


Abstract
Background: Circulating mRNA is a less invasive and more easily accessed source of samples for biomedical research and clinical applications. However, it is of poor quality. We explored and compared the ability of two high-throughput platforms for the profiling of circulating mRNA regarding their ability to retrieve useful information out of this type of samples. Materials and Methods: Circulating mRNAs from three non-small cell lung cancer patients and three healthy controls were analyzed by the cDNA-mediated annealing, selection, extension, and ligation (DASL) assay and high-throughput RNA sequencing (RSEQ). Twelve genes were selected for further confirmation by reverse transcription-quantitative polymerase chain reaction (RT-qPCR). Results: The overall expression profiles derived from the two platforms showed modest-to-moderate correlation. Genes with higher expression levels had higher cross-platform concordance than those of medium- and low-expression levels. In addition, the pathway signatures identified by gene set enrichment analysis from both platforms were in agreement. The RT-q PCR results for the selected genes correlated well with that of RSEQ. Conclusion: Genes with higher expression levels have cross-platform concordance and can be potential biomarkers. Furthermore, RSEQ is a better tool for profiling circulating mRNAs.
As an important component of liquid biopsies from peripheral blood and other body fluids, circulating RNAs, have recently attracted interest because they are a less invasive, easily-accessed source of samples for biomedical research or clinical applications (1, 2). Circulating RNA is protected in membrane vesicles released from many tissues of the body, including cancer tissues. Because circulating RNA reflects the status of its parent cells, specific circulating RNA species have the potential to serve as biomarkers for cancer diagnosis or therapeutic monitoring. Among these RNA species, small non-coding RNAs, including microRNAs (miRNAs) and small nuclear RNAs (snRNAs), are more abundant and stable, making them easier to detect. It has been suggested that several small non-coding RNAs in circulation are associated with cancerous diseases (3, 4).
Through systemic RNA profiling with high-throughput techniques, such as microarray analysis or next-generation sequencing, many circulating RNA transcripts have been found to be highly correlated with cancers and are considered potential biomarkers for diagnostics (5-7), prognostics (8), long-term surveillance (9), and evaluation of therapeutic efficacy (10). However, while most achievements on circulating RNA profiling focus on small RNA species, few reports are addressing mRNA because it is more diverse, fragmented, and less abundant in the circulation, making it relatively more difficult to detect.
Few methods can be applied to detect low-abundant and fragmented RNA. One is the cDNA-mediated annealing, selection, extension, and ligation (DASL) assay. Another is high-throughput RNA sequencing (RSEQ). The microarray-based DASL assay was originally designed for profiling RNA from formalin-fixed, paraffin-embedded (FFPE) tissues. The fragmented RNAs are first reverse transcribed into cDNA with designed oligomers and then amplified with polymerase chain reaction (PCR). The PCR products are hybridized to gene-specific probes on a beadchip for signal detection. This strategy can enhance opportunities for detecting low levels of fragmented RNA molecules from FFPE tissues (11-14). These studies demonstrate the potential of DASL assay in profiling circulating RNAs.
RSEQ is widely used to survey transcriptomic profiles in different types of samples. In addition to digital gene expression, it can also identify alternatively spliced transcripts as well as strand-specific expression. RSEQ has been used for profiling FFPE samples (15) and liquid biopsy (16). Many novel biomarkers in different RNA species have been identified through this approach (5, 17-20).
In the present study, we explore the ability of two methods to profile circulating mRNA. The expressing data derived from both platforms are compared and validated with reverse transcription (RT)-quantitative PCR (qPCR). The DASL assay and RSEQ have better concordance in genes that have high expression levels than in the genes with medium and low expression levels. In addition, RSEQ correlates better with RT-qPCR.
Materials and Methods
Sample preparation. Plasma samples from three non-small cell lung cancer (NSCLC) patients and three healthy volunteers were used. Informed consent was obtained from each attendee. This study was approved by the Institutional Review Board of Chang Gung Memorial Hospital (approval number: 100-2065A3).
In order to prepare the plasma samples, 10 ml of venous blood from each attendee was drawn into a blood-collecting tube with K3EDTA as an anti-coagulant. After centrifugation at 1,600 × g, the plasma was collected and stored at −80°C until use. Circulating RNAs in the samples were extracted with TRIzol-LS reagent (Thermo Fisher Scientific, MA, USA) according to the manufacturer's instructions. The RNA concentration was quantitated with NanoDrop-1000 (Thermo Fisher Scientific). The RNA quality, especially integrity, was measured with a Bio-Analyzer 2100 (Agilent, CA, USA).
DASL assay and RSEQ assay. Aliquots of 500 ng of circulating RNA were used for each DASL and RSEQ assay. The assays were outsourced to an Illumina certificated service provider, Genetech Biotech (Taipei, Taiwan). For the whole-genome DASL assay, the RNA samples were reverse transcribed, amplified, and hybridized to Human HT-12 v4 Expression BeadChip, which contains probes for 23,811 coding transcripts. For the RSEQ assay, the RNA samples were subjected to library construction with the ScriptSeq-V2 kit (Epicentre) except that the fragmentation step was skipped in the procedures before ribosomal RNA depletion. The libraries were then sequenced using a MiSeq sequencer. More than 10 million single-end reads with 100-bp in length were generated for each sample. The sequencing data were analyzed by the TopHat-Cufflinks pipeline (21). Briefly, five nucleotides and ten nucleotides were trimmed from the 5’- and 3’-ends of each read, respectively. The trimmed reads were mapped to human genome (hg19, UCSC) with TopHat. Then, the expression level of each transcript was calculated as reads per kilobase per million mapped reads (RPKM) with Cufflinks.
Gene set enrichment analysis. To identify the biosignatures of circulating RNAs, gene set enrichment analysis (GSEA) was introduced. The gene expression level determined through either the RSEQ or DASL assay was processed in the GSEA module of GenePattern software (22). During analysis, the permutation type was assigned as “Gene Set”, which were based on the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (c2.cp.kegg.V4.0.symbols). The differences in the expression for each gene between the NSCLS patients and healthy volunteers were ranked according to the signal-to-noise ratio (SNR). The higher SNR value indicate a higher expression level in NSCLC patients than in healthy volunteers. According to the SNR of each gene, the KEGG pathways that were overexpressed in either patients or healthy volunteers were selected. The normalized enrich score (NES) represented activation in NSCL patients.
Reverse-transcription and quantitative PCR. Twelve gene transcripts were selected for further confirmation by RT-qPCR, including five genes that showed high-expression levels in RSEQ and low expression levels in the DASL assay, four genes that showed low expression levels in RSEQ and high expression levels in the DASL assay, and three housekeeping genes that showed high expression levels in both platforms. The detailed information is shown in Table I. Reverse-transcription of RNA was conducted using random hexamers andM-MLV reverse transcriptase in the supplied buffer and dNTPs at 42°C for 90 min. The resulting cDNAs were used as templates for qPCR for specific genes. The PCR mixture contained the cDNA templates and specific primers, 50 mM Tris-HCl (pH8.5), 2% BSA, 3 mM MgCl2, 200 μM of each dNTP, 0.5X SYBR green I, and 0.5 U Platinum Taq. PCR was conducted on a LightCycler 480 (Roche, Basel, Switzerland). The thermal profile used for gene amplification was as follows: a pre-incubation step at 95°C for 3min, which was followed by 45 amplification cycles consisting of 3 steps: 95°C, 5 s; 60°C, 15 s; and 72°C, 30 s. During PCR, fluorescent acquisition of SYBR green I was conducted at the end of 60°C incubation of each cycle.
Statistics. The Pearson's correlation coefficient was applied to evaluate the correlations between the following: (1) the expression profiles generated by DASL and RSEQ assays, (2) the pathways identified from the data of DASL and RSEQ assays, and (3) the relative expression levels of selected genes between RT-qPCR and either the DASL or RSEQ assay.
The correlation between expression profiles derived from the DASL and RSEQ assays was based on the quantile-normalized gene expression level of both platforms. The correlation between pathways identified by the two platforms was according to the NES values calculated by GSEA. The correlation between the results of RT-qPCR and either DASL or RSEQ assays was based on the CT value of qPCR and the correspondent log2-transformed expression level of each platform.
Results
RNAs from the plasma of three non-small cell lung cancer (NSCLC) patients and three age-matched healthy volunteers were extracted using TRIzol-LS protocol. RNA yields of the samples were sufficient, with a range from 1 to 3 μg, but the integrity of the RNAs were far below the demand of most high-throughput assays. These samples were used for mRNA profiling with RSEQ and DASL assay.
Primer sets for candidate genes.
Data analysis for RSEQ and the DASL assay. The sequencing results contained 10 to 13 million 100-bp single-end reads for each sample. To quickly access the read distribution, we mapped the reads to human RNA (GRCh37, NCBI) and DNA sequences (hg19, UCSC) using Borrows-Wheeler Aligner (BWA). We found that approximately 20-42% of the reads could be matched to human RNA and 51-70% to human genomic DNA; the other 5-10% could not be matched to either sequences. Among the matched RNA reads, most (around 85%) were duplicate reads, suggesting a potential bias due to excess amplification from the low RNA input. The reads were then analyzed with the TopHat-Cufflinks pipeline, which is commonly used for computing gene expression profile. The expression level of each transcript was represented in RPKM format. The transcripts recorded in the Ensemble database were analyzed. A total of 41,888 transcripts were accessed in at least one sample, which included mRNAs, microRNAs, and non-coding RNAs. The RSEQ data were available in Sequence Read Archive (SRA) database (PRJNA286036). The DASL contains probes for 29,285 transcripts. There were 20,817 transcripts identified in at least one sample in this study. The expression levels of these transcripts were computed using Genome Studio Data Analysis Software (Illumina) and have been uploaded to Gene Expression Omnibus (GEO) database (GSE69732).
Genes with higher-expression levels showed cross-platform concordance. There were 14,686 transcripts detected in both platforms and were further analyzed for cross-platform concordance. First, the expression levels of these transcripts in each sample were quantile-normalized. The correlations between the DASL and RSEQ results are shown as a scatter plot with Pearson's correlation coefficients. In the example of N11 and P08, the Pearson's correlation coefficients were 0.6028 and 0.4628, respectively, representing moderate correlations (Figure 1). Overall, there were 4 in 6 samples that had coefficients ranging from 0.4-0.7 (Figure 2). Leading to further dividing the genes into three groups, low-expression (the bottom 33%), medium-expression (the meddle 33%), and high-expression (the top 33%), based on the data of the DASL assay. The Pearson's correlation coefficients were much higher in the high expression groups than that in the medium and low expression groups (Figures 1 and 2).

Scatter plot of gene expression level determined by the DASL assay and RNA sequencing. RNA expression profiles for samples N11 and P08 are demonstrated in this Figure. Based on the results of the DASL assay, all genes were separated into three groups: the high-expression, medium-expression, and low-expression group. The quantile-normalized gene expression level derived from DASL assay (QTLDASL) and RNA-sequencing (QTLRSEQ) for each group was plotted. The Pearson's correlation coefficient between the two platforms in each group was shown on the top of each plot.
Most enriched pathways were in agreement in the expression profiles determined by both platforms. The expression profiles in the three cancer patients and three healthy controls determined by either platform were then analyzed for pathway signatures using GSEA. The NES of each pathway was calculated based on signal-to-noise ratio. A positive NES indicated that the pathway was activated in the cancer group, whereas a negative one indicated that the pathway was down-regulated. The distribution of NES in most pathways were concordant from the data of both platforms (110 had negative NES and 21 had positive NES), only 32 pathways were discordant, with a Pearson's correlation coefficient of 0.6459 (Figure 3). These results suggested that most pathway signatures determined by the two platforms were in agreement.
Gene expression level determined by RSEQ showed higher concordance to that determined with RT-qPCR. RT-qPCR was used to confirm the gene expression determined by the two platforms. Twelve genes were selected as targets; five of them showed high-expression levels in RSEQ and low-expression levels in the DASL assay, four of them showed low expression levels in RSEQ and high-expression levels in the DASL assay, and the other three were housekeeping genes that showed high-expression levels in both platforms. The correlations of the expression levels determined using the two methods in each sample were shown in Figure 4A and 4B. The Pearson's correlation coefficient between the RT-qPCR and DASL assays ranged from 0.23 to 0.54 (modest to moderate correlation), without significance. The correlation between RT-qPCR and RSEQ ranged from 0.56 to 0.70 (moderate-to-high correlation) with statistical significance (p-value <0.05). These results demonstrated a higher concordance between RT-qPCR and RSEQ.
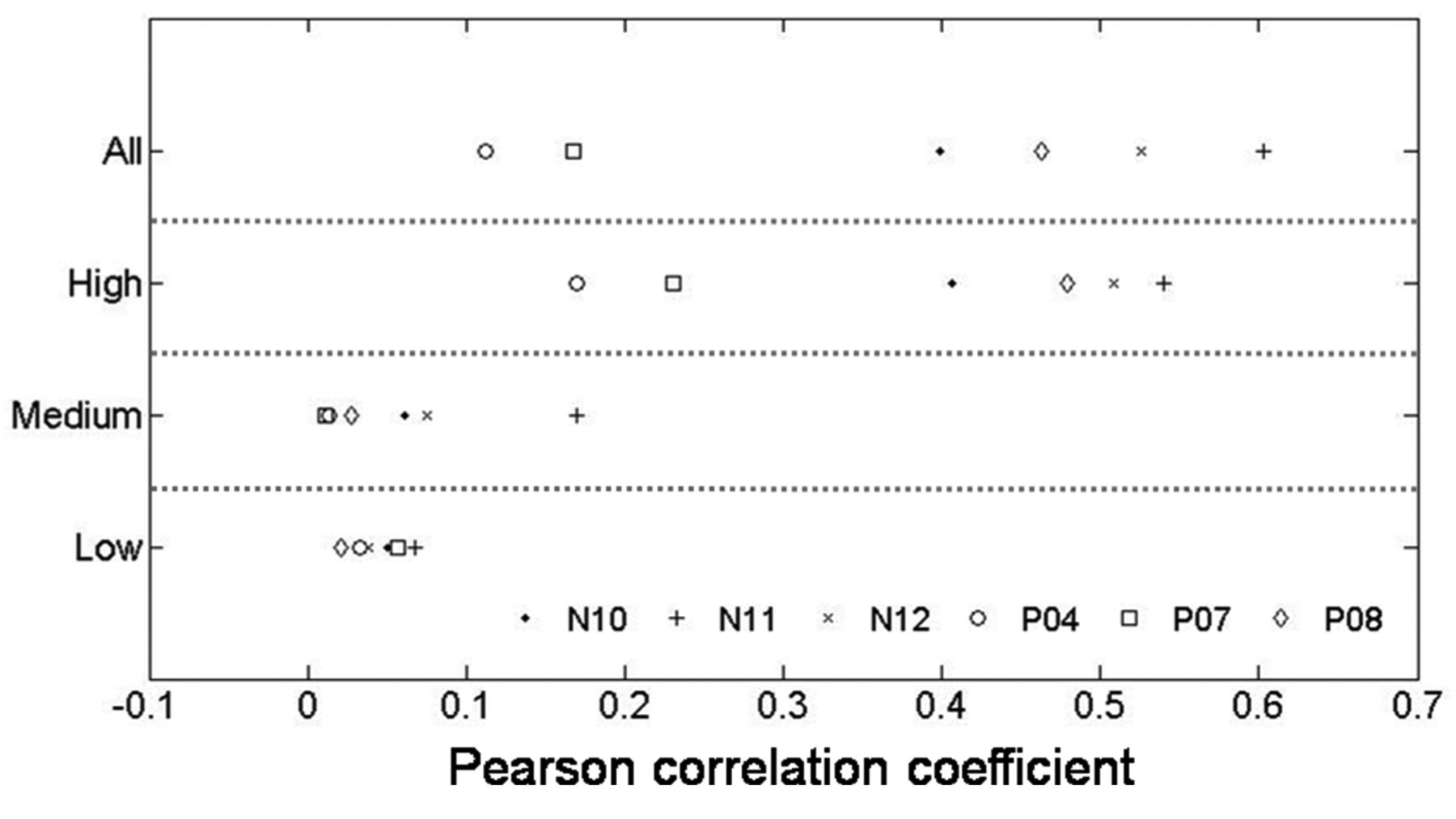
Correlation of the expression data determined by the DASL assay and RNA sequencing. The Pearson's correlation coefficients for genes with different expression levels in each sample were plotted.

Correlation of the pathway signatures derived from the two platforms. The pathways or gene sets that were enriched in the expression data derived from the two platforms were analyzed by GSEA. The normalized enrichment score (NES) of each pathway in both platforms was plotted.
Discussion
In clinical practice, 5-10 ml volume is regular for a single blood draw. The quantity and quality of circulating mRNA in the above blood sample is low. To test if high-throughput methods can be applied to profile mRNA and retrieve useful information from this type of samples, we explored and compared the ability of two platforms, the microarray-based DASL assay and high-throughput sequencing-based RSEQ. The reason is that both methods have a potential to accept fragmented RNAs: The DASL assay is designed for degraded RNA from FFPE tissues; RSEQ has a RNA fragmentation step during library preparation, so an input of degraded RNA might be processed directly. In addition, both methods have a PCR step so a small amount of RNA can be amplified before signal acquisition.
We were pleased to find that the expression profiles derived from both methods in most samples had moderate correlations. The expression profiles in the high-expression genes had a stronger correlation than that in the medium- and low-expression genes. In addition, the pathway signatures identified from the profiles also had good concordance between both platforms. We also found that the RSEQ results were more concordant to RT-qPCR than the DASL results, suggesting that RSEQ and RT-qPCR have similar efficiency and dynamic range for the quantitation of circulating mRNA.

Correlation of the expression level determined by RT-qPCR and each platform. Expression levels of 12 genes were determined by RT-qPCR to confirm the data derived from the DASL and RSEQ assays. A, The expression level of each gene determined by both platforms and the corresponding CT value determined by RT-qPCR. B, The summary of Pearson's correlation coefficients between RT-qPCR and each platform in all patients is shown.
The fact that both platforms identified similar pathway signatures suggests that finding cancer-related pathways and biomarkers from circulating mRNA may be possible. In the present study, the most markedly enriched pathways or gene sets are those involved in olfactory receptors, the WNT pathway, amino acid metabolism, neuron-associated receptors, and cytochrome-related genes. All the pathways or biological functions have been reported to be associated with cancer development (23-28), further supporting this argument.
Taken together the above facts, we have demonstrated that circulating mRNA can be profiled using the DASL and RSEQ assays. Genes with higher expression levels have better cross-platform concordance. The expression profiles determined by the high-throughput methods have the potential to identify cancer-related pathways and biomarkers in circulation.
Acknowledgments
This work was supported by the Ministry of Science and Technology, ROC (NSC 97-2320-B-182-016-MY3) and Chang Gung Memorial Hospital (CMRPD1B0091).
Footnotes
↵* These Authors contributed equally to this study.
- Received June 12, 2015.
- Revision received July 25, 2015.
- Accepted August 7, 2015.
- Copyright© 2015, International Institute of Anticancer Research (Dr. John G. Delinasios), All rights reserved




{kind=link}
{kind=link}
{kind=link}
{kind=link}