


Abstract
Background/Aim: Non-small cell lung cancer (NSCLC) patients with activating somatic mutations in the epidermal growth factor receptor (EGFR) have better outcomes with tyrosine kinase inhibitors (TKIs) than with chemotherapy. However, even with the most effective therapies, not all patients respond. The presence of concurrent pathogenic mutations could play a role in resistance. The objective of this study was to analyze the impact of concurrent mutations in genes other than EGFR on survival outcomes of patients treated with TKIs for EGFR-mutated NSCLC. Patients and Methods: We conducted a retrospective cohort analysis of patients with advanced NSCLC treated with TKIs in our center between January 2016 and December 2019. Clinical and pathological characteristics, EGFR mutational status, presence of co-occurring genetic alterations, overall (OS) and progression-free survival (PFS) were evaluated. Results: Of the 42 patients with advanced NSCLC harboring EGFR mutations who received TKIs in our center, 22 (52%) had no concurrent mutations, 15 (36%) had a non-pathogenic, non-resistance co-mutation, and 5 (12%) had a concurrent resistance mutation. The median OS of the global population was 14.9 months, with a shorter OS in the group harboring a concurrent resistance mutation (7.7 vs. 18.1 months, p=0.002). Concurrent mutations possibly associated with resistance were found in PIK3CA, KRAS and PTEN genes. Conclusion: Concurrent resistance mutations in genes other than EGFR influenced the outcome of patients with NSCLC, while non-resistance mutations did not alter survival, compared to the absence of co-mutations. This evidence highlights the importance of a careful interpretation of molecular findings. The best treatment options for these patients should be studied in randomized controlled trials.
Ranking among the most frequent malignancies worldwide, lung cancer can be divided into two main categories: non-small cell lung cancer (NSCLC) and small cell lung cancer (SCLC), representing 80-85% and 15-20% of cases, respectively (1). The former comprises 3 main histological subtypes: adenocarcinoma (60%), squamous cell carcinoma (35%), and large cell carcinoma (5%); each subtype has a distinct molecular presentation (2). The carcinogenesis of adenocarcinomas is driven by known oncogenic alterations in up to 66% of cases, while the same is rare (13%) in squamous cell carcinoma (3). The most common oncogenic drivers in adenocarcinoma are Kirsten rat sarcoma viral oncogene homolog (KRAS) (4), epidermal growth factor receptor (EGFR), anaplastic lymphoma kinase (ALK) (5, 6), and proto-oncogene tyrosine-protein kinase ROS1, found in 25%, 15%, 5%, 2% of cases, respectively (7). Certain molecular alterations are mutually exclusive while others can be co-existent.
In this paper, we focus on patients with lung adenocarcinoma harboring mutations that lead to excessive EGFR activity. These mutations are most common among non-smokers, and younger, female, Asian lung cancer patients (7). Ninety percent of the activating mutations in the tyrosine kinase domain of EGFR consist of exon 19 deletions or L858R point mutations within exon 21. These mutations result in constitutional activation of EGFR without the need for ligand-induced stimulation, promoting cell proliferation, survival, and dissemination (8, 9).
TKIs have been the standard front-line therapy for metastatic EGFR mutant NSCLC for almost a decade, ever since the efficacy of first-generation (erlotinib, gefitinib) and second-generation (afatinib) TKIs demonstrated improved outcomes and lower toxicity compared to standard platinum-based chemotherapy (10). Nevertheless, primary or acquired resistance to TKIs occurs in all patients, with a progression-free survival (PFS) ranging from 9 to 15 months (11, 12). Resistance mechanisms such as the development of a T790M mutation in EGFR exon 20 prompted the development of a third-generation TKI (osimertinib).
Osimertinib has proven to be superior to first and second generation EGFR TKIs in terms of PFS and overall survival (OS), when used as front-line therapy in patients with exon 19 deletions or L858R point mutations within exon 21 (13). Unfortunately, acquired resistance has been shown to occur in virtually all patients, with a median PFS of 19 months and OS of 38.6 months (12). The substitution of cysteine with serine at codon 797 is the most common on-target secondary mutation conferring resistance to osimertinib (14). Trials are ongoing to evaluate treatments for this alteration, as no treatment has been proven effective in clinical practice so far. Not all patients derive similar benefit from osimertinib (15). The variable outcomes could be related to resistance mechanisms, both on- and off-target, but may also stem from co-occurring genetic alterations, which appear to impact both PFS and OS (16).
Several retrospective analyses have shown a worse outcome for patients with EGFR-mutant NSCLC harboring concomitant mutations in genes such as phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA) (10, 17), KRAS (18), phosphatase and tensin homolog (PTEN) (19), retinoblastoma 1 (RB1), FAT tumor suppressor homolog 1 (FAT1), or ATP-binding cassette sub-family B member 1 (ABCB1) mutations, but not tumor protein p53 (TP53) mutations (10). Concomitant ALK (20) or SMAD4 mutations may also influence survival outcomes (21).
In an analysis of 423 patients with EGFR mutations treated with first-generation TKIs, an unfavorable group was seen with PIK3CA, RB1, FAT1, or ABCB1 mutations (10). In a large database study, metastatic NSCLC patients with EGFR/PIK3CA and EGFR/KRAS co-mutations experienced worse PFS with TKIs compared to patients who did not harbor pathogenic mutations other than EGFR in a panel of 50 cancer-related genes (18). The same pattern was described with concomitant homozygous PTEN deletions (19). In this single center, retrospective study, we investigated the impact of co-mutations on survival of patients with EGFR-mutated advanced NSCLC.
Patients and Methods
Patients. In this retrospective study, all patients who received targeted therapy (gefitinib, erlotinib, afatinib, osimertinib) for advanced EGFR-mutated NSCLC in our center, between January 2016 and December 2019 were identified. A total of 42 patients were identified and their medical records were reviewed to extract histologic and molecular data (from pathology records), clinical and radiological outcomes (from dedicated reports), date of death if occurred and date of last follow-up visit. All patients enrolled signed a general research consent form providing access to their medical records. The study was approved by the local Ethics Committee (CCER 2020-01628).
Data evaluation. At the time of diagnosis, tumor DNA was extracted with QIAGEN AllPrep DNA/RNA Mini Kit (ref#80204, Qiagen, Hilden, Germany) and analyzed on the Ion Proton® sequencer (Thermo Fisher Scientific, Basel, Switzerland) using the Ion Ampliseq™ Cancer Hostpot Panel v2 (Thermo Fisher Scientific, ref# 4475346), which includes mutations from 50 oncogenes and tumor suppressor genes. Genes of interest other than EGFR were pre-specified, including adenomatous polyposis coli (APC), ataxia telangiectasia mutated (ATM), cyclin-dependent kinase inhibitor 2A (CDKN2A), KRAS, MET, PIK3CA, PTEN, SMAD4, TP53. The classification of mutations and their contribution to resistance was performed before statistical analyses. We evaluated the pathogenicity of mutations (i.e. their impact on protein function) using online international databases, namely ClinVar, Catalog of Somatic Mutations in Cancer (COSMIC) and oncoKB, and their potential impact on treatment resistance based on the existing medical literature. Patients were classified into 3 cohorts: absence of co-mutations, presence of co-mutations judged to be non-pathogenic or non-informative (“non-resistance”), and presence of co-mutations that could plausibly induce resistance to treatment (“resistance”).
Regarding clinical outcomes, OS (primary) and PFS (secondary) were assessed. PFS was calculated from the date of TKI initiation to date of radiological progression, or death. OS was calculated from the date of TKI initiation to date of death, based on the vital status in February, 2020. Patient characteristics included gender, age at diagnosis, smoking status, performance status (PS) and presence of brain metastases at diagnosis.
Statistical analysis. Statistical analysis was performed with the R language for statistics (v3.6.0 https://cran.r-project.org/). Kaplan Meier curves were plotted with the R survminer package. We used Cox's proportional hazards model and the log rank and log ratio tests to examine hypotheses. For statistical purposes and in order to conserve power, all tests compared patients with resistance mutations to patients without resistance mutations, including both patients without any mutation and patients with non-resistance mutations. All tests were two-sided and an alpha cutoff of 0.05 was used. Fisher's exact test was used to test the distribution of categorical variables (response vs. mutation class).
Results
Clinical characteristics. Forty-three patients were identified but one was excluded due to early loss at follow-up (less than 3 months). Molecular analyses were carried out at the time of diagnosis. Twenty-two (52%) patients had no co-mutations (EGFR mutations only), 15 (36%) had a non-resistance co-mutation, and 5 (12%) had a resistance co-mutation mutation. Twenty-five (60%) were female and 17 (40%) were male (17, 40%) and median age was 69 years (range=45-84). Nineteen (45%) patients never smoked, 7 (17%) were current smokers and 16 (38%) former smokers. Fifteen (36%) patients had brain metastases at diagnosis, 32% in the EGFR mutation only cohort (7/22), 27% in the non-resistance mutation (4/15) group and 80% among resistance co-mutation patients (4/5). Patient characteristics are summarized in Table I. Median follow-up was approximately 32 months [962 days, 95% CI=565-not reached (NR)].
Mutational and histologic characteristics. All patients with EGFR-mutated NSCLC had adenocarcinoma. The most frequent mutations of the EGFR gene were typical mutations in exon 19 and exon 21, in 26 (62%) and 12 (29%) patients, respectively. Three patients with atypical mutations were observed, 2 in exon 20 and one in exon 18. One mutation was unspecified.
Clinical and demographic characteristics of patients (n=42).
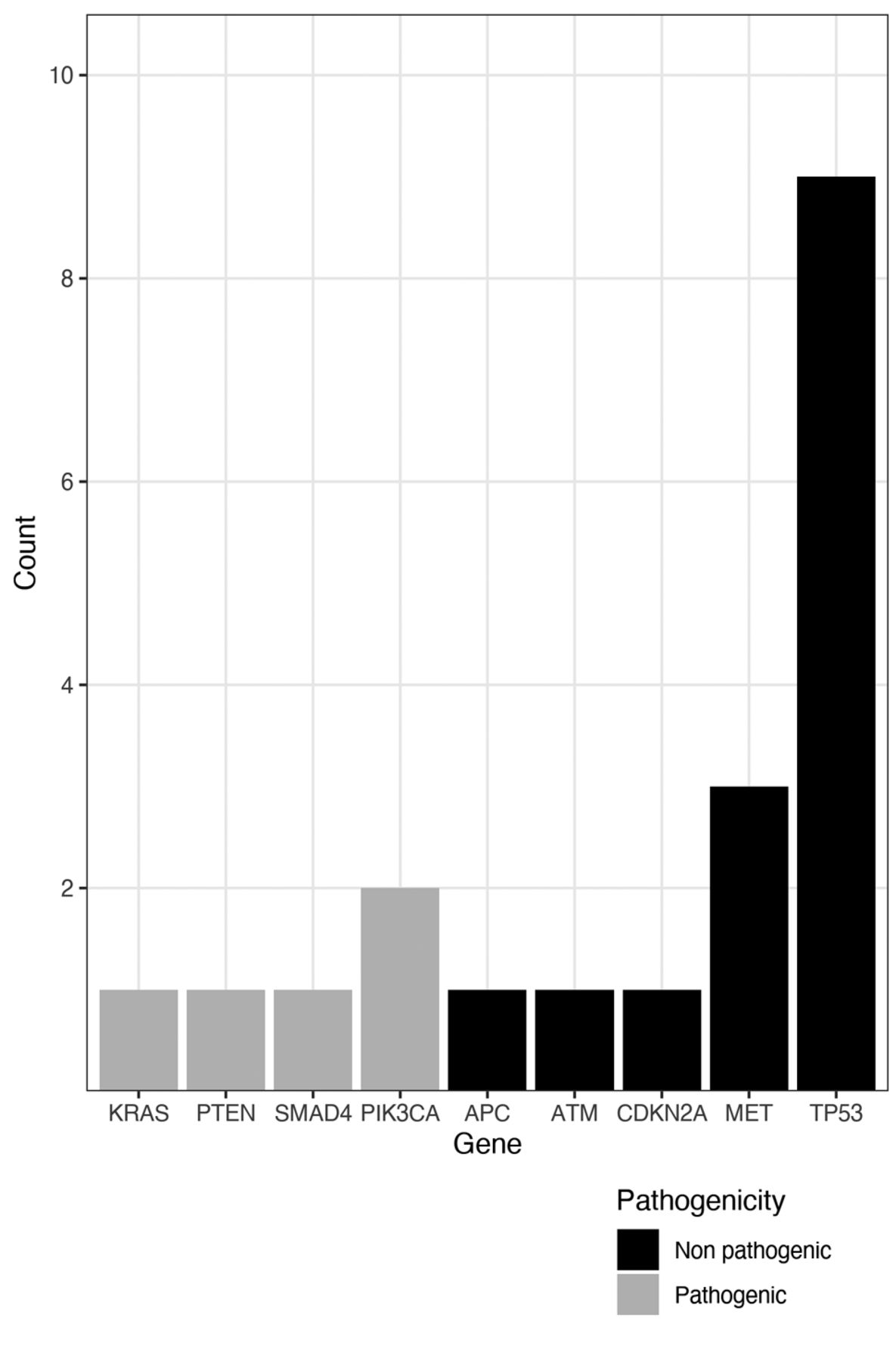
Regarding patients with concurrent mutations, 15 (36%) had non-resistance co-mutations in APC, ATM, CDKN2A, MET, TP53, and 5 (12%) had resistance concurrent mutations in KRAS, PIK3CA, PTEN and SMAD4 (Figure 1).
Survival outcomes. The median OS for all patients was 14.9 months (95% CI=11.4-30.1). Median OS in the absence of co-mutations and in the presence of non-resistance mutations was 18.1 and 16.8 months, respectively, while it was significantly reduced in the group with resistance mutations (7.7 months, HR=4.6, 95% CI=1.56-13.3, p=0.002, compared to the other two groups). At 1 year, all patients with resistance concurrent mutations were deceased, while 14 of 22 (63%) patients without any mutation and 7 of 15 (47%) patients with non-resistance co-mutations were still alive. At 2 years, 10 out of 37 (27%) patients without resistance co-mutations were still alive, including 8 of 22 (36%) patients without co-mutations and 2 of 15 (15%) patients with non-resistance co-mutations (Figure 2).
Among the other clinical variables, only the occurrence of an objective response and male gender were significantly associated with survival. In a bivariable model including response and resistance co-mutations, an objective response (HR=0.31, 95% CI=0.14-0.68, p<0.01) as well as the presence of resistance co-mutations (HR=3.30, 95% CI=1.12 to 9.72, p=0.03) correlated with overall survival. Similarly, in a bivariable model including gender and resistance co-mutations, male patients exhibited a trend towards poorer OS (HR=2.11, 95% CI=0.98-4.56, p=0.06), but the effect of resistance co-mutations was maintained (HR=3.70, 95% CI=1.25-10.97, p=0.02). The impact of resistance mutations on OS remained significant in bivariable models including, age at diagnosis (p<0.01), tobacco exposure (p<0.01), and performance status (p<0.01). In conclusion, the resistance co-mutation class remained associated with OS even when other variables were considered (Table II).

Number of concurrent mutations found and their pathogenicity.
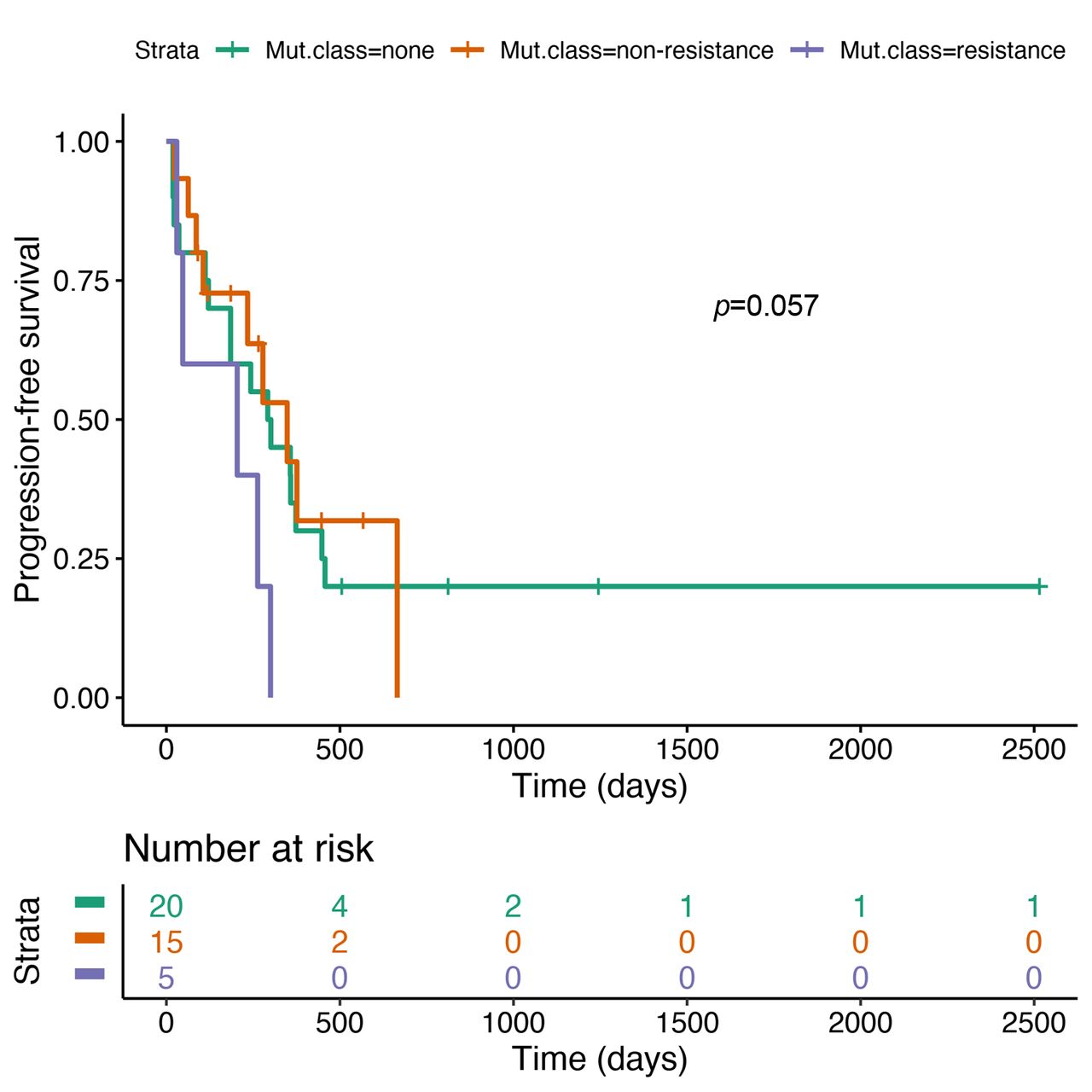
Median PFS was 9.8 months (95% CI=6.2-15.2) for patients without any co-mutation, 11.6 months (95% CI=7.8-NR) for patients with non-resistance co-mutations and 6.8 months (95% CI=1.6-NR) for patients with resistance co-mutations (Figure 3). The difference between patients with resistance mutations and the remaining patients did not reach statistical significance (HR=2.67, 95% CI=0.97-7.31, p=0.057).
Discussion
We identified a similar proportion of concurrent pathogenic mutations that are expected to induce resistance to treatment in patients with EGFR-mutated NSCLC compared to previous retrospective analyses in this population (8, 15, 16, 21, 22). Worse survival outcomes have been reported among patients with advanced NSCLC harboring EGFR mutations and concomitant pathogenic alterations in genes PIK3CA (10, 17), KRAS (18), PTEN (19), RB1, FAT1, ABCB1, ALK (20), and SMAD4 (10).

Overall survival by mutation status.
Our findings are in accordance with previous results (10, 18, 20). We found that specific resistance co-mutations predicted a poorer outcome, with a significantly shorter median OS and a statistical trend toward shorter PFS. The mutations predictive of poor survival in our cohort were found in PIK3CA, PTEN, KRAS and SMAD4. Other mutations, in genes such as TP53 or MET, were classified as non-resistance based on their impact on protein function, and appeared not to influence response and survival. It is important to note that the MET alterations identified in our patients were known to be non-pathogenic according to international databases, however other MET alterations can be pathogenic (22).
Bivariable model of the impact of resistance co-mutations.
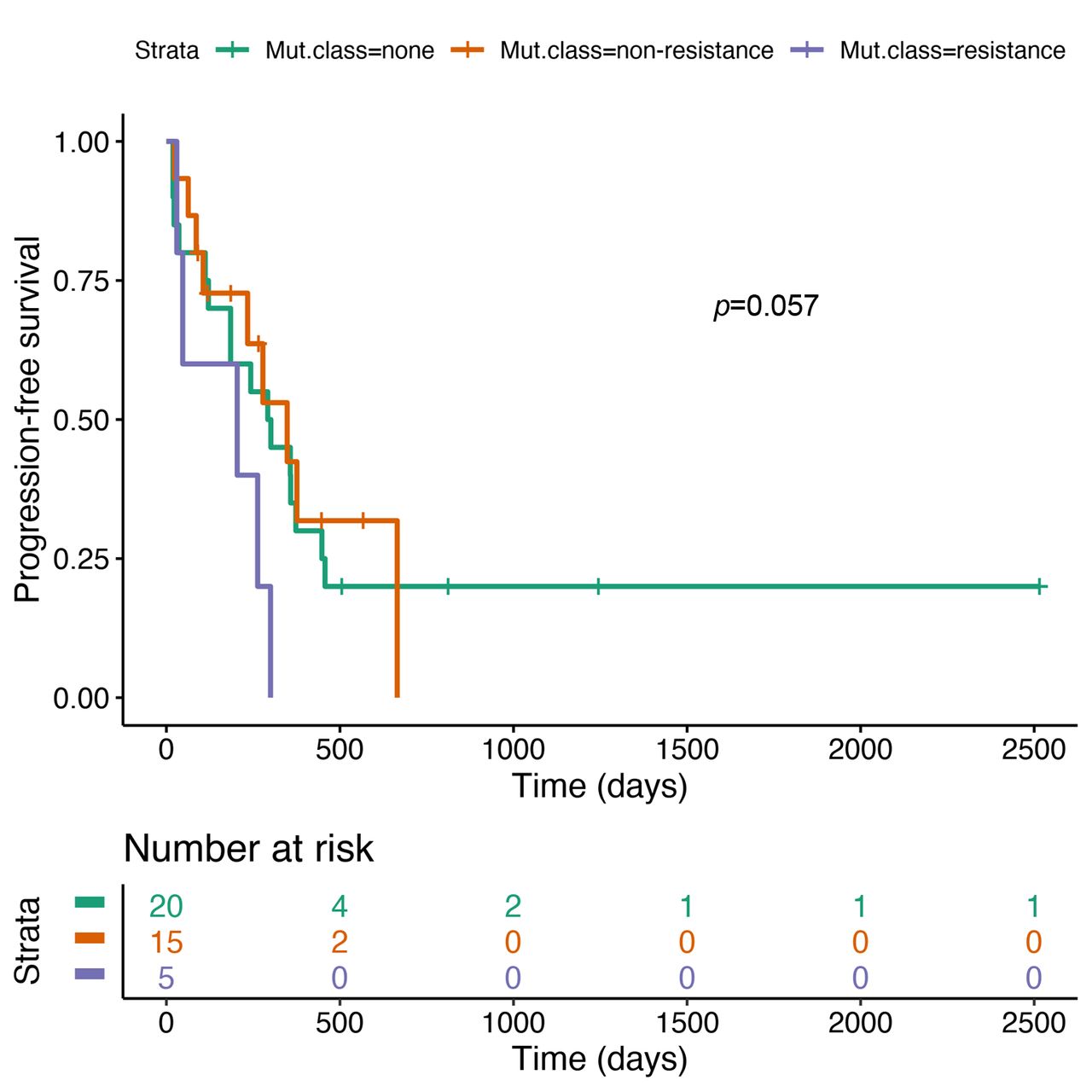
Progression-free survival by mutation status.
OS was similar between patients without concurrent mutations and those with non-resistance co-mutations such as TP53. The strength of our study is the pre-analysis assessment of each concurrent alteration and the classification of the latter according to the expected biological role, thus allowing for a more accurate analysis. The OS difference in patients with resistance co-mutations is not limited to the PFS difference on front-line treatment. This suggests that co-mutations also affect the response to subsequent therapies.
Given the retrospective nature of our trial, there are inherent limitations. The PFS could be over-estimated, as it corresponds to the date of pre-planned radiological evaluations, usually performed every three months. However, if a physician opts to perform scans less frequently in the absence of symptoms, it could artificially prolong the PFS by under-diagnosing progression. Subsequent treatments were not standardized and could result in survival bias as well. Furthermore, our cohort is small in size and mutational data could be influenced by the evolution of NGS techniques and panels used over the 4-year treatment period, though there was no major change in the 50 gene panel routinely used for screening at our center. In addition, there was treatment heterogeneity among our patients; those in the last 2 years were more likely to receive upfront third-generation TKIs, while those in the first 2 years started with first-generation TKIs. However, all patients harboring co-occurring resistance mutations were treated with upfront osimertinib; hence, any bias would probably under-estimate the detrimental influence of co-mutations. It is well known that smoking impairs the efficacy of anti-EGFR TKIs (23), but this does not explain our results, as none of the patients harboring co-mutations was an active smoker (9). Furthermore, the imbalance in brain metastases in diagnosis did not affect survival in our bivariable analyses.
In our study, pathogenic mutations in PIK3CA, PTEN, KRAS and SMAD4 were predictive of shorter survival, presumably through resistance to treatment (8). The possibility of combining EGFR-directed therapy with a targeted treatment for druggable co-existing genetic alterations requires further investigation. For example, PI3K inhibitors and Akt inhibitors could have a role in the treatment of NSCLC with EGFR alterations and concurrent PIK3CA and PTEN mutations, respectively (17, 19). Reports have shown interesting results in advanced EGFR mutant NSCLC from simultaneously targeting EGFR and other alterations, such as fusions involving RET (24),, BRAF (25, 26) or ALK (27). To date, there is no randomized trial among this subset of EGFR mutant NSCLC patients harboring co-mutations.
The ongoing ORCHARD Phase 2 trial (NCT03944772) aims to explore treatment options following disease progression on first-line osimertinib, by targeting resistance mechanisms based on molecular profiles at progression. If this yields positive results, it could prompt the evaluation of a similar approach in the front-line setting.
Novel approaches for patients with NSCLC harboring an EGFR-sensitizing mutation and co-occurring genetic alterations are needed. Whether the optimal treatment will be a combination targeting EGFR and the co-mutation, remains uncertain. Other combinations are also possible, like the addition of an anti-angiogenic antibody or chemotherapy to an early generation EGFR TKI, which has shown its safety and efficacy in the front-line setting (28).
Conclusion
Within the subgroup of patients with lung adenocarcinoma who have an activating EGFR mutation, a wide range in survival rates was observed in our study, as well as in other larger trials (13) Based on our results, we conclude that this could be partially explained by the presence of concomitant pathogenic mutations in cancer-related genes. Interestingly, the presence of mutations that are not expected to confer resistance did not appear to influence survival, highlighting the importance of a careful interpretation of the molecular findings. Hence, the impact of concomitant mutations on the clinical outcomes of EGFR-mutated NSCLC patients underscores the need for tailored therapies. Future prospective, larger studies are needed to establish the best treatment approach in this population.
Acknowledgements
The work of PT is funded by the Ligue Genevoise Contre le Cancer.
Footnotes
Authors' Contributions
All Authors participated in drafting and editing the manuscript. The concept was from AA and AF. Statistical support was provided by PT.
This article is freely accessible online.
Conflicts of Interest
MC declares no potential conflicts of interest. PT has received compensation for participation in advisory boards from Bristol-Myers-Squibb, Roche, Merck Sharpe & Dohme and Sanofi-Aventis and travel expenses from Lilly and Sanofi-Aventis. AA has received compensation from Bristol-Myers Squibb, AstraZeneca, Merck Sharpe & Dohme, Takeda, Pfizer, Roche and Boehringer Ingelheim for participating on advisory boards. AF has received compensation from Roche, Pfizer, Merck Sharpe & Dohme, and Bristol-Myers Squibb for participating in advisory boards.
- Received May 9, 2020.
- Revision received June 30, 2020.
- Accepted July 2, 2020.
- Copyright© 2020, International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved
References
In this issue




{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.