


Abstract
Follistatin (FST), as a single-chain glycosylated protein, has two major isoforms, FST288 and FST315. The FST315 isoform is the predominant form whilst the FST288 variant accounts for less than 5% of the encoded mRNA. FST is differentially expressed in human tissues and aberrant expression has been observed in a variety of solid tumours, including gonadal, gastric and lung cancer, hepatocellular carcinoma, basal cell carcinoma and melanoma. Based on the current evidence, FST is an antagonist of transforming growth factor beta family members, such as activin and bone morphogenetic proteins (BMPs). FST plays a role in tumourigenesis, metastasis and angiogenesis of solid tumours through its interaction with activin and BMPs, thus resulting in pathophysiological function. In terms of diagnosis, prognosis and therapy, FST has shown strong promise. Through a better understanding of its biological functions, potential clinical applications may yet emerge.
Follistatin (FST), which was first isolated from porcine and bovine follicular fluid (1, 2), was initially described as a protein involved in the regulation of the secretion of follicle-stimulating hormone. This monomeric glycosylated polypeptide chain is encoded by a single gene located on the long arm chromosome 5q11.2 (3), which through alternative splicing may be transcribed into the mRNA precursors, FST317 and FST344. From these precursors, three major FST isoforms may be produced, namely FST288 (from pre-FST317), FST315 (from pre-FST344) and a third FST isoform, FST303, produced from the post-translational truncation of the FST315 C-terminus. These three main FST isoforms can also be glycosylated to yield six further FST isoforms that were previously identified in bovine (4) and porcine (5, 6) follicular fluid. At the core, all FST isoforms contain a 63 residue N-terminal domain and three follistatin domains, termed FSD1, FSD2 and FSD3. These domains comprise 73-77 amino acid residues and are characterised by an arrangement of 10 conserved cysteine residues (7). Both FST288 and FST315 are differentially expressed in human tissues (6-9). FST315 is the predominant isoform, whilst the FST288 isoform accounts for less than 5% of the encoded mRNA (10, 11). Some molecules such as activin, transforming growth factor-beta (TGFβ), forkhead domain transcription factor L2 (12, 13), gonadotropin-releasing hormone (14), zinc finger protein (GLI2) (15), dexamethasone (16), androgens (17), activators of wingless-related integration site (WNT) signalling (18, 19) and 1,25-dihydroxyvitamin D (20) have been shown to regulate the transcription of the FST gene. In addition, down-regulation of FST gene expression by peroxisome proliferator-activated receptor gamma (PPARγ) or the transcription factor epiprofin (21, 22) has been shown.
As an antagonist of TGFβ superfamily member activin, FST seems to primarily function in relation to the role that activin plays. Along with FST, activin acts as a pleiotropic growth factor system, which is involved in proliferation, differentiation, and apoptosis of a number of cell types (23-28). As such, the functions of the FST isoforms are therefore linked to their binding affinity for activin (6, 10, 11, 29-37). In fact, this critical binding and neutralisation of activin, either partial or complete, is based on the order of at least two of the FST cysteine domains and its N-terminal (38), conferring a difference in affinities for activin between the FST isoforms. The binding complex between activin and FST generally occurs in a 1:2 ratio, where the two FST molecules, connected C-terminus to N-terminus, envelop the activin molecule, thus blocking access to both the type I and type II receptor binding sites (9). The binding complex handicaps the interaction of activin with its specific receptors, leading to the inhibition of most of its the biological effects in various cells and tissues (5, 6, 11, 32, 35). An important contribution of the FSD1 region to FST localisation and hence, bioactivity, lies in its ability to bind to plasma membrane heparin sulfate proteoglycans. For example, FST288 binds to heparin sulfate proteoglycans with a high affinity and may function as a local regulator by potentially preventing the autocrine, endocrine and paracrine actions of activin (39). Additionally, FST288-activin binding complex can interact with cell-surface proteoglycans followed by the internalization and subsequent degradation of this complex with lysosomal enzymes (3, 5, 6). Conversely, FST315 is predominantly localised in the circulation, consistent by its low binding affinity for heparin, and its binding to activin appears to be irreversible (5).
Activin seems to be able to exert context-dependent and cell type-specific inhibiting and activating functions (40). There is evidence that activin can both have an inhibitory or proliferative effect on cell proliferation; dependent on the cell type (41-47). In vivo studies have shown that FST knockout mice died soon after birth due to a variety of skeletal and cutaneous defects amongst other defects seen in the activin-deficient mice (48). Lin et al. also demonstrated that mice with an FST315-expressing construct could overcome this neonatal lethality but still had severe biological defects. Overexpression of mouse FST using the mouse metallothionein I promoter in transgenic mice led to gonadal defects and eventual infertility, primarily due to local effects of FST in these tissues (50). Dermatological defects were present in both FST-knockout and -overexpressing mice, but there was no evidence of cancer (51). Some of these defects were similar to those that were seen in bone morphogenetic protein 5 (Bmp5)-mutant mice (52) and TGFβ-overexpressing mice (53). These similarities suggested that FST might be a modulator of other TGFβ-related proteins apart from activin or might function independently (48). Some other studies have demonstrated that FST may also bind to other molecules such as BMPs, especially BMP-2, -4, -5, -6, -7 and -11 (4, 54-58), myostatin (59) and TGFß3 (60). Table I shows the expression profile of FST and its targeting ligands in various tissues and organs of the human body. In addition, FST may also have an interaction with other non-TGFβ family proteins, such as angiogenin, known as a pro-angiogenic factor (61), and α2-macroglobulin, known as a serum pan-protease inhibitor (62). However, further investigations are required to explain the functional value of the interplay between FST and these molecules.
Follistatin in the Development and Progression of Solid Tumours
A study on the molecular mechanisms underlying antral gastric tumours, which develop in gastrin-deficient mice, demonstrated elevated FST expression in the proliferative neck zone of hyperplastic antrum, a trend which has also been observed in human gastric cancer (63). These findings indicated that antral hyperplasia in gastrin-deficient mice involves amplification of mucous cell lineages, in which FST may play a role. In addition, strong FST expression was found in basal cell carcinoma and seemed to be mainly activated by transcription factor GLI2, which as a Hedgehog signalling mediator, has been shown to be up-regulated by the expression of FST (15, 64). This phenomenon is suggestive of the engagement of FST in the development of basal cell carcinoma, which has yet to be fully elucidated.
Stove et al. found that all melanoma cell lines expressed activin receptors. Treatment of these cell lines with activin also resulted in the phosphorylation of SMAD signal transduction molecules (65). Secretion of FST, either native or after retroviral transduction, efficiently prevented SMAD activation or activation of an activin-responsive luciferase reporter construct. In melanocytes, activin treatment led to growth inhibition and induction of apoptosis, processes which were counteracted by an addition of FST. Thus this study suggests that in melanoma, FST is involved in neutralising the effects of activin.
Activin also appears to be a major regulatory factor in liver tissue homeostasis, controlling cell proliferation and apoptosis. Both activin and TGFβ itself may inhibit DNA synthesis and induce apoptosis in vitro as well as in vivo (66-72). It has been reported that experimentally administered FST acted as an inducer of hepatocellular DNA synthesis (73-76) through reduction in the local bioavailability of activin. Additionally, overexpression of FST was also found in rodent liver tumours (77), of which most were hepatocellular carcinomas, when compared to surrounding liver tissue; the level of overexpression of FST was different but independent of the carcinogen treatment that rodents had received and the histological stage of malignancy. Differential expression of FST and its interacting ligands in different malignancies is shown in Table II.
In addition, there is also some indirect proof regarding the growth-promoting effect of FST on prostate cancer cells. Rapamycin (78) and soy isoflavones (79), plant-derived polyphenolic compounds with estrogen-like properties, inhibit prostate cancer cell growth and are associated with a decrease in the expression of FST. Type III TGFβ receptor has been reported to play an inhibitory role in the growth of prostate cancer cells (80). FST has been shown to directly bind to and completely block type III TGFβ receptor-induced epithelial–mesenchymal transition of normal murine mammary gland epithelial cell line in vitro (60) and thus might promote prostate cell proliferation by targeting this receptor.
Expression profile of follistatin (FST) and its target molecules in different organs and tissues.
It was found that exogenously added activin A and B induced an inhibitory effect on the growth of two human prostate cancer cell lines, LNCaP and DU145, which was completely reversed by FST (24). Overexpression of FST was observed in human LNCaP cells (81), which is in line with the theory of FST overproduction favouring the growth of prostate cancer by inhibiting signalling of activin (23). However, another human prostate cancer cell line, PC3, is resistant to the growth-inhibiting effects of activin A. Intriguingly, this phenomenon is further explained by the study of McPherson et al. which showed that only PC3 cells produced the FST288 isoform, while LNCaP and DU145 cells predominantly produced the FST315 isoform. Blockade of FST288 activity with a neutralizing antibody rendered PC3 cells responsive to activin A, as measured by inhibition of proliferation (25). These results indicate that the resistance of PC3 cells to activin A is more likely due to predominantly expressed of FST288 in comparison with LNCaP and DU145 cells. Sidis et al. found that inhibition of endogenous activin bioactivity, irrespective of the FST isoforms being administered endogenously or exogenously, was correlated closely with its surface-binding activity (36). These findings suggest that the cell-surface dwelling FST288 isoform inhibits activin-mediated downstream signal transduction pathways more efficiently than the FST315 isoform. Further investigations will shed light on the interaction of the FST288 isoform with activin receptors and, consequently, the biological effects induced by these molecules.
Expression of follistatin (FST) and its target genes in malignancies.
In some cases, however, FST may also inhibit tumour growth. An in vitro study showed that the growth of inhibin-deficient gonadal tumour cells from mice could be stimulated in culture via an autocrine mechanism of activin, while FST added to the culture medium slowed the growth rate of the tumour cells (82). Another in vitro study observed that for four ovarian cell lines not synthesizing FST, treatment with activin (1-100 ng/ml) resulted in an increase of proliferation, whereas FST treatment (1-100 ng/ml) inhibited their proliferation (83). This inhibitory function was further verified in an in vivo study, showing inhibin-deficient mice that carry the mouse metallothionein I follistatin transgene exhibited a less severe activin-mediated cancer cachexia-like wasting syndrome, lower serum activin levels, and a statistically significant prolonged survival in a number of cases compared with the mice deficient in inhibin alone (51).
Follistatin in Tumour-associated Angiogenesis
The effect of FST on tumour angiogenesis was found in a study by Ogino et al. (84), in which two human small cell lung cancer cell lines, SBC-3 and SBC-5, were transfected with the FST gene and subsequently inoculated into the natural killer cell-depleted severe combined immunodeficient (SCID) mice. Histological analysis results revealed that the number of proliferating tumour cells and the tumour-associated microvessel density were significantly reduced in the lesions produced by FST transfectants, compared with the parental SBC-5 or mock control, which was consistent with the results seen in liver-metastatic lesions produced by FST transfectants. These observations in both lung- and liver-derived tumours is suggestive of FST inhibiting tumour angiogenesis. However, no obvious effect on cell proliferation, motility, invasion, or adhesion were observed for FST in endothelial cells in vitro nor on tumour growth and metastasis in vivo (84).
Unlike the findings by Ogino et al. (84), there is also some evidence indicating that FST may play a promotory role in the progression of tumour angiogenesis. Kozian et al. found that FST implantation in the cornea in combination with subcritical concentrations of fibroblast growth factor (bFGF) induced a strong angiogenic response (85), which demonstrates that FST by itself, but particularly in synergy with bFGF, induces angiogenesis. This may be primarily due to the action of FST antagonising activin, which inhibits bFGF-induced sprouting angiogenesis in vitro and in vivo (40). Other evidence that FST may have a promotory effect on tumour angiogenesis is the study by Krneta et al. in which SCID mice were inoculated with R30C mammary carcinoma cells transfected with FST288 or activin. The FST-expressing tumours had a denser network of small-diameter capillaries than activin-expressing tumours (40). FST288 may be required for the promotion of angiogenesis in a mouse model (49). Collectively, FST, especially the FST288 isoform, seems to be involved in tumour angiogenesis, at least partially through attenuating activin signalling.
Follistatin in Solid Tumour Metastasis
In the study by Ogino et al., FST transfectants produced significantly fewer metastatic colonies in multiple organs, including lung, liver, and bone in natural killer cell-depleted SCID mice when compared with their parental cells or vector control clones (84), supporting the potential role of FST in controlling tumour metastasis.
BMP2, -4 and -7, together with their inhibitors, including FST, have been shown to be involved in osteoblastogenesis (86-88). The activin/FST system was reported to play a regulatory role in normal bone homeostasis (89-92). It has been speculated that FST might play a role in the pathogenesis of bone metastasis through interaction with BMPs and activin.
A study demonstrated that ribozyme transgene targeting BMP7 expression in the PC3 prostate cancer cell line resulted in increased invasiveness and motility, which appeared to be facilitated by changes in the level of the BMP antagonists noggin and FST (58). Although these findings indicate the role of FST in solid tumours, more evidence is needed for greater clarification of its involvement in the metastases associated with solid tumours.
Value of Follistatin in Diagnosis, Prognosis and of Solid Tumours
FST is expressed in multiple tissues (10, 77, 93-97), and its expression levels are altered in tumour tissues (25, 90, 98-100) and in body fluids (101-104) of patients with solid tumors. These findings provoked studies investigating FST as a diagnostic or prognostic marker for solid tumours. One study showed that serum FST and ovarian carcinoma antigen CA125 levels were significantly increased in patients with ovarian cancer when compared with healthy individuals and patients with benign ovarian cysts, which suggests that the serum FST level has potential diagnostic value, especially when combined with CA125 detection in order to reduce the number of false-positive results (105). Serum levels of FST were also found to be elevated in patients with hepatocellular cancer (HCC) (21, 106). Survival of patients with HCC with high FST levels was significantly shorter than for those with low serum levels of FST (106). Multivariate analysis revealed that in addition to large tumour size and presence of portal vein thrombus, high FST levels were independently correlated with poor prognosis (106). Therefore, a promising potential is indicated for FST in HCC prognosis. A survival analysis of FST in breast, lung, ovarian and gastric cancer was also performed using an online Kaplan–Meier analysis (http://kmplot.com/analysis/) (Figure 1) (107). An increased expression of FST was associated with poorer overall survival of patients with ovarian, gastric and lung cancer. These data also correlate with the previously mentioned studies of serum level in ovarian cancer and HCC. In contrast, a reduced expression of FST was correlated with shorter relapse-free survival (Figure 1B), although no correlation was seen between FST expression and overall survival of patients with breast cancer (data not shown).
Blank et al. reported FST in serum was negatively correlated and was decreased in both distant and lymph node metastasis associated with gastric cancer compared to patients without metastasis. In contrast, they noted that tissue from the patients with metastasis exhibited higher FST levels. Additionally, neoadjuvantly treated patients with lymph node metastasis still exhibited lower FST levels, whilst those with distant metastasis and poorly differentiated tumors (G3/4) exhibited elevated FST serum levels. Prognostically, a tumour tissue level of FST below the median was associated with better survival. However, in primary resected and neoadjuvantly treated patients, FST levels in tissue had no impact (108). Therefore FST levels appear to have a further prognostic significance in gastric cancer.
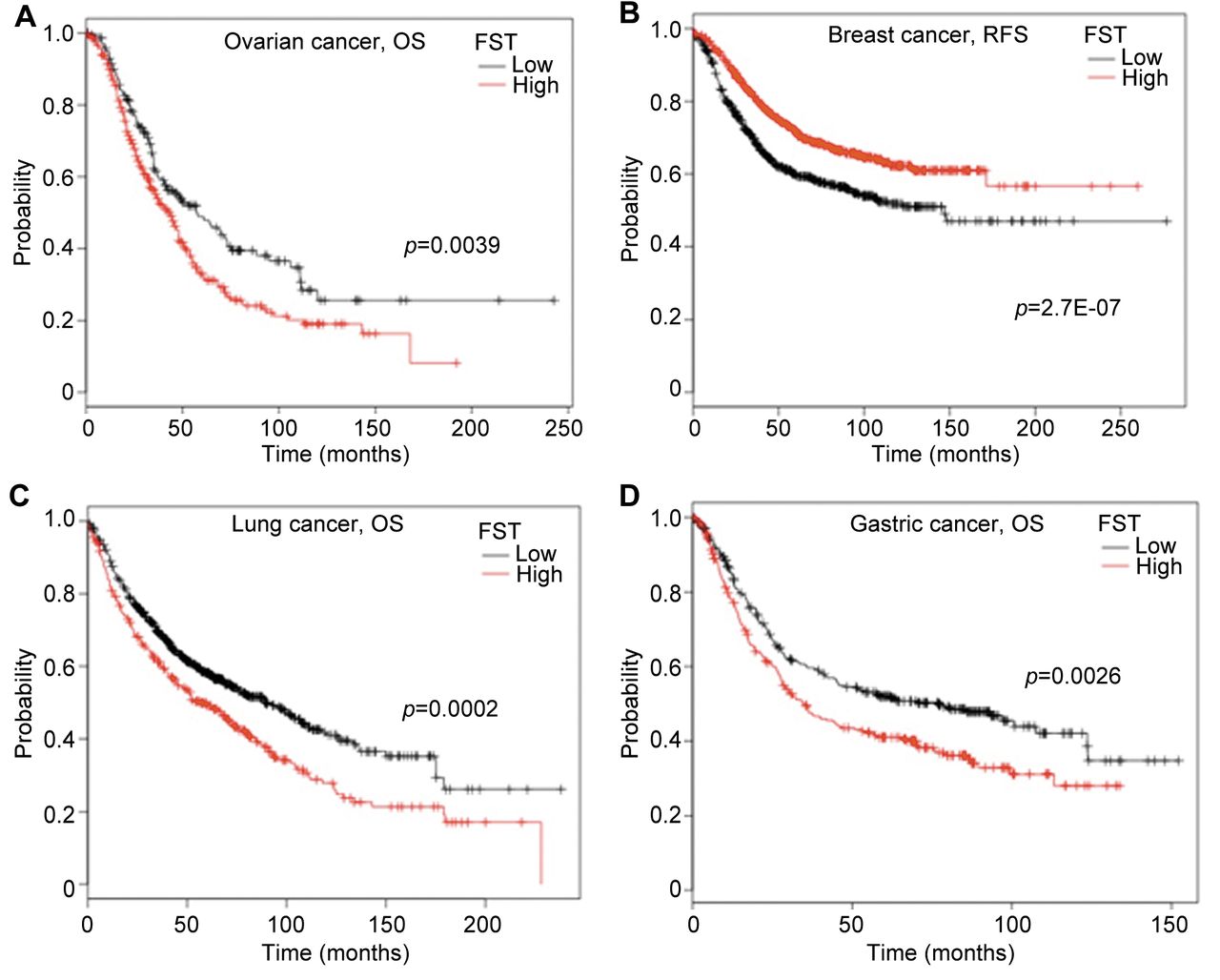
Expression of follistatin (FST) is associated with survival of patients with cancer. The association with survival of patients with ovarian, breast, lung and gastric cancer was analyzed in a combined cohort of these cancer types using an online Kaplan–Meier survival analysis tool (KMplot, http://kmplot.com/analysis/) (107). A: High expression of FST was found to be associated with shorter overall survival (OS) of patients with ovarian cancer. B: High FST expression was associated with relapse-free survival (RFS) of patients with breast cancer. C: High expression of FST was associated with shorter OS of patients with lung cancer. D: High FST expression was associated with shorter OS of patients with gastric cancer. Number of patients of each cohort are provided in Table III. The cut-off levels were auto-selected using the KMplot and number of patients in each group are listed in Table III.
Number of patients and cut-off levels of follistatin (FST) for the Kaplan–Meier survival analyses.
Other than the potential from diagnostic and prognostic perspectives, FST might also be considered as a therapeutic target. Grusch et al. reported that FST was overexpressed while both activin subunits were down-regulated in the majority of rat and human liver tumours studied (109). Treatment of normal and pre-neoplastic hepatocytes with FST stimulated DNA synthesis preferentially in pre-neoplastic rat hepatocytes, whereas activin A repressed it. This study suggests that the balanced expression of FST and activin becomes deregulated during hepatocarcinogenesis and the sensitivity of pre-neoplastichepatocytes to activin signals suggests the activin/FST system might be a promising target for therapeutic intervention.
FST was indicated to have potential in clinical use (81, 104) in prostate cancer, but its diagnostic effectiveness was found not to be significantly superior to that of activin A or prostate-specific antigen (110), although it could still be useful as a biomarker for follow-up of patients with prostate cancer undergoing antimetastatic treatments. Together with another possibility that FST may contribute to fostering bone metastasis formation in patients with prostate cancer (58, 86-88, 111), FST may be regarded as a potential, molecular target in the treatment of metastatic bone disease.
Conclusion
Based on current findings, FST plays a role in tumourigenesis, progression, metastasis and angiogenesis of solid tumours. This molecule seems to exert its effect mainly by interaction with TGFβ family members, such as activin and BMPs. As an antagonist of activin, FST seems primarily to function depending on the role that activin plays, exerting context-dependent and cell type-specific inhibition and activation (40), including a stimulating effect observed in activion of the FST-stimulating effect on gastric (63, 112) and prostate (23-25, 36, 81) cancer, while playing an inhibitory role in gonadal tumours (81, 82). FST288 has more inhibitory effects on activin-induced biological action than FST315, which has been supported by a series of studies (23-25, 36, 81). Future investigations can elucidate the exact role played by the FST288 isoform in solid tumours and also the corresponding application in diagnosis and treatment.
Acknowledgements
The Authors wish to thank Cancer Research Wales, Welsh Life Science Network (NRN, Ser Cymru) and the Wales Metastasis Platform for their support. Dr Shi was a recipient of the China Medical Scholarship from Cardiff University.
- Received September 5, 2016.
- Revision received October 1, 2016.
- Accepted October 5, 2016.
- Copyright© 2016, International Institute of Anticancer Research (Dr. John G. Delinasios), All rights reserved
References
In this issue




{kind=link}
Jump to section
Related Articles
Cited By...
- Genetically Defined Syngeneic Mouse Models of Ovarian Cancer as Tools for the Discovery of Combination Immunotherapy
- Patient-derived tumor-like cell clusters for drug testing in cancer therapy
- Surgical Resection and Outcome of Synchronous and Metachronous Primary Lung Cancer in Breast Cancer Patients
- Increased Expression of Follistatin in Breast Cancer Reduces Invasiveness and Clinically Correlates with Better Survival