


Abstract
Background: WNT inducible secreted protein 2 (WISP2) has been linked with a variety of human cancer types and may contribute to cancer metastasis. The current study investigated the importance of WISP2 in colorectal cancer cells, examining the impact of targeting WISP2 on Caco-2 cell invasion and motility together with potential mechanisms of action. Materials and Methods: WISP2 expression was targeted in Caco-2 cells using a ribozyme transgene system and successful knockdown was verified using reverse transcription-polymerase chain reaction (RT-PCR). The impact of WISP2 knockout (Caco-2WISP2 KO) on cell growth, adhesion, motility and invasion was examined using a number of in vitro functional assays. In vitro invasion assays were repeated in the presence of wingless-type MMTV integration site family (WNT) inhibitors (FH535 and IWP-2) to investigate the role of the WNT-signalling pathway in the regulation of cell invasion by WISP2. Quantitative-PCR was conducted to measure matrix metalloproteinase (MMP) expression in control [wild-type (Caco-2WT) and cells containing the empty pEF6 plasmid (Caco-2pEF6)] and Caco-2WISP2 KO cells. Results: WISP2 knockout resulted in a significant increase in Caco-2 cell invasion and motility (p<0.05 in comparison to wild-type and plasmid control Caco-2 cells). WISP2 knockout had no significant effect on Caco-2 cell growth rate in 3- and 5-day incubation and no significant impact on Caco-2 cell-matrix adhesion rates (p>0.05). Expression analysis of a number of MMPs indicated an insignificant up-regulation of MMP2, MMP9 (p>0.05) but significant up-regulation of MMP7 (p=0.025) in Caco-2WISP2 KO cells compared to controls. Inhibition of WNT signalling using FH535 and IWP-2 brought about a significant or borderline significant decrease in Caco-2WISP2 KO cell invasion (FH535 p=0.065) and (IWP-2 p=0.002) and negated the pro-invasive effect of targeting WISP2 in Caco-2 cells. Conclusion: WISP2 knockout significantly increased Caco-2 cell invasion and motility. Up-regulation of MMP2, -7 and -9 may indicate that WISP2 regulates invasion and motility through MMPs. Regulation of invasion by WISP2 may involve the WNT signalling pathway.
Colorectal cancer (CRC) is the second most common cause of cancer death in Europe and the third most commonly diagnosed cancer in the U.K. (1). Patients with CRC usually die due to metastasis, most commonly to the liver. Complex molecular mechanisms are required for CRC metastasis. Studies have shown that signalling molecules and pathways involved in adhesion, invasion, motility and growth of cancer cells may be important in the metastatic process (2). Wingless-type MMTV integration site family, member 1 (WNT1) induced secreted proteins (WISPs) are members of the cysteine-rich 61, connective tissue growth factor, nephroblastoma overexpressed (CCN) group of proteins, involved in cell migration, cell mitosis, angiogenesis, adhesion and apoptosis (3). WISP1, -2 and -3 are downstream components of the WNT signalling pathway and are commonly up-regulated in WNT1 transformed cells (4). CRC tumourigenesis is closely associated with mutations in genes which code for proteins of the β-catenin and WNT signalling pathway. Mutations such as these can result in aberrant activation of WNT signalling in the absence of WNT ligand binding (5, 6). All three WISP molecules have been implicated in tumourigenesis and metaplasia in numerous tissues.
WISP1 is thought to act to promote cancer progression in CRC, with elevated WISP1 expression being associated with higher grade, poorly differentiated colorectal tumours and a worse prognostic outcome (4). Tian et al. also noted that levels of WISP1 expression in surgically excised rectal tumours were associated with Dukes’ staging, differentiation grade and lymph node status (7). Increased expression of WISP1 has also been observed in numerous CRC cell lines (SW40, COLO, 320DM, HT-19, HT-29, WiDr and SW403) (11). Studies by Pennica et al. and Khor et al. have demonstrated elevated levels of WISP1 expression in surgical specimens of CRC compared to normal tissues (8, 9).
In contrast WISP2 may act as a tumour suppressor gene in CRC. Significantly lower levels of WISP2 expression have been found in CRC tissue compared to normal colorectal tissue (4, 8). The role of WISP3 in CRC is yet to be defined but evidence suggests that it may act to promote the aggressiveness of the cancer. Although Davies et al. observed higher levels of WISP3 expression in CRC specimens compared to normal colorectal tissue, these results were not significant (4). Pennica et al. also found overexpression of WISP3 in 63% of colorectal tumours examined, reporting overexpression ranging from 4- to 40-fold compared to normal adjacent mucosa (8).
The WISP2 gene is situated on 20q12-20q13 and has been strongly implicated in carcinogenesis. The role of WISP2 seems to differ depending on the cancer tissue type involved, possibly because its expression may become modulated depending upon the particular cellular context (10). WISP2 may also act to promote cancer aggression in prostate cancer and one study suggested that suppression of WISP2 in prostate cancer cells reduced tumour metastatic potential (11). WISP2 ma, however, act as a tumour suppressor gene in pancreatic adenocarcinoma, human salivary gland tumours and hepatocellular carcinoma (12-14). In pancreatic adenocarcinoma, reduced WISP2 expression was associated with more undifferentiated tumours and a higher rate of tumour progression (13). Additionally, in salivary gland tumours, loss and down-regulation of WISP2 may be associated with tumour development (14). WISP2 may, however, act as a protective factor in breast cancer progression. Banerjee et al. reported a biphasic pattern of WISP2 expression, with undetectable levels in normal mammary tissues, increased expression in non-invasive breast lesions, and decreased levels of WISP2 expression in invasive, poorly differentiated breast adenocarcinomas (15). Results of this study also indicated that higher levels of WISP2 were detectable in well-differentiated and moderately differentiated tumours compared to poorly differentiated tumours.
WISP2 may perform its role as a protective factor in breast cancer by negatively regulating cell migration and invasion, as well as controlling cell adhesion and motility (15). Studies suggest WISP2 has a role in preventing the process of epithelial-to-mesenchymal transition (EMT) in breast cancer (16). Loss of WISP2 expression, as observed in the progression of non-invasive lesions to invasive breast cancer, may therefore lead to increased breast tumour cell invasiveness, growth and migration (15). The mechanism through which WISP2 plays these roles in cancer remains to be elucidated fully, although a number of potential mechanisms have been identified. WISP2 may act as a transcriptional repressor of genes associated with EMT in breast cancer. WISP2 has also been shown to restrict transcription of the transforming growth factor receptor-beta receptor II (TGF-βRII) and therefore reduce TGF-β signalling. Loss of WISP2 expression in breast cancer may therefore lead to increased TGF-β signalling, cell invasiveness and EMT (17). WISP2 may also be linked to the p53 pathway. One study showed that p53 overexpression in pancreatic adenocarcinoma cells was directly correlated with loss of WISP2 expression. Loss of WISP2 expression was found to be closely associated with pancreatic cancer progression as WISP2 is thought to have a protective role against EMT (13). Dhar et al. reported an inverse association between the level of mutant p53 and WISP2 expression in breast cancer cells and suggest that WISP2 inactivation may be essential for the development of an invasiveness phenotype in p53-mutant breast cancer cells (18).
There has been little research to investigate the role of WISP2 in CRC. The current study aimed to investigate the effect of WISP2 knockout on Caco-2 CRC cell growth, adhesion, invasion and motility. Additionally, the study aimed to assess matrix metalloprotease (MMP) and WNT-signalling pathways as potential mechanisms for WISP2 regulation of Caco-2 cell motility and invasion.
Materials and Methods
Materials. Caco-2, HRT-18 and RKO CRC cell lines were purchased from the American Type Culture Collection (ATCC, Rockville, MD, USA). Wild-type cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% foetal calf serum (FCS), penicillin, streptomycin and amphotericin B. Cells transfected with control pEF6 plasmids (Invitrogen, Paisley, UK) or pEF6 plasmids containing WISP2 ribozyme transgenes were maintained in blasticidin maintenance medium (Dulbecco’s modified Eagle’s medium, 10% FCS, penicillin, streptomycin, amphotericin B and blasticidin 0.5 μg/ml).
IWP-2 (Tocris Bioscience, Bristol, UK), at concentrations of 2.5 nM and 25 nM or FH535 (Tocris Bioscience), at concentrations of 0.1 μM, 1 μM and 10 μM, were used to inhibit WNT-signalling and establish the involvement of this pathway in the regulation of CRC cell invasion and motility by WISP2.
Generation of WISP2 knockdown cells using a ribozyme transgene system. Ribozyme transgenes specific for WISP2 were synthesised and cloned into pEF6/V5- His TOPO TA expression plasmids in line with the manufacturers protocol (Invitrogen, Paisley, UK). WISP2 ribozyme transgene plasmids and control pEF6 plasmids were subsequently transfected into Caco-2 cells using an electroporator (Easyjet, Flowgene, Surrey, UK). Cells were placed in normal medium overnight to allow recovery and then in selection medium (normal medium containing blasticidin 5 μg/ml) for eight days to select for cells containing the plasmid. Following selection, cells were placed into a maintenance medium (0.5 μg/ml blasticidin) and reverse transcription polymerase chain reaction (RT-PCR) was used to confirm WISP2 knockout in cells transfected with WISP2 ribozyme transgene plasmids. These cells (Caco-2WISP2 KO) were subsequently used in the in vitro studies.
Primers used in the study.
Quantitative-polymerase chain reaction (Q-PCR) primers used in the study.
RNA isolation and RT-PCR. RNA was generated from confluent cells in a 25 cm2 flask in accordance with the supplied protocol (Sigma, Dorset, UK). Once extracted, RNA was quantified using a UV1101 Biotech Photometer (WPA, Cambridge, UK). Samples were standardised to 500 ng and reverse transcription undertaken using iScript cDNA synthesis kit (BioRad, Hertfordshire, UK) to generate cDNA. The quality of the cDNA was assessed using glyceraldehyde-3-phosphate dehydrogenase (GAPDH). WISP2 expression was also probed (see Table I for list of primers). PCR was conducted using REDTaq® ReadyMix™ PCR Reaction Mix (Sigma) in a T-Cy Thermocycler (Creacon Technologies Ltd., the Netherlands). Reaction conditions were: 94°C for 5 minutes (initial denaturation), followed by 36 cycles of 94°C for 40 seconds, 55°C for 40 seconds and 72°C for 60 seconds. Final extension was 10 minutes at 72°C. PCR products were loaded onto an agarose gel and electrophoresis was conducted. The gel was stained with ethidium bromide and visualised using a UV light.
Quantitative polymerase chain reaction (Q-PCR). The level of MMP2, MMP7 and MMP9 transcripts were determined using real-time Q-PCR (see Table II for Q-PCR primer sequences). Q-PCR was carried out using the iCycler iQ™ system (Bio-Rad) and custom hot-start Q-PCR master mix using a method previously described (19). The reactions conditions were: 95°C for 15 minutes, followed by 50 cycles of 95°C for 15 seconds, 55°C for 4 seconds and 72° C for 15 seconds. The levels of MMP transcripts were normalised using GAPDH.
In vitro growth assay. Growth assays were carried out using a method previously described (20). Cells were seeded at a density of 3,000 cells per well. Three replicate plates were prepared (overnight, day 3 and day 5). The plates were incubated for the appropriate period of time before being fixed with 4% formaldehyde [v/v in balanced salt solution (BSS)], stained with 0.5% (w/v) crystal violet and washed. The stain was extracted using 10% (v/v) acetic acid and absorbencies calculated using a plate spectrophotometer (EL×800, Bio-Tek; Wolf Laboratories, York, UK). Cell growth percentage increase was calculated using the overnight plates as a reference.
In vitro Matrigel adhesion assay. Adhesion assays were conducted using a method previously described (21). Wells were pre-coated with 5 μg of Matrigel Matrix Basement Membrane (BD Biosciences, Oxford, UK) and rehydrated prior to use. A total of 45,000 cells were seeded onto the Matrigel and incubated for 45 minutes. Following incubation, unadhered cells were removed through washes with BSS. Adherent cells were subsequently fixed with 4% formaldehyde (v/v in BSS), stained with 0.5% (w/v) crystal violet and washed. Randomly selected fields per well were photographed under a microscope (×200 magnification) and the number of adhered cells counted.
In vitro Matrigel invasion assay. Invasion assays were carried out using a method previously described (22). Culture plate inserts containing 8 μm pores were pre-coated with 50 μg of Matrigel Matrix Basement Membrane (BD Biosciences) and rehydrated briefly before seeding 30,000 cells. The plate was incubated for three days. Cells which invaded to the underside of the inserts were fixed with 4% formaldehyde (v/v in BSS), stained with 0.5% (w/v) crystal violet and washed. Randomly selected fields per insert were photographed under a microscope (×200 magnification) and the number of cells observed counted.

A: Reverse transcription-polymerase chain reaction (RT-PCR) capture demonstrating WNT1 inducible signalling pathway protein 2 (WISP2) expression profile in RKO, Caco-2 and HRT-18 colorectal cancer cells. B: RT-PCR showing successful knockdown of WISP2 in Caco-2 cells transfected with the WISP2 ribozyme transgene in comparison to wild-type and empty plasmid controls. NC: Negative control.
In vitro cytodex bead motility assay. Motility assays were carried out using a method previously described (23, 24). A total of 1×106 cells were added into a universal container and topped up with normal medium. Cytodex-2 carrier beads were added before incubating overnight. The cell/bead pellet formed was washed twice, through centrifugation and removal of the supernatant. After the final wash, the cell/bead pellet was resuspended in medium before being aliquoted into a 96-well plate and incubated for four hours. The plate was washed twice with BSS to remove unadhered cells and beads before adhered cells were fixed with 4% formaldehyde (v/v in BSS), stained with 0.5% (w/v) crystal violet and washed. The stain was extracted using 10% (v/v) acetic acid and absorbencies calculated using a plate spectrophotometer (EL×800, Bio-Tek; Wolf Laboratories).
Statistical analysis. At least three independent repetitions were carried out for all in vitro assays. Statistical analysis was carried out using the SigmaPlot statistical software package (Systat Software Inc., London, UK). Data with a normal distribution and equal variances were compared using either a two-sample, two-tailed t-test or a one-way analysis of variance (Holm-Sidak method). Data which were not normalised were analysed using a Mann-Whitney U-test or a Kruskal-Wallis one-way analysis of variance on ranks. Results were considered significant at p≤0.05.
Results
Caco-2 cells express high levels of WISP2. RKO, Caco-2 and HRT18 cells were screened for WISP2 expression using conventional PCR. WISP2 was found to be expressed in Caco-2 cells and to a much lesser extent RKO (Figure 1A). Uniform GAPDH expression was seen in all cell types.
Verification of WISP2 knockdown in Caco-2 colorectal cells. Caco-2 cells demonstrated a strong expression of WISP2 and were subsequently chosen as a cell model to target WISP2 expression. Following transfection with the ribozyme transgene targeted to WISP2 and selection of cells containing the plasmid, RT-PCR was used to verify knockdown of WISP2. RT-PCR confirmed successful WISP2 knockout in Caco-2 cells transfected with the WISP2 ribozyme transgene (Caco-2WISP2 KO) and expression of WISP2 in this cell line was substantially reduced in comparison to cells containing a closed plasmid only (Caco-2pEF6) and to wild-type cells (Caco-2WT) (Figure 1B).
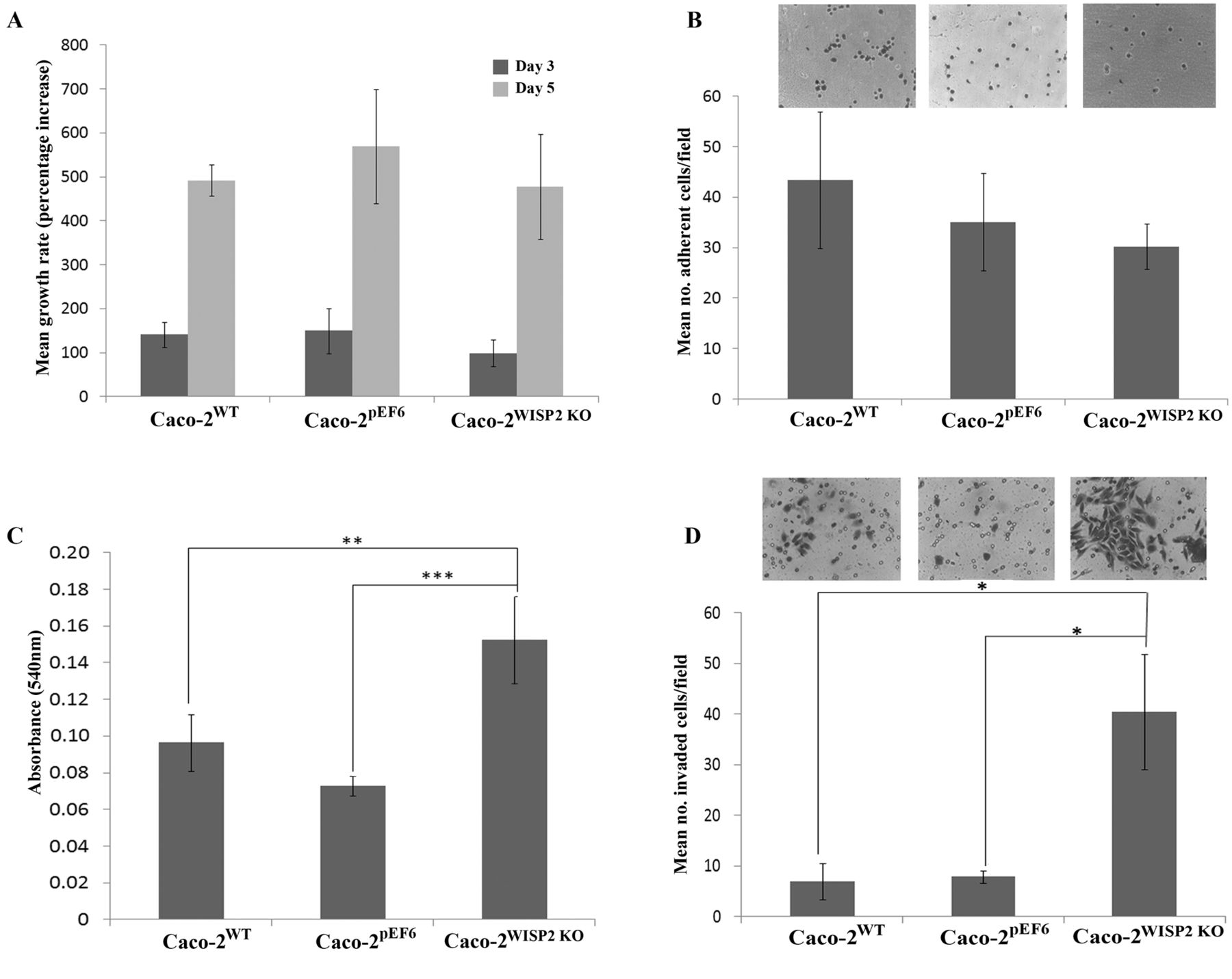
WISP2 knockout does not affect Caco-2 cell growth. Growth assays were used to assess the effect of WISP2 knockout on Caco-2 cell growth rates. There was no significant difference in growth rate at day 3 between any of the cell types (p=0.481) (Figure 2A), nor was there any significant difference in growth rate observed between the cell types following 5-day incubation (p=0.623) (Figure 2A).
WISP2 knockout does not affect Caco-2 cell adhesion. The effects of WISP2 knockout on Caco-2 cell adherence to an artificial Matrigel matrix was assessed using a cell adhesion assay. There was no significant difference in the number of adherent cells (per field) between any of the cell types (p=0.443) (Figure 2B).
WISP2 knockout enhances Caco-2 cell motility. Cytodex bead cell motility assays were used to investigate the effect of WISP2 knockout on Caco-2 cell motility. The numbers of cells which had become disseminated from Cytodex beads and adhered to the tissue plate surface were quantified by measuring absorbance, following cell fixation and staining. Knockdown of WISP2 in Caco-2 cells enhanced their motility with significant increases in absorbance readings for Caco-2WISP2 KO cells (p<0.001) compared to the wild-type and pEF6 empty plasmid control cells (p=0.001) (Figure 2C).
WISP2 knockout increases Caco-2 cell invasiveness. The effect of WISP2 knockout on Caco-2 cell invasiveness was assessed using an in vitro Matrigel invasion assay. The number of cells which effectively invaded through an artificial basement membrane (Matrigel) to become adhered to the underside of the invasion insert were quantified in each cell line. Targeting of WISP2 in this cell line appeared to significantly impact on the invasiveness of the cells. There was a significant increase in the numbers of invading cells per field in Caco-2WISP2 KO cells compared to wild-type (p=0.014) and pEF6 control cells (p=0.016) (Figure 2D).

Impact of WNT1 inducible signalling pathway protein 2 (WISP2) knockdown on Caco-2 cell function. A: WISP2 knockout did not significantly affect Caco-2 cell growth rate and there was no significant difference in mean growth rate at day 3 (p=0.481) or day 5 (p=0.623) compared to the control cell lines. B: WISP2 knockdown did not significantly affect Caco-2 cell adhesion (p=0.43). C: Knockdown of WISP2 significantly impacted on Caco-2 cell motility with a significant increase in cell motility compared to Caco-2WT cells (p<0.01) and Caco-2pEF6 cells (p=0.001). D: Similarly, knockdown of WISP2 impacted on Caco-2 invasiveness with a significant increase in cell invasiveness in Caco-2WISP2 KO cells compared to Caco-2WT (p=0.014) and Caco-2pEF6 cells (p=0.016). Data shown represent mean values from a minimum of three independent repeats, error bars represent SEM. Images shown are representative. *p<0.05; **p≤0.01; ***p<0.001.
WISP2 knockout may increase Caco-2 cell invasiveness and motility due to up-regulation of MMPs. As a possible mechanism to account for the enhanced cell invasion and motility seen in WISP2-targeted Caco-2 cells, the expression levels of several MMPs were examined. Transcript levels of MMP2, MMP7 and MMP9 were analyzed in Caco-2WT, Caco-2pEF6 and Caco-2WISP2 KO cells using Q-PCR (Figure 3). The levels of MMP2 and MMP9 transcripts were found to be similar in Caco-2WT and Caco-2pEF6 cells but consistently elevated in Caco-2WISP2 KO cells. However, despite this trend the elevation in both MMP-2 and -9 in Caco-2WISP2 KO cells was not found to be significant and no statistical significances were seen within the group (p>0.05) (Figure 3A and B). MMP 7 expression was also found to be much lower in both Caco-2WT and Caco-2pEF6 cells than in those transfected with the WISP2 ribozyme transgene. Levels of MMP7 transcript were significantly elevated in Caco-2WISP2 KO cells compared to Caco-2WT cells and significant differences were observed within the group (p=0.025) (Figure 3C).

Impact of WNT1 inducible signalling pathway protein 2 (WISP2) knockout on matrix metalloproteinase (MMP) expression. A: Increased levels of MMP2 transcript were observed in Caco-2WISP2 KO cells compared to Caco-2WT and Caco-2pEF6 cells, however this was not significant (p>0.05). B: Similarly, knockdown of WISP2 also resulted in an increased expression of MMP9, however again this observation was not significant (p>0.05). C: Significantly elevated levels of MMP7 transcript were observed in Caco-2WISP2 KO cells compared to Caco-2WT cells (p=0.025). Data shown are from a representative set of results, error bars represent SD; *p<0.05.

Impact of the wingless-type MMTV integration site family (WNT) inhibitors on Caco-2 cell growth. A: No significant differences in growth rate at day 3 was observed in any of the FH535 WNT inhibitor treatment groups (p=0.07) compared to untreated cells. B: There was a significant decrease in growth rates between the control and groups treated with 2.5 nM IWP-2 (p=0.007) and 25 nM IWP-2 (p=0.001) at day 3. Data shown are of a representative set of results. Error bars represent SD. **p<0.01;***p≤0.001.
Impact of IWP-2 and FH535 WNT inhibitors on Caco-2 cells function. To further explore the potential mechanism through which WISP2 exerts its effect on Caco-2 cells, a number of functional assays were conducted using the inhibitors IWP-2 (inhibits WNT processing and signalling) and FH535 (inhibits WNT/β-catenin signalling). The cytotoxicity of these inhibitors was firstly examined in the Caco-2 cell line over a 3-day incubation period. No significant difference in mean growth rate at day 3 between any of the FH535 treatment groups were observed (p=0.07) (Figure 4A). However, a significant decrease in growth rates between the control and 2.5 nM (p <0.001) and 25 nM (p<0.001) IWP-2 treatment groups was seen following the 3-day incubation (Figure 4B).
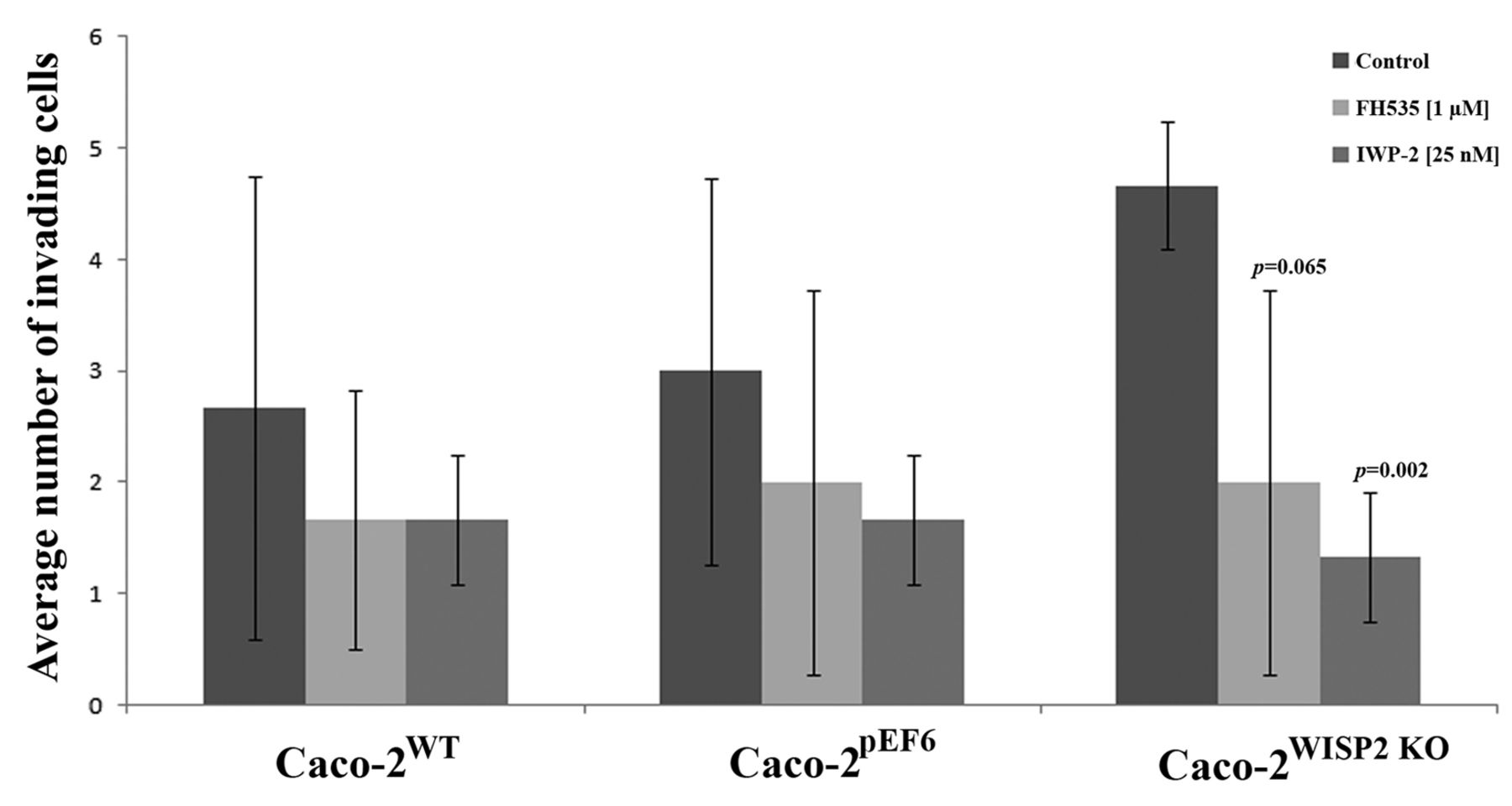
Effect of wingless-type MMTV integration site family (WNT) inhibition on Caco-2 cell invasiveness. Treatment with either FH535 or IWP-2 resulted in only slight reductions in the invasive potential of Caco-2WT and Caco-2pEF6 control cells. Treatment of Caco-2WISP2 KO cells with either inhibitor brought about substantial reductions in cellular invasion and resulted in significant (p=0.002 following IWP-2 treatment) and borderline significant (p=0.065 following FH535 treatment) reduction in the invasive potential of Caco-2WISP2 KO cells. Data shown are from a representative set of results. Error bars represent SD.
Inhibiting WNT signalling reduces WISP2 knockout cell invasiveness. In vitro invasion assays were carried out with the addition of either FH535 (1 μM) or IWP-2 (25 nM). In keeping with the basic functional data, knockdown of WISP2 in Caco-2 cells enhanced cell invasion. Following treatment with either FH535 or IWP-2 (Figure 5), a general reduction in cell invasion was observed in all cell types. However, treatment with either inhibitor appeared to have the most substantial effect on Caco-2WISP2 KO cells, with significant or close to significant decreases in invasion (FH535, p=0.065; IWP-2, p=0.002). Treatment of Caco-2WISP2 KO cells with these inhibitors appeared to negate the enhanced invasiveness seen following knockdown of WISP2, returning the level of invasion to that of the Caco-2WT and Caco-2pEF6 controls similarly treated with the inhibitors.
Discussion
Studies in breast cancer cells have suggested that WISP2 expression is required for tumour cell proliferation. Banerjee et al. reported that in MCF-7 breast tumour cells, disruption of WISP2 signalling caused a significant reduction in tumour cell proliferation (5). In a separate study, they found that inhibition of WISP2 expression also reduced proliferation of oestrogen receptor (ER)- and epidermal growth factor receptor (EGFR)-positive MCF-7 tumour cells (25). It has been proposed that complex molecular cross-talk between the phosphatidylinositol 3-kinase/v-akt murine thymoma viral oncogene (AKT) and mitogen-activated protein kinase signalling pathways and WISP2 may regulate breast tumour cell proliferation (25). Our results indicate that WISP2 may not be key in CRC cell growth, as knockdown of WISP2 in Caco-2 CRC cells did not bring about any significant impact on the growth rates of this cell line. The increased tumour cell proliferation upon WISP2 knockout in breast cancer cells and no observable effect on proliferation in CRC cells suggests that WISP2 may have different signalling roles in the two cell types. Further studies using other CRC cells lines are required to further understand the role played by WISP2 in CRC cell growth and to identify other mechanisms by which cell proliferation may be regulated by WISP2 in CRC and breast cancer cells.
Ray et al. showed that forced expression of WISP2 in breast cancer cells can result in reduced cell proliferation, migration and invasion (10). These findings are consistent with the proposed idea that loss of WISP2 expression is associated with breast cancer progression (15). In keeping with this Banerjee et al. suggested that WISP2 may negatively regulate migration and invasion in breast cancer cells through the modulation of SNAIL-E-cadherin signalling and expression of MMP2 and -9 (15). They showed that blocking WISP2 expression in MCF-7 cells resulted in the suppression of E-cadherin expression through activation of SNAIL. Increased MMP2 and MMP9 expression was also observed. Blocking of WISP2 expression in MCF-7 cells subsequently resulted in increased cell motility and invasion (15). Our results indicate that knocking down WISP2 in the Caco-2 CRC cell line had no effect on tumour cell-matrix adhesion but resulted in increased tumour cell invasiveness and motility. Our findings are consistent with those reported in breast cancer cells by Banerjee et al. (15). Our results indicate that WISP2 may be a negative regulator of MMP2, -7 and -9 in CRC cells, where targeting of WISP2 in Caco-2 cells brought about a general elevation in the expression of these MMPs. Up-regulation of these enzymes may contribute to the observed increase in cell invasiveness in WISP2 knockout cells compared to controls.
MMPs are commonly associated with cancer invasion, migration, tumourgenesis and metastasis. Degradation of extracellular matrix components (including collagen, proteoglycans, fibronectin, laminin and other glycoproteins) by MMPs facilitates invasive growth and metastasis of tumour cells, via blood and lymphatic routes around the body (26). MMPs are secreted as inactive pro-enzymes which undergo proteolytic cleavage to form active enzymes. MMP2 (gelatinase A) and MMP9 (gelatinase B) are important in the degradation of type IV collagen and gelatine (two major components of the extracellular matrix). Both experimental and clinical studies have shown that higher levels of MMP2 and MMP9 expression are associated with increased tumour aggression and cancer cell invasion (26). A recent study identified that overexpression of MMP2 protein in CRC tumours was closely associated with tumour size, lymph node metastasis, distant metastasis, Dukes’ staging and tumour invasion (27). Similarly, MMP9 expression in CRC specimens has been closely associated with CRC progression, with significantly elevated levels of MMP9 found in colorectal tumours which had metastasised to the liver (28). Higher levels of MMP2 and MMP9 expression in colorectal tumours have been associated with a poorer prognostic outcome (27, 28). Overexpression of MMP7 has also been reported in both benign and malignant colorectal tumours. Like MMP2 and MMP9, higher levels of MMP7 expression have been closely associated with more advanced CRC staging. Overexpression of MMP7 in CRC liver metastases has been commonly reported in cell lines, animal models and human tissues samples. One study also identified significantly higher levels of the activated form of MMP7 in CRC liver metastases (28). MMPs play an important role in CRC progression and specifically in cell invasion and metastasis. Understanding the role of WISP2 in the regulation of MMP2, -7 and -9 may allow a greater understanding of the invasive phenotype observed when the protective effect of WISP2 is lost in advanced CRC.
Cytotoxicity tests conducted on Caco-2 cells using the two WNT inhibitors, IWP-2 and FH535, demonstrated a significant decrease in cell growth rates following the addition of IWP-2 (2.5 nM and 25 nM) after three days incubation. This indicates that IWP-2 has some cytotoxicity at these concentrations. FH535 did not appear to have significant cytotoxic effects on Caco-2 cells and no significant differences in growth rates were seen at any test concentration over the three days incubation. For further work with these inhibitors, concentrations close to the median inhibition concentration (IC-50) values were chosen, namely 25 nM for IWP-2 and 1 μM for FH535.
Addition of either WNT inhibitor appeared to have a differential effect on cellular invasiveness across control and transfected Caco-2 cell lines. Treatment with either inhibitor brought about a slight decrease in the invasiveness of the control Caco-2WT and Caco-2pEF6 cells. Treatment of Caco-2WISP2 KO with these inhibitors substantially reduced invasiveness, negating the previous pro-invasive impact of targeting WISP2 in Caco-2 cells and returned levels of invasion of Caco-2WISP2 KO cells to that of the control. However, as indicated previously, there may be some cytotoxicity inherent with the 3-day treatment of IWP-2 and this may somewhat contribute to the reduced invasiveness brought about by IWP-2 over this 3-day invasion assay. As both control and WISP2 knockdown cells were treated for the same period of time, however, it is likely that the trend outlined in this assay is due to the inhibition of WNT. This is further implied by the invasion data following treatment with the inhibitor FH535, which similarly impact on WNT signalling but do not appear to display the significant cytotoxicity of IWP-2. These data again demonstrate that the pro-invasive effect of targeting WISP2 in Caco-2 cells can be negated and levels returned to those of treated control cells following treatment with FH535. Taken together these facts may imply that regulation of invasion by WISP2 is linked to the WNT signalling pathway. Both IWP-2 and FH535 are inhibitors of the WNT/β-catenin signalling pathway (29, 30). This implies that WISP2 regulates CRC cell invasion through the canonical WNT signalling pathway as opposed to the non-canonical WNT signalling pathways (which can include the WNT/Ca2+ pathway and the WNT/jun proto-oncogene (c-Jun) N-terminal kinase pathway).
Interestingly, two studies have proposed that activation of the WNT/β-catenin signalling pathway in CRC cells may result in increased cell invasion due to the up-regulation of MMP7. One study showed that β-catenin can activate the MMP7 promoter by interacting with transcription factor-4 (TCF4). Results showed that levels of β-catenin and MMP7 expression were closely correlated in the CRC specimens examined (31). Similar results were also published by Crawford et al. who observed up-regulation of the MMP7 promoter by β-catenin in various CRC cell lines (32). Taken together these studies may indicate a mechanism by which WISP2 regulates CRC cell invasiveness, namely targeting WISP2 may result in increased WNT-signalling which causes up-regulation of MMP7 expression. However, as yet we have no evidence to suggest that targeting WISP2 causes increased WNT signalling. In fact, this is doubtful as targeting WISP2 did not result in increased CRC cell growth. It would be expected that if knocking-down WISP2 resulted in increased WNT signalling, an increase in cell growth rate in the WISP2 knockout cells would have been observed. Further research is therefore required to understand the molecular mechanisms by which WISP2 regulates both invasion and motility in CRC cells.
More research is required to further understand the role of WISP2 in CRC progression. It is hoped that in the future WISP2 may be a potential therapeutic target for the treatment of CRC metastasis.
Acknowledgments
The Authors would like to thank the Royal College of Surgeons England for their support in funding this study. The Authors are also grateful to Cancer Research Wales for their support.
- Received May 29, 2013.
- Revision received June 14, 2013.
- Accepted June 18, 2013.
- Copyright© 2013 International Institute of Anticancer Research (Dr. John G. Delinasios), All rights reserved




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}