancer (30-32). Data from The Cancer Genome Atlas (TCGA) indicated that the steady-state RNA level of miR-133 was down-regulated in GC tissues, however, miR-7 was up-regulated in tumor tissues (Figure 2).

Down-regulated miRs targeting transmembrane receptors with activity in gastric cancer-related in vivo models. Targets and downstream effectors are shown. A: miR-7, miR-133, miR-27-3p, miR-302b, miR-7-5p, miR-22 and miR-29c; B: miR-148a, miR-218, miR-338, miR-381, miR-573, miR-874 and miR-993. AKT: Serine-threonine AKT; Angio: angiogenesis; AQP3: aquaporin 3; BCL2: BCL2 apoptosis regulator; CCK-BR: cholecystokinin receptor B; COX2: cyclo-oxygenase 2; EPHA2: erythropoietin-producing hepatocellular carcinoma receptor A2; ERK1/2: extracellular signal-regulated kinase; FGF18: fibroblast growth factor 18; GLI: zinc finger protein GLI; IGFR-1R: insulin growth factor receptor 1; ITGβ1: integrin β1; JAK2: Janus kinase 2; MAPK: mitogen-activated protein kinase; MEK: MAPK kinase; MMP2,9: matrix metalloproteinase 2,9; MT1-MMP: membrane-type matrix metalloproteinase-1; MTDH: metadherin; NFκB: nuclear factor κB; PI3K: phosphatidylinositol-4,5-bisphosphate 3-kinase; PLCγ: phospholipase Cγ; RAF: rapidly accelerated fibrosarcoma; RAS: rat sarcoma; ROBO1: roundabout1; ROR1: tyrosine kinase-like orphan receptor 1; SLC34A2: solute carrier family 34, member 2; SMD: smoothened; STAT3: signal transducer and activator of transcription 3; TGFβ: transforming growth factor β; TMEM16A: transmembrane channel 16A; TSPAN1: tetraspanin 1; WNT: WNT signaling.

Expression of selected miRs in gastric adenocarcinoma compared to normal tissues. Data are shown for miR-133, miR-148a, miR-218, miR-22, miR-27a, miR-29c, miR-338, miR-381, miR-573, miR-7 and miR-874. Data from 436 tumor samples and 41 normal stomach samples derived from The Cancer Genome Atlas are shown. miR expression was quantified by RNA sequencing and is shown as log2 of normalized read counts. The red lines indicate lower versus higher expression around 100 counts. Expression data are shown as box plots. The line represents the median value, the rectangles show the upper and lower 25% quartiles, and 50% of all data points are included in the greater rectangle. All of the data points, except for the outliers are located within the upper and lower whiskers.
miR-27-3p. miR-27-3p (Figure 1A) was shown to suppress cell proliferation and induce cell-cycle arrest in BGC-823 cells by targeting receptor tyrosine kinase-like orphan receptor 1 (ROR1) (33). AGS GC cells co-transfected with a miR-27-3p inhibitor gave rise to larger tumors after subcutaneous implantation into nude mice (33). Tumorigenicity of BGC-823 cells was suppressed after transfection with a miR-27-3p mimic and subcutaneous implantation into nude mice (33). It was shown that ROR1 induces the SRC/signal transducer and activator of transcription 3 (STAT3) signaling pathway, which activates c-MYC, cyclin D1 (CCND1) and subsequently proliferation of GC cells (33). ROR1 promoted the G0/G1 to G1/S transition in GC cells and was up-regulated in GC tissues compared to paired adjacent tissues (33). ROR1 is an oncofetal antigen for tumor therapy and acts as a survival factor for cancer cells (34). Hematological cancers have been the focus as a target indication of ROR1 inhibitors (35). ROR1 is highly expressed in GC (36). More comprehensive ROR1-related target assessment would provide a clearer picture of the validity of this target for the treatment of GC. Data from TGCA indicate that miR-27a was up-regulated in GC tissues in comparison to corresponding normal tissues (Figure 2).
miR-302b. miR-302b (Figure 1A) inhibited cell-cycle progression and Matrigel-based invasion of AGS and SGC-7901 GC cells by targeting transmembrane tyrosine kinase erythropoietin-producing hepatocellular carcinoma receptor A2 (EPHA2) (37). Down-regulation of EPHA2 by miR-302b suppressed EMT (37). miR-302b negatively regulated EPHA2/WNT/β catenin signaling (37). Smaller tumors were observed for SGC-7901 cells transfected with miR-320b after subcutaneous implantation into nude mice and after tail injection, and inhibition of lung metastasis was observed (37). Activation of EMT by EPHA2 in GC has been described independently (38). EPHA2 was shown to be associated with poor survival in patients with GC (39).
Other Transmembrane Receptors
miR-7-5p. miR-7-5p (Figure 1A) was shown to be down-regulated in GC stem cells positive for cluster of differentiation 44 (40). Overexpression of miR-7-5p in GC stem cells led to inhibition of colony formation and reduced invasion and tumor growth inhibition in a xenograft model in nude mice (40). Smoothened and transcription factor HES1 were identified as targets of miR-7-5p (40, 41). Smoothened has been identified as a G-protein-related transmembrane receptor which plays a role in neoplastic transformation, GC development and cancer–stroma interaction (42, 43).
miR-22. miR-22 (Figure 1A) was shown to be down-regulated in GC and associated with advanced clinical progression and lymph node metastasis (44). Metadherin (MTDH) was identified as a direct target of miR-22 (44). Expression of MTDH was inversely correlated with miR-22 levels in GC (44). miR-22 inhibited GC cell proliferation and invasion by targeting MTDH in SGC-7901 cells (44). SGC-7911 cells expressing miR-22 gave rise to less lung metastatic nodules after tail vein injection in nude mice (44). MTDH was shown to be overexpressed in several types of cancer and to modulate pathways such as phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K)/AKT, nuclear factor κB (NFκB), mitogen-activated protein kinase (MAPK) and WNT/β catenin signaling (45, 46). In GC, MTDH promoted the malignant phenotype and induced EMT (47). Data derived from TCGA did not confirm that miR-22 was down-regulated at the RNA level in GC tissues (Figure 2).
miR-29c. Expression of integrin β1 (ITGβ1) was found to be reduced in GC in comparison to corresponding normal tissues (48). Ectopic expression of miR-29c (Figure 1A) in GC cell lines inhibited proliferation, adhesion, invasion, migration and suppressed xenograft tumor growth in nude mice (48). miR-29c expression stepwise decreased during hyperplasia–dysplasia transition in transgenic mice models of GC (48). Loss of ITGβ1 expression is an early step in GC carcinogenesis (48). ITGβ1 was shown to play an important role in angiogenesis, sustained proliferative signaling, resistance to cell death, evasion of immune destruction and metastasis of several types of cancer (49-51). Data derived from TGCA have confirmed that miR-29c was down-regulated in GC tissues at the RNA level in comparison to corresponding normal tissues (Figure 2).
miR-148a. miR-148a (Figure 1B) was shown to be down-regulated in GC tissues and cell lines in comparison to corresponding controls (52). miR-148a inhibits proliferation and migration in NCI-N87, SGC-7901 and MKN-45 GC cell lines (52). Ectopic expression of miR-148a attenuated tumorigenicity of GC cell lines after their subcutaneous implantation in nude mice (52). Receptor for cholecystokinin B (CCK-BR) was identified as a target for miR-148a (52). Its ligand, cholecystokinin B is a peptide hormone secreted by enteroendocrine cells which, together with gastrin, binds to CCK-AR and CCK-BR in the gastrointestinal-tract and the brain (53, 54). CCK-BR is overexpressed by many GCs and when activated by gastrin stimulates tumor growth by an autocrine mechanism (53, 54). Gastrin acting on CCK-BR induces cyclo-oxygenase 2 (COX2) and janus kinase 2 (JAK2)/STAT3/PI3K/AKT signaling (55). Data derived from TCGA did not reveal a difference in RNA steady-state levels of miR-148a in GC tissues in comparison to corresponding normal tissues (Figure 2).
miR-218. miR-218 (Figure 1B) was found to be decreased in GC, and was associated with advanced clinical stage, lymph node metastasis and poor patient prognosis (56). In MKN28 GC cells transfected with miR-218, no effect on proliferation or the cell cycle was noticed, however, cell migration and invasion were inhibited (56). After tail vein injection, complete inhibition of liver and lung metastases was observed in nude mice (56). Roundabout 1 (ROBO1) was identified as a direct target of miR-218 (56). ROBO1 is part of a gene family containing four members with five cytoplasmic immunoglobulin domains each, three fibronectin type III repeats, a transmembrane domain and four conserved motifs in the cytoplasmic domain. The ROBO- family interacts with three types of SLIT ligands (57). The SLIT/ROBO pathway is involved in axonal repulsion, axon guidance, neuronal migration in the nervous system and formation of the vascular system (57). ROBO1 was found to be overexpressed in cancer cells and to promote tumor angiogenesis and metastasis via interaction with SLIT ligands (58). In GC, contradictory functions for SLIT/ROBO signaling have been reported by different groups (59, 60). The role of the SLIT/ROBO pathway as a target for development of anticancer drugs is under active investigation (61). Data derived from TGCA revealed that miR-218 is down-regulated in GC tissues in comparison to corresponding normal tissues (Figure 2).
miR-338. miR-338 (Figure 1B) was shown to be down-regulated in GC tissues and cell lines (62). miR-338 directly targeted neuropilin1 (NRP1), inhibited proliferation, migration, invasion and promoted apoptosis in AGS and MKN-45 GC cells (62). miR-338 inhibited EMT and phosphorylation of extracellular regulated kinase 1/2 (ERK1/2), p38 MAPK and AKT via NRP1 (62). miR-338 ectopically expressed in AGS and MKN-45 GC cells reduced tumor growth which was restored by expression of NRP1 (62). NRP1 is a multifactor non-tyrosine kinase receptor which is involved in development of the nervous system and which acts as a receptor for VEGFA in both endothelial and tumor cells and affects tumor growth as a co-receptor for VEGF receptor and transforming growth factor β receptor I (TGFβRI) and TGFβRII, platelet-derived growth factor, receptor tyrosine kinase c-MET and semaphorin 3A. NRP1 promotes proliferation, migration, invasion and survival of tumors cells, as well as angiogenesis (63, 64). A mAb directed against NRP1 is presently in clinical studies in patients with solid tumors (65). Data derived from TCGA revealed that miR-338 was slightly down-regulated at the RNA level in GC tissues in comparison to corresponding normal tissues (Figure 2).
miR-381. miR-381 (Figure 1B) was found to decreased in GC tissues and was associated with adverse clinicopathological features and poor prognosis (66). miR-381 inhibited proliferation, invasion and migration of AGS and BGC-823 GC cells, down-regulates the EMT phenotype and suppresses transforming growth factor β (TGFβ) signaling (66). Transmembrane channel 16A (TMEM 16A) has been identified as a target of miR-381 (66). In vivo, miR-381 inhibited tumor growth of AGS and BGC-823 GC cells after subcutaneous implantation into nude mice and lung metastasis after tail vein injection (66). TMEM 16A is a voltage-gated calcium-activated anion channel which acts as a chloride channel (67). TMEM 16A mediates cell invasion by induction of TGFβ secretion, EMT and MAPK signaling in GC and several types of cancer (68-70). TMEM 16A is also involved in cell proliferation, transepithelial iron transport, neuronal excitability and many other physiological functions. The structure of TMEM 16A has not yet been resolved (71). Data derived from TCGA revealed that miR-381 was slightly down-regulated at the RNA level in GC tissues in comparison to corresponding normal tissues (Figure 2).
miR-573. Knock-down of tetraspanin 1 (TSPAN1) led to inhibition of proliferation and invasion of BGC-23 and HGC-27 GC cells (72). BGC-823 and HGC-27 cells expressing miR-573 (Figure 1B) suppressed tumorigenicity after subcutaneous implantation into nude mice (72). TSPAN1 is up-regulated in GC and TSPANs plays a central role in this type of tumor (73, 74). TSPAN1 promoted EMT and metastasis of cholangiocarcinoma via PI3K/AKT signaling (75). TSPANs are associated with proliferative status and metastasis of cancer cells (76, 77). Targeting of TSPANs is subject of several drug discovery efforts (78). Data derived from TCGA indicated very low expression of miR-573 and equal RNA steady-state levels in GC tissues and corresponding normal tissues (Figure 2).
miR-874. miR-874 (Figure 1B) was shown to target aquaporin 3 (AQP3) and its ectopic expression in GC cell lines suppressed growth, migration, invasion and promotes apoptosis in vitro (79). In vivo, miR-874 suppressed tumorigenicity of GC cells after subcutaneous implantation into nude mice (79). Concomitantly, miR-874 down-regulated BCL2 apoptosis regulator (BCL2), membrane-type matrix metalloproteinase 1 (MT1-MMP) and matrix metalloproteinases 2 and 9 (MMP2 and -9) (79). AQPs consist of monomers possessing six transmembrane domains and form homotetramers mediating water transport, membrane permeability and energy homeostasis (79). AQP3 is found at the basolateral plasma membrane of human gastric mucosal tissues, mediates cancer cell proliferation, migration, angiogenesis, metastasis and promotes stem-like properties of human GC cells by activating WNT/β catenin signaling (80-82). AQP3 expression was associated with poor prognosis of GC (83). The downstream effects of AQP3 need to be resolved in more detail to identify markers for drug discovery projects. Data derived from TCGA indicate that mR-874 was slightly more highly expressed in GC tissues in comparison to corresponding normal tissues (Figure 2).
miR-993. miR-993 (Figure 1B) was found to be down-regulated in GC and low expression was correlated with poor clinical outcome (84). miR-993 suppressed proliferation, invasion and migration of SGC-7901, MNK-45 and AGS GC cell lines in vitro (84). In an orthotopic xenograft model, miR-993 inhibited growth of miR-993-transfected SGC-7901 cells, attenuated phosphorylation of ERK1/2 and the rapidly accelerated fibrosarcoma (RAF)/MAPK kinase (MEK)/ERK pathway (84). Solute carrier family 34 member 2 (SLC34A2) was identified as a direct target of miR-993 (84). SLC34A2 is a transmembrane receptor and transports inorganic phosphate into epithelial cells via sodium ion cotransport (85). The oncogenic role of SLC34A2 remains to be resolved in more detail.
Secreted Factors
miR-24. miR-24 (Figure 3A) was shown to be down-regulated in GC tissues compared to non-matched tumor tissues (86). miR-24 expression in SGC-7901 cells inhibited proliferation, and led to cell-cycle arrest in G0/G1 phase and apoptosis (86). Regenerating island-derived protein (REG4) was identified as a direct target of miR-24 (86). miR-24 overexpression inhibited tumorigenicity of SGC-7901 cells after subcutaneous implantation into nude mice (86). REG4 is a small secretory protein expressed in gastrointestinal organs and up-regulated in gastrointestinal tumors (87, 88). REGs belong to the calcium-dependent lectin (C-type) superfamily and REG4 contains a single carbohydrate recognition domain (86). REG4 induces proliferation, regeneration, carcinogenesis, survival, activates epidermal growth factor receptor (EGFR)/AKT and is associated with poor prognosis in GC (87, 88). Identification of receptors and components of downstream signaling should be resolved for REG4 target validation in GC.
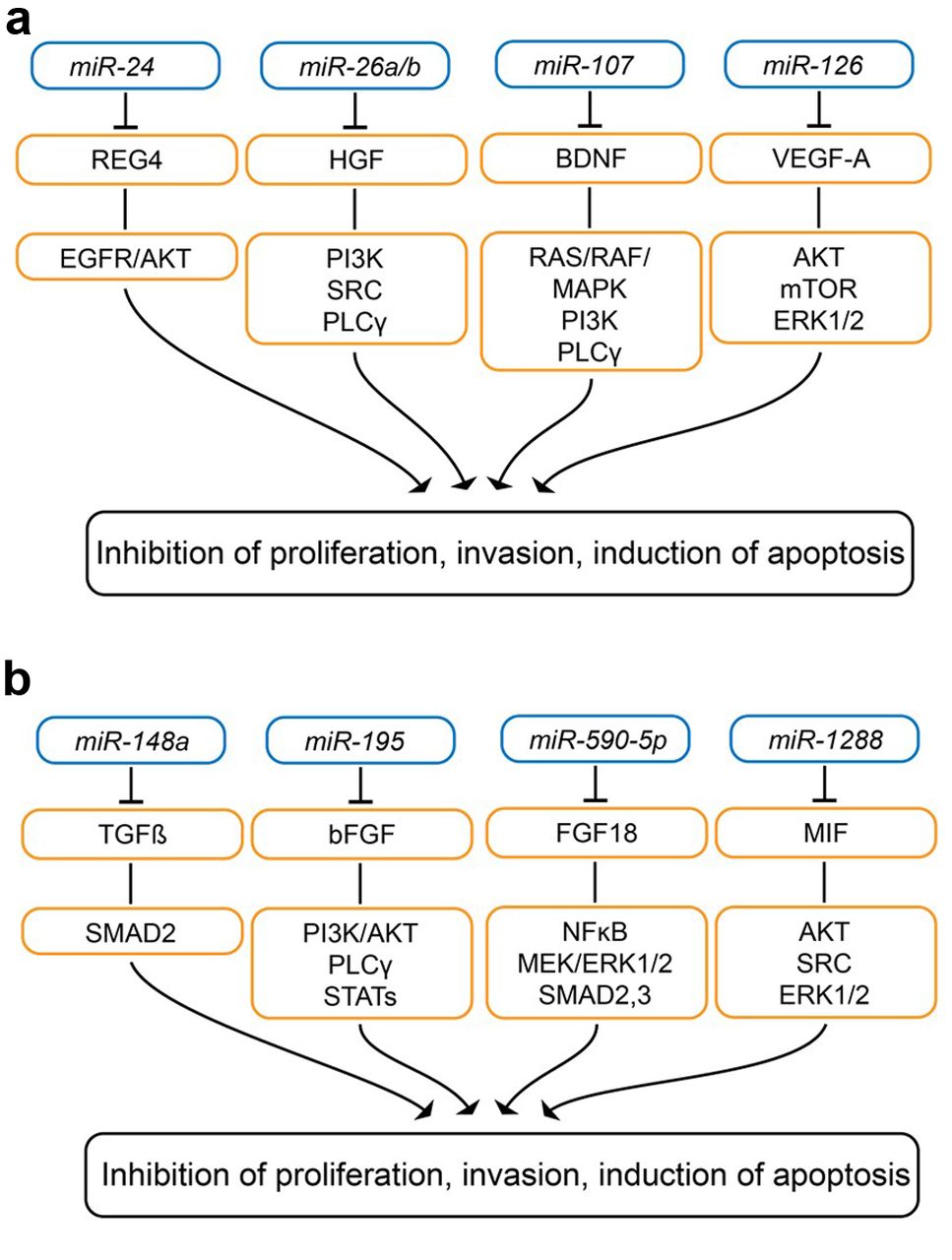
Down-regulated miRs targeting secreted factors with activity in gastric-cancer related in vivo models. Targets and downstream effectors are shown. A: miR-24, mIR-26a/b, miR-107 and miR-126; B: miR-148a, miR-195, miR-590-5p, miR-338 and miR-1288. AKT: v-AKT murine thyoma viral oncogene homolog ; BDNF: brain-derived neurotrophic factor; bFGF: basic fibroblast growth factor; EGFR: epidermal growth factor receptor; ERK 1/2: extracellular signal-regulated kinase; MAPK: mitogen-activated protein kinase; MIF: macrophage inhibitory factor; mTOR: mechanistic target of rapamycin; NFκB: nuclear factor κB; NRP1: neuropilin; p38 MAPK: p38 mitogen-activated protein kinase; PI3K: phospho-inosite-3-kinase; PLCγ: phospholipase Cγ; RAS: rat sarcoma; REG4: regenerating islet-derived protein 4; SMAD2,3: small mothers against decapentaplegic homolog 2/3 ; SRC: tyrosine kinase SRC; STAT: signal transducer and activator of transcription; TGFβ: transforming growth factor β; VEGF-A: vascular endothelial growth factor A.
miR-26a/b. miR-26a/b (Figure 3A) was shown to be down-regulated in serum and tissues of patients with GC (89). miR-26a/b inhibited proliferation and migration of MGC-803 and SGC-7901 GC cells (89). Hepatocyte growth factor (HGF) was identified as a target of miR-26a/b (88). Overexpression of HGF in GC cells stimulated increase in VEGF expression (89). miR-26a/b ectopically expressed in MGC-803 GC cells inhibited tumorigenicity and vessel growth in nude mice after subcutaneous implantation, whereas HGF strongly boosted vessel and tumor growth (89). HGF–c-MET interaction plays a pivotal role in the growth, survival and invasiveness of GC and aberrant activation of this pathway is correlated with poor clinical outcome (90). Several mAbs directed against HGF and c-MET, as well as selective c-MET tyrosine kinase small-molecule inhibitors, are presently under clinical investigation in patients with GC (90-93).
miR-107. Transfection of SGC-7901 cells with miR-107 (Figure 3A) reduced cell proliferation, wound healing and migration in transwell assays, and inhibited tumor growth of these cells in nude mice after subcutaneous implantation (94). Brain-derived neurotrophic factor was identified as a target of miR-107 (94), interacts with tropomyosin receptor tyrosine kinase B and mediates cancer cell growth, proliferation, survival and EMT by stimulating PI3K, RAS/RAF/MAPK and phospholipase C signaling, and transactivation of EGFR (95). Several compounds targeting TRKB are under clinical investigation in several types of cancer (95).
miR-126. miR-126 (Figure 3A) inhibited proliferation of SGC-7901 GC cells and ectopic expression of miR-126 in SGC-7901 GC cells attenuated tumorigenesis in vivo (96).
miR-126 was inversely correlated with VEGF-A protein and microvessel density in GC tissue (96). VEGF-A was identified as a direct target of miR-126 (96). In SGC-7901, MKN-28 and MKN-45 GC cells, miR-126 also was shown to inhibit VEGF-A downstream targets such as AKT, mechanistic target of rapamycin (mTOR) and ERK1/2 (96). VEGF-A affects division, proliferation and migration of endothelial cells (97, 98). Ramucirumab, which inhibits binding of VEGF-A, -C and -D to VEGF receptor 2, is approved for treatment of advanced and metastatic GC (99).
miR-148a. miR-148a (Figure 3B) reduced proliferation, invasion and migration of AGS, YCL3 and SCA GC cells (100). TGFβ2 and small mothers against decapapentaplegic homolog 2 (SMAD2) were identified as direct targets of miR-148a (99). N-Methyl-N-9-nitro-N-nitrosoguanidine induced GC in rats and increased expression of TGFβ2 and SMAD2 in rat gastric tissues (100). Tumor growth of BGC-823 GC cells ectopically expressing miR-148a was reduced after subcutaneous implantation into nude mice (100). SMAD2 is a downstream effector of TGFβ2, therefore miR-148 increased inhibition of TGFβ2 signaling (101, 102). TGFβ1 and -2 play dual roles in gastrointestinal tumor development and progression and can act both as a tumor promoter and TS depending on the stage of carcinogenesis (103). Elevated levels of TGFβ1/2 were correlated with lymph node metastasis, poor prognosis and worse survival in patients with GC (104).
miR-195 and miR-590-5p. miR-195 (Figure 3B) inhibited migration and invasion by SNU-1 and KATO3 GC cell lines by targeting basic fibroblast growth factor (bFGF), also known as fibroblast growth factor 2 (105). Ectopically expressed miR-195 inhibited tumorigenesis in a xenograft mouse model, which was restored by re-expression of bFGF (105). An inverse correlation of expression has been noted between miR-195-5p and bFGF in human GC tissues (105). bFGF binds to FGF receptors 1, -2 and -4, acts as a mitogen for tumor and stromal cells, stimulates angiogenesis and activates rat sarcoma (RAS)/MAPK, PI3K/AKT, phospholipase Cγ and STATs (106, 107). bFGF has been identified as a prognostic factor for patients with GC and several antagonists (mAbs and small molecules) are presently under clinical investigation in several types of cancer (106-108).
miR-590-5p inhibited proliferation of AGS and MKN28 GC cell lines (109). Xenograft formation was inhibited in miR-590-5p transfectants of these cell lines (109). FGF18 was identified as the target of miR-590-5p (109). Autocrine secretion of FGF18 promoted tumor growth of GC cell lines (109). FGF18 is a glycosylated protein which interacts with FGFR3 and FGFR4 and is abundant in GC (109). FGF18 is up-regulated in GC and is correlated with poor prognosis (109). FGF18 activates angiogenesis, NFκB, MEK-ERK signaling and SMAD2/3, key effectors of TGFβ signaling, resulting in cancer cell proliferation and migration (109-111). Several therapeutic approaches for inhibition of FGF signaling including receptor tyrosine kinase inhibitors, receptor neutralizing mAbs and FGF-ligand traps have been pre-clinically validated and are under clinical investigation (112-114).
miR-1288. miR-1288 (Figure 3B) was found to be increased in G0/G1 phase cells in GC cells and to reduce VEGF secretion (115). Conditioned media from miR-1288-overexpressing GC cells reduce the capacity and to promote endothelial cell migration and tube formation in comparison to the media of control cells (115). Macrophage inhibitory factor (MIF) was identified as a target of miR-1288 (115). In nude mice, coexpression of MIF with miR-1288 increased tumor growth and microvascular density (115). MIF interacts with cluster of differentiation 74 (CD74) as a receptor, forms a signaling complex with CD44 and SRC and activates ERK1/2, MAPK and WNT signaling, inhibits apoptosis and reduces expression of E-cadherin (116). Furthermore, MIF induces an antitumor immune response by increasing secretion of inflammatory cytokines such as tumor necrosis factor α, interferon-γ, interleukin-1β and -12 (116). MIF has at least two distinct catalytic activities as a keto-enol tautomerase and as a thiol-protein oxidoreductase, which are both druggable. The contribution of these activities to functions as described above needs still to be resolved (116). MIF is a poor prognosis factor in GC and several MIF-inhibitory agents are under preclinical evaluation as anticancer agents, but for GC, more target validation experiments are required (117).
miRs Targeting Enzymes
Ser-Thr Kinases
miR-137. miR-137 (Figure 4A) was shown to be down-regulated in GC cells and clinical samples (118). miR-137 inhibited proliferation, migration and induces apoptosis of GC cell lines HGC-27 and SGC-7901 (118). miR-137 attenuated tumor growth of HGC-27 and SGC-7901 cells ectopically expressing miR-137 after subcutaneous implantation into nude mice and lung metastases after tail vein injection (118). v-AKT murine thymoma viral oncogene homolog 2 (AKT2) was identified as a direct target of miR-137 (118). miR-137 also down-regulated AKT2 effectors glycogen-synthase 3β and BCL2-antagonist-of-cell-death (BAD) (118). AKT2 is activated by PI3K or phosphoinosite-dependent kinases, as well as growth factors, inflammation and DNA damage, and is a mediator of survival, proliferation, migration and angiogenesis of tumor cells (119). Several small-molecule AKT inhibitors are undergoing clinical studies in cancer-related indications (119, 120).

Down-regulated miRs targeting enzymes with activity in gastric cancer-related in vivo models. Targets and downstream effectors are shown. A: miR-137, miR-199b/a-3p, miR-203, miR-140-5p, miR-375, miR-1224; B: miR-146, miR-338-3p, miR-1182, miR-1207-5p, miR-1266, miR-124, miR-330-3p, miR-137, miR-516-3p and miR-5590-3p. AKT2: v-AKT murine thyoma viral oncogene homolog 2 (AKT2); BAD: BCL2 antagonist of cell death; c-MYC: transcription factor c-MYC; COX2: cyclo-oxygenase 2; DDX5: dead box protein 5; ERK1/2: extracellular signal regulated kinase 1,2; FAK1: focal adhesion kinase 1; FOXOM1: forkhead box protein M1; GSK3β: glycogen synthase 3β; JAK2: janus kinase 2; MAPK: mitogen-activated protein kinase; MEK: mitogen-activated protein kinase kinase; mTOR: mechanistic target of rapamycin; NFκB: nuclear factor κB; p21: cyclin-dependent kinase inhibitor; PAK4: p21-activated protein kinase 4; pAKT: phospho-serine-threonine kinase AKT; pERK1/2: phospho-extracellular signal regulated kinase 1,2; PI3K: phosphatidylinositol-4,5-bisphosphate 3-kinase; protein p27: cell-cycle regulatory protein; PTP-1B: protein tyrosine phosphatase 1B; SPHK1: sphingosine kinase 1; STAT3,5: signal transducer and activator of transcription 3,5; TERT: telomere reverse transcriptase; WNT: WNT signaling; YES1: protein tyrosine kinase 1.
miR-199b/a-3p. Expression of miR-199b/a-3p (Figure 4A) was shown to be reduced in GC compared to corresponding normal tissues and inhibited proliferation of MGC-803 and SGC-7901 GC cells (121). p21 activated kinase 4 (PAK4) was identified as a target of miR-199b/a-3p (121). miR-199b/a-3p mimics transfected into MGC-803 cells inhibited tumor growth and reduced the PAK4 level after subcutaneous transplantation into nude mice (121). miR-199b/a-3p suppressed PAK4/MEK/ERK signaling in MGC-803 and SGC-7901 cells (121). PAK4 silencing inhibited proliferation of MGC-803 and SGC-7901 cells (121). Furthermore, it has been shown that PAK4 binds to translation elongation factor eEF1A1 to promote GC cell migration and invasion (122). PAK4 has been shown to mediate cancer cell proliferation, invasion, metastasis, EMT, drug resistance, WNT/β-catenin signaling, to activate cAMP response element-binding protein and to play a role in actin/cytoskeletal organization (123). In patients with GC, high PAK4 expression is associated with poorer disease-specific survival (124, 125).
miR-203. Expression of miR-203 (Figure 4A) was shown to be decreased in GC (126). Ectopic expression of miR-203 in SGC-7901 cells inhibited invasion and motility, and reduced expression of phospho-ERK1/2 and SLUG (126). In nude mice, miR-203 inhibited peritoneal metastasis of SGC-7901 cells (126). ERK1/2 contributes to MEK-ERK-MAPK signaling, which plays a role in proliferation and progression of many types of tumors, and several inhibitors of this pathway have reached clinical trials (127, 128).
Non-receptor Tyrosine Kinases
miR-140-5p. miR-140-5p (Figure 4A) was shown to be down-regulated in GC tissues and down-regulation was correlated with poor patient survival (129). miR-140-5p suppressed proliferation, invasion and migration of AGS and BGC-823 GC cells in vitro (129). Proto-oncogene tyrosine kinase 1 (YES1) was identified as a target of miR-140-5p (129). Reconstitution of YES1 rescued miR-140-5p-mediates inhibition of GC cells (129). miR-140-5p suppressed growth of BGC-823 xenografts in nude mice (129). YES1 is member of the SRC-family of non-receptor kinases which is recruited to receptor tyrosine kinases and phosphorylates its substrates to activate signaling pathways. YES1 acts as a proto-oncogene and stimulates PI3K and WNT/β-catenin signaling (130). YES1 is also involved in regulation of the cell-cycle and cytokinesis (131). Gene amplification of YES1 has been observed in cancer tissues (132). In GC, more detailed investigations uncovering the role of YES1 should be performed.
miR-375. miR-375 (Figure 4A) was shown to be frequently down-regulated in GC and to inhibit proliferation of AGS and MGC-803 GC cells (133). Ectopic expression of miR-375 in MGC-803 cells suppressed growth as xenografts in nude mice (133). JAK2 was identified as a target of miR-375 (133). miR-375 down-regulation was associated with increase in JAK2 protein in GC (133). V617F mutation of JAK2 is involved in the pathogenesis and therapy of myeloproliferative disorders such as polycythemia vera, essential thrombocythemia and primary myelofibrosis (134, 135). JAK2 is a mediator of signaling of the type II cytokine receptor family, such as granulocyte-macrophage colony stimulating factor receptor, interleukin 6 receptor and erythropoietin receptor and small-molecule inhibitors of JAK2 have been generated for treatment of myeloproliferative neoplasms (136). In GC, it has been shown that matrix vitamin K-dependent carboxylation/gamma-carboxyglutamic domain protein leads to tumor progression by JAK2/STAT5 signaling (137).
miR-1224. miR-1224 (Figure 4A) was shown to be down-regulated in GC and this was correlated with lymph-node metastasis and poor prognosis (138). In MKN-7 and MKN-28 GC cells, miR-1224 inhibited cell migration and EMT (138). MKN-7 cells transfected with a miR-1224 agomir exhibited reduced lung metastasis after tail vein injection into nude mice (138). Focal adhesion kinase 1 (FAK1) was identified as a target of miR-1224 (138). miR-1224-FAK1 interaction suppressed migration and EMT of GC cells by inhibition of STAT3 and NFκB pathways (138). FAK1 is a non-receptor tyrosine kinase that mediates integrin signaling, proliferation and survival (139). The options for therapeutic intervention are small-molecule kinase inhibitors or inhibitors interfering with kinase-independent scaffolding functions. Several inhibitors are in clinical development for treatment of cancer (139-142). FAK1 has been shown to be involved in invasion and metastasis of GC (143) and inhibition of FAK1 was shown to induce apoptosis of GC cells (144).
miR-146b and miR-338-3p. An inverse correlation between miR-146b (Figure 4B) and its target protein tyrosine phosphatase 1B (PTP-1B) has been uncovered in GC tissues (145). miR-146b inhibited growth of GC cells and induced apoptosis which can be reversed by PTP-1B (145). In xenografts of miR-146-expressing GC cells, tumor growth was inhibited and expression of PTP-1B reduced (145). miR-338-3p (Figure 4B) has been shown to target PTP-1B, as well as attenuating migration and inducing apoptosis of GC cell lines MKN-45 and MGC-803 (146). In an orthotopic xenograft model in nude mice, tumor growth of MKN-45 cells transfected with miR-338-3p was inhibited. Metastasis of these cells was blocked in a dissemination model in nude mice after injection into the peritoneal cavity (146). In GC cells, PTP-1B overexpression elevated the levels of pAKT and pERK1/2 (146). PTP-1B is a non-membrane tyrosine phosphatase with tumor-suppressing and -promoting effects depending on the substrates involved and the cellular context (147). PTP-1B is a negative regulator of the insulin signaling pathway and is a potential target for treatment of type 2 diabetes (148). For GC, the role of PTP-1B remains to be worked out in more detail.
miR-1182, miR-1207-5p and miR-1266. miR-1182 (Figure 4B) targeted telomerase reverse transcriptase (TERT) by binding to its open reading frame, whereas miR-1207-5p and miR-1266 bind to its 3’-untranslated region (149, 150). These miRs inhibited proliferation, induced cell-cycle arrest in SGC-7901 and U2OS GC cells; these effects were rescued by their antagomirs (149, 150). miR-1182 attenuated the proliferative and metastatic potential of SGC-7901 GC cells in nude mice (149). miR-1207-5p and -1266 inhibited growth of transplanted SGC-7901 cells in nude mice (149). An inverse correlation between miR-1182 and TERT expression has been noted in patients with GC (150). The telomerase complex consists of TERT and the RNA component. The loss of telomerase maintenance results in cell death and its inhibition led to improved outcomes in cancer-related xenograft models (151). The clinical development of small-molecule TERT inhibitors is hampered by efficacy issues (152, 153). Immunotherapeutic approaches such as vaccination against TERT are also being pursued (154). TERT expression in GC was found to have prognostic value because it is correlated with pathological variables and lymph node metastasis (155). Inhibition of TERT through reconstitution of miR-1182, miR-1207-5p or miR-1266 (Figure 4B) is an innovative approach for treatment of GC but more POC experiments should be performed.
Other Enzymes
miR-124 and miR330-3p. Sphingosin kinase 1 (SPHK1), the target of miR-124 was found to be up-regulated in GC tissues and to be associated with shorter survival of patients (156, 157). miR-124 (Figure 4B) was inversely correlated with SPHK1 expression in GC samples (156). miR-124 inhibited proliferation of GC cells in vitro and in vivo in nude mice (156). miR-124 induced downstream cell-cycle inhibitory proteins p21 and p27, suppressed AKT and enhanced the transcriptional activity of forkhead box O1 (156). miR-330-3p (Figure 4B) was shown to target SPHK1 and sphingosin1-phosphate receptor 1 (S1PR1) (157). Ectopic expression of miR-330-3p inhibited proliferation, migration, in vitro invasion, and tumor growth of MKN1 GC cells after subcutaneous implantation into nude mice (157). In MKN1 and KATO3 cells, inhibition of SPHK1 by short-hairpin RNA suppressed c-MYC and increased expression of p21 and p27 (157). miR-330-3p inhibited the ERK/AKT pathway in GC cells (156). SPHK1 and S1PR1 were shown to be activated in GC (158). In cancer cells, ceramide and sphingosine inhibited cell proliferation and induce apoptosis, while S1P has the opposite effect (159, 160). However, the issue is complicated since S1P can bind to five G-protein-coupled receptors with different outcomes regarding cell invasion. The physiological function of S1P may therefore be dependent on the S1PR receptor profile of the corresponding tumor cell (159, 160). Therefore, for GC more target validation experiments are necessary concerning the role of SPHK1 as a therapeutic target.
miR-137. miR-137 (Figure 4B) was found to be underexpressed in patients with GC and cell lines in comparison to corresponding controls (161). miR-137 reduced cell proliferation, and impaired migration and invasion in MGC-803 and HGC-27 GC cell lines (161). miR-137 suppressed growth of MGC-803 xenografts (161). Cyclo-oxygenase 2 (COX2) has been identified as a direct target of miR-137 (161). Ectopic expression of miR-137 in GC cells suppressed expression of p-AKT (161). The COX2–prostaglandin pathway is activated in several types of cancer and is correlated with aggressiveness and metastasis (162). In GC, the COX2–prostaglandin pathway induces inflammatory cytokines such as IL11, chemokine (C-X-C) motif ligands 1, 2, 5 (CXCL1, 2, 5) which have tumor-promoting functions by different mechanisms (163). COX2 and miR-137 deserve further target validation in GC.
miR-516-3p. A highly metastatic variant of scirrhous GC, cell line 44As3, was used to identify sulfatase-1 (SULF1) as a direct target of miR-516-3p (Figure 4B), which shows reduced expression in GC tissues (164). Scirrhous GC exhibits a high frequency of metastasis to the peritoneum (165). Stable overexpression of miR-516-3p in 44As3 cells reduced proliferation, migration and invasion in vitro (164). Longer overall survival and less ascites fluid were noticed in orthotopic 44A3 tumors intratumorally injected with an expression vector for miR-516-3p (164). SULF1 is an extracellular sulfatase which removes internal glucosamine-6-sulfate from heparan sulfate proteoglycans, thereby modulating interactions with various signaling molecules (165, 166). SULF1 promotes WNT signaling by liberating WNT ligands which bind to frizzled receptors by an autocrine mechanism (167-169). Further target validation experiments for miR-516-3p and SULF1 in GC should be performed.
miR-5590-3p. miR-5590-3p (Figure 4B) was found to be down-regulated in GC tissues and to target portable ATP-dependent helicase DEAD box protein 5 (DDX5) (170). It suppressed GC cell proliferation in vitro and in vivo through the DDX5/AKT/mTOR pathway and inhibition of downstream CCND1 and cyclin-dependent kinase 2 expression (169). DDX5 is a member of the family of DEAD-box helicases (170, 171). DDX5 is involved in tumorigenesis, proliferation, metastasis and regulates several cancer-related pathways (172). DDX5 resolves G-quadruplexes and mediates c-MYC gene transcriptonal activation (173). The role of miR-5590-3p and DDX5 in GC remains to be further elucidated and validated.
Conclusion and Perspectives
In this review, we did not discuss down-regulated miRs targeting MMPs with efficacy in preclinical in vivo models of GC because studies of MMP inhibitors in cancer-related indications have devalidated these targets. Identification of a down-regulated miR defines targets for therapeutic intervention which can be inhibited with small molecules, mAb-related moieties or immunological intervention. We identified 38 miRs covering tractable targets such as transmembrane receptors (n=13), secreted factors (n=9) and enzymes (n=16). All of the identified targets need more target-validation experiments to support their role as targets for treatment of GC. A critical issue is that several targets have tumor-promoting as well as tumor-suppressive functions in a context-dependent manner as described in the preceding sections. An important issue is the reconstitution of functions such as proliferation, invasion and survival in GC cell lines ectopically expressing the corresponding miRs through reconstitution of expression of the corresponding targets. Investigation of target-related pathways for identification of pharmaco-kinetic and pharmaco-dynamic markers is another crucial issue. The prevalence of expression of the identified targets should also be investigated in more detail.
Another option is therapeutic intervention by reconstitution therapy resulting in expression of the corresponding miR or synthetic double-stranded miR mimics in GC tumors. miRs can be administered intra-tumorally, systemically in appropriate formulations, or as plasmids or viral vectors (13, 174-176). Adeno-associated virus-based vectors have emerged as preferred vehicles for replacement therapy (177, 178). POC experiments in mouse lung cancer models have shown that reconstitution of Let-7 (179), miR-29b (180) and miR-708-5p (181) inhibits tumor growth. Clinical approaches for reconstitution therapy have relied on miR-34a and miR-16. miR-34 inhibits crucial oncogenic pathways such as EMT, NOTCH, WNT and TGFβ/SMAD and its reconstitution leads to inhibition of tumor growth in several xenograft models (182). However, a clinical phase I trial in patients with different tumors involving administration of lipid nanoparticles filled with miR-34 mimetics had to be closed due to immune-related side-effects (182, 183). In another clinical study, EGFR-coated bacterial minicells expressing miR-16 mimics were injected into patients with metastatic pleural mesothelioma (184). miR-16 mediates a tumor-suppressive effect by down-regulation of BCL2 and CCND1 (185). In a phase I study with 27 patients with malignant pleural mesothelioma, one objective response and stabilization of disease in 15 patients was observed (185). Taken together, clinical POC for miR reconstitution therapy has not yet been achieved in patients with cancer.
From a preclinical point of view, many critical issues have to be resolved. These issues have to be tackled case-by-case and are not discussed in detail in this review. Among the critical issues are: The development of efficient delivery systems, pharmaco-kinetic and pharmaco-dynamic issues, cytokine-release syndrome, hematological and hepatic toxicity, removal of complexed nucleic acids by the reticulo-endothelial system, entry into the target cell and endosomal escape (12, 186-192).
The next couple of years will tell us whether POC of miR-based reconstitution therapy in patients with cancer can be achieved in a clinical setting.
Footnotes
Authors’ Contributions
FB, SA, UB and UHW jointly prepared the article and the corresponding Figures.
This article is freely accessible online.
Conflicts of Interest
FB, SA and UB are employees of Roche; UHW has been an employee of Roche.
- Received March 25, 2021.
- Revision received April 30, 2021.
- Accepted May 5, 2021.
- Copyright© 2021, International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved
References
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.