


Abstract
Therapies targeting p53 mostly concentrate on (re)activation of the p53 protein, to further induce apoptosis in cancer cells. In the present investigations, the focus was on the identification of small molecules that block the DNA-binding domain of p53 and thus inhibit its function. Using high-throughput in silico screening of approximately 300,000 compounds, we identified eight putatively interacting with the DNA-binding domain of p53. Subsequently, HCT116 p53 wild-type (p53+/+) and knockout (p53–/–) cells were irradiated with 16 Gy and treated with these compounds. Among the eight compounds, NSC 23175 offered the best protection against γ-irradiation-mediated injury. Microarray-based mRNA expression profiling revealed many downstream p53-dependent genes in irradiated and NSC 23175-treated p53+/+ cells. Using a luciferase reporter assay, we showed that NSC 23175 suppressed p53 binding to the promoter of EGR1, a p53-regulated gene. The fact that NSC 23175 protected p53+/+ cells implicates a putative protective effect of the compound during radiotherapy of p53-mutated tumors. The role of NSC 23175 as protecting agent, in reducing radiotherapy-related side-effects merits future investigation.
- p53
- molecular docking
- pharmacogenomics
- radioprotection
- tumor suppressor
- virtual drug screening
- in silico screening
Cancer is frequently correlated with the altered expression of p53, discovered in 1979 as oncogene and re-discovered as a tumor suppressor in 1989 (1-4). p53 acts as an on-off switch for a variety of downstream processes, including apoptosis and growth arrest, after activation by post-translational modifications such as phosphorylation, acetylation, and methylation (5-9). The mechanisms of apoptosis induced by p53 consist of transcriptional activation of FAS, KILLER/DR5, and the mitochondrial pathways (6, 10, 11). Furthermore, genes promoting cell survival such as BCL2, IGFR, MCL-1, survivin and PIK3CA are inhibited by the activated form of p53 through several different mechanisms (12-16). p21, GADD45, 14-3-3σ, and PTGFβ are also involved in the p53 function via transactivation and direct DNA binding (10, 17). In contrast to its producing apoptotic effects, p53 can also act by promoting cell survival (12, 18). Indeed, p53 has become one of the most investigated proteins. It is mutated in more than 50% of all human carcinomas (http://www.iarc.fr/p53). Most of the mutations occur in the DNA-binding domain and lead either to misfolding of the protein (e.g. if residues 175H, 249S or 281G are mutated) or to disruption of the DNA-binding ability (e.g. through mutations at 248W and 273H) (19). Mutations in the gene encoding p53 induced by radiation lead to a conformational change of the p53 protein, in many cases causing loss of function. The loss of its apoptotic function can lead to the development of radio- and drug-resistant cancer cells (20-24).
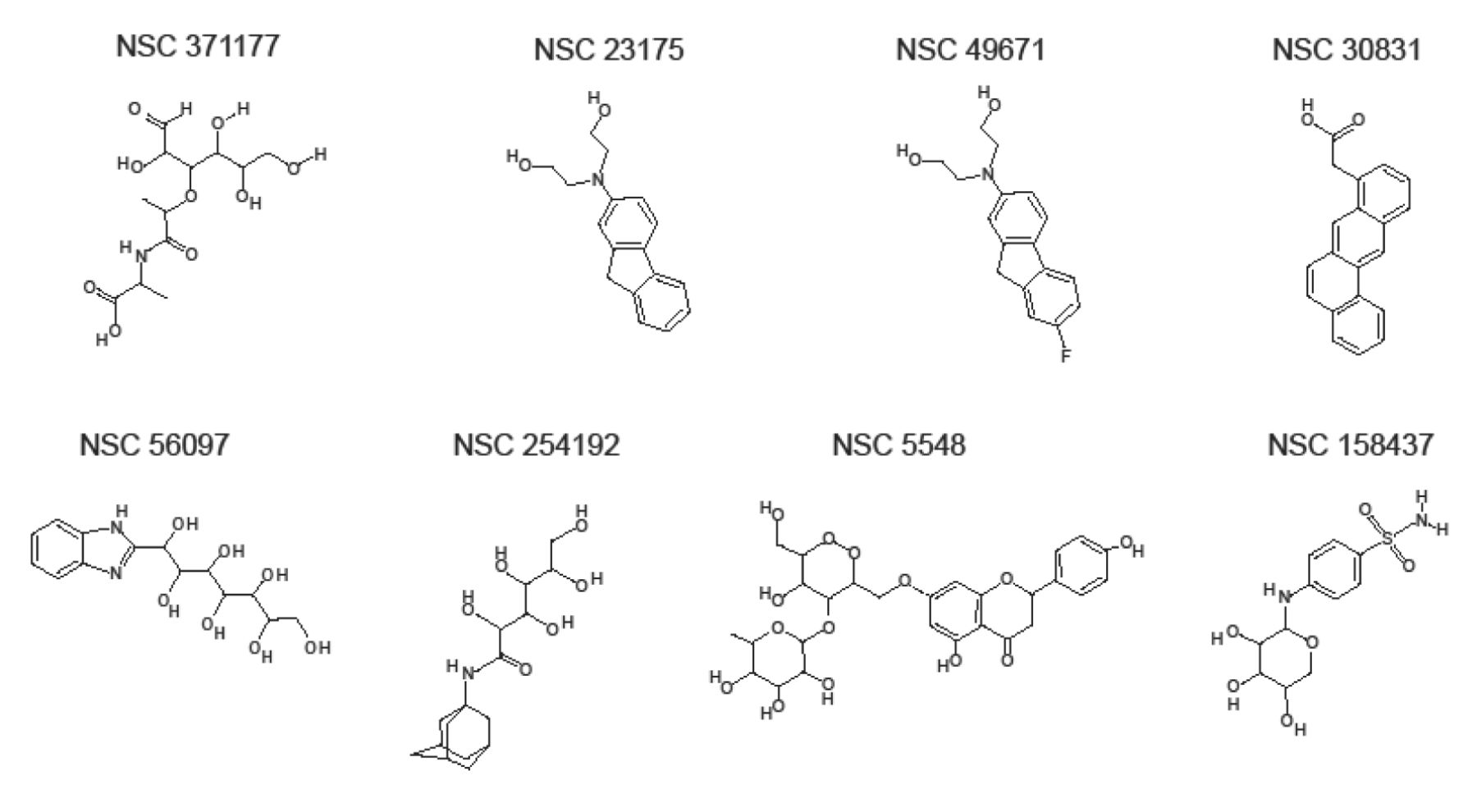
Structural formula of the eight chemicals bioinformatically-identified as putative p53-interacting compounds.
The effect of radiation on p53 activation and p53-induced cell death was first observed in mouse thymocytes (25, 26). A number of subsequent reports confirmed the crucial role of p53 as a mediator of apoptosis, and tremendous efforts were undertaken to develop therapies targeting this key molecule in order to restore its function in cancer cells (27).
Considering the facts discussed above, the plausibility of pharmacological inhibition of p53 may seem questionable. However, transient inhibition of p53 would be highly desirable in order to reduce side-effects of radiation or chemotherapy. Successful inhibition has been demonstrated by pifithrin (PFT)-α and PFT-μ, two small molecules that directly target the DNA-binding domain of p53. These compounds protected mice from carcinogenesis driven by irradiation-induced p53 mutations when subjected to high doses of irradiation (28, 29). Although mutations in p53 foster cancer development and therapy resistance in many tumor types, this is not true for all cancer types. In nasopharyngeal carcinoma, p53 was found to exist mostly in its wild-type form, and wild-type p53 is correlated with radioresistance of tumors and poor prognosis for patients (30, 31). Moreover, in colon cancer, a direct relationship was observed between p53 deficiency and radiosensitization (32). In the past years, accumulated evidence has created indications of a new role of p53 as an anti-apoptotic and pro-survival player in cancer (18, 33).
In the present investigation, we identified novel inhibitors that target the DNA-binding domain of p53. We screened for the chemical compound library of the National Cancer Institute (NCI), USA, consisting of approximately 300,000 individual chemical structures and tautomeric forms by an in silico approach. As a second step, identified candidate compounds were tested in combination with irradiation in p53 wild-type and knockout HCT116 colon cancer cells to determine the putative therapeutic effects of p53 targeting. Furthermore, microarray-based gene expression profiling was performed to investigate whether expression of downstream p53-regulated genes is affected by these compounds.
Materials and Methods
In silico screening. Glide (Grid-based ligand docking energetics) software (Schrödinger, Mannheim, Germany) was used for in silico screening, and was run from the graphical interface of Maestro (Schrödinger, Mannheim, Germany). The drug library of the NCI (Bethesda, MA, USA) consisting of approximately 300,000 individual structures was downloaded (http://cactus.nci.nih.gov/download/nci/) and prepared using the LigPrep task to produce structural variations, perform corrections, exclude undesirable structures, generate tautomers, add hydrogen atoms, neutralize charged groups and optimize ligand structures for a pH range from 5 to 9.
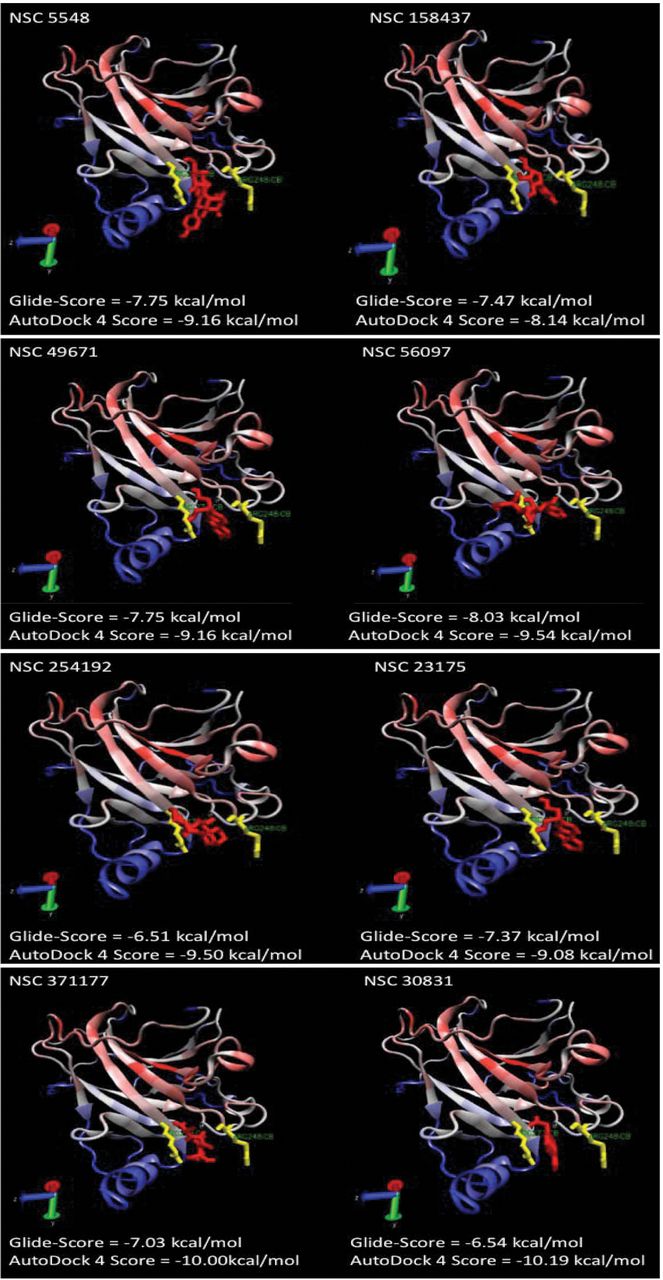
The receptor grid box was set directly on the DNA-binding domain of the p53 crystal structure 1TUP (chain C) from the PDB database (www.pdb.org) after extraction of the included DNA molecule. The amino acids R248 and R273 were covered by the grid box, as they are known to be important for DNA binding (34). For the first run, the grid files were chosen and the high-throughput virtual screening HTVS option was selected. For the second run, a new drug library was generated consisting of the 10% best-evaluated structures from the first run. The settings for screening were switched from HTVS to standard precision (SP) with post-docking minimization. The best 10% of the evaluated structures were transferred to the final run at extra precision (XP). The results of this run were manually evaluated and 25 compounds were chosen for cross validation using a different docking algorithm, AutoDock 4 (35). Finally, eight compounds with results in both docking programs were chosen for further experimental analysis. These compounds were obtained from the Drug Synthesis and Chemistry Branch of the national Cancer Institute (Bethesda, Maryland, USA). Their chemical structures are shown in Figure 1.
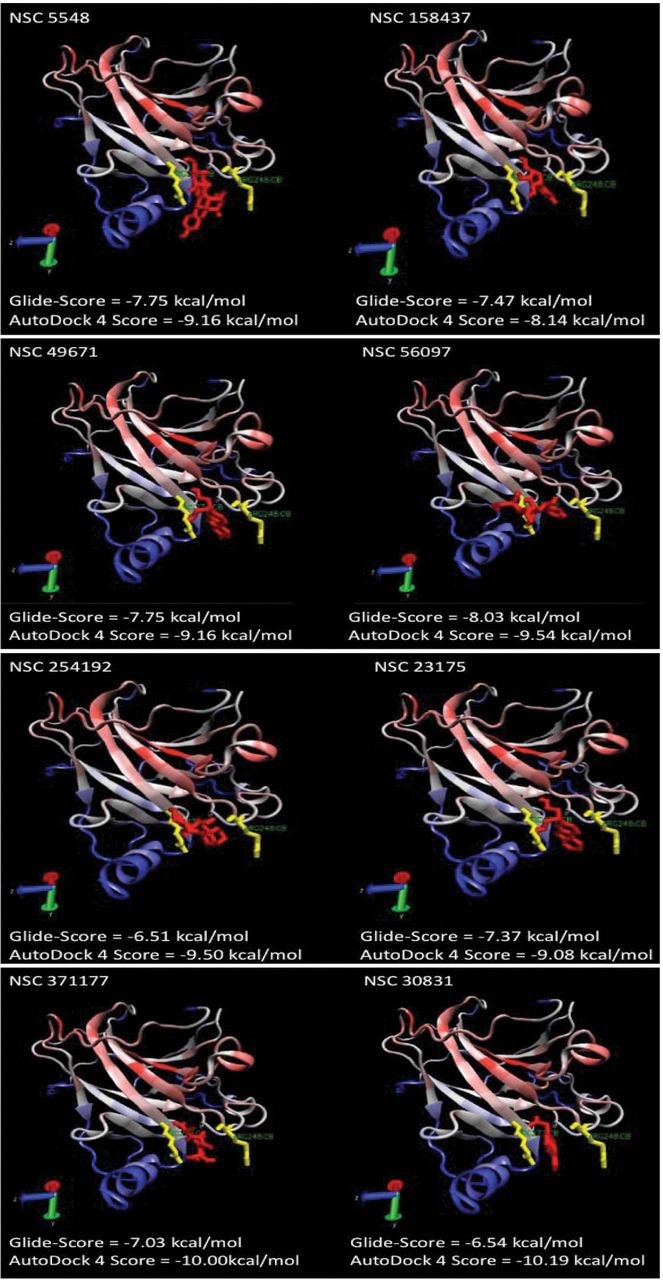
Putative orientation of the candidate compounds in the DNA-binding cavity of p53. The residues important for p53 activity, R248 (right) and R273 (left), are highlighted in yellow. The compounds are shown in red. In all dockings, the corresponding compound blocked the DNA-binding site flanked by R248 and R273.
Cell lines. Human wild-type HCT116 colon cancer cells p53+/+, as well as knockout clones p53–/– derived by homologous recombination (36, 37) were a generous gift from Dr B. Vogelstein and H. Hermeking (Howard Hughes Medical Institute, Baltimore, MD, USA). The cells were cultured in RPMI-1640 medium without L-glutamine, supplemented with 10% (v/v) fetal bovine serum and 100 μg/ml penicillin/streptomycin (all from Invitrogen, Darmstadt, Germany).
XTT proliferation assay. Cell proliferation was assessed using the 2,3-bis[2-methoxy-4-nitro-5-sulfophenyl]-2H-tetrazolium-5-carboxanilide inner salt (XTT) assay (Roche Diagnostics, Mannheim, Germany), which measures the metabolic activity of viable cells (38). This assay is based on cleavage of the yellow XTT by dehydrogenases ubiquitous in viable cells, which leads to the formation of orange formazan dye. The amount of formazan dye is commensurate to the number of metabolically active cells. The assay was performed as previously described (39). The cytotoxic effect of the treatment was determined by calculating the percentage of viable cells as compared to untreated control wells. Microsoft Excel 2007 software was used for analysis.
Eight compounds (NCS 371177, NSC 23175, NSC 49671, NSC 30831, NSC 56097, NSC 254192, NSC 5548, NSC 158437) were obtained from the Drug Repository of the Development Therapeutics Program of the NCI (http:/dtp.nci.nih.gov) (Figure 1).
Transfection and luciferase assay. D-Luciferin was obtained from Applichem (Darmstadt, Germany), CoA from Sigma-Aldrich (Taufkirchen, Germany), ATP from Roth (Karlsruhe, Germany), and coelenterazine from Biotrend (Köln, Germany). A total of 2×104 HCT-116 cells were seeded in 96-well CellBind plates (Costar/Corning, Amsterdam, the Netherlands). On the following day they were transiently transfected with 0.25 μg pFW-EBSI4-luc and 0.25 μg pRL-TK renilla (Promega, Heidelberg, Germany) using FuGENE® HD transfection reagent (Roche, Mannheim, Germany) according to manufacturer’s instructions. pFW-EBSI4-luc contains four EGR1 binding sites and was a gift from Prof. Gerald Thiel (Saarland University Medical Center, Homburg, Germany) (40). After 16 h, cells were treated with the test compounds and lysed in 1× passive lysis buffer (Promega, Heidelberg, Germany) at –80°C. Lysates were transferred into white 96-well plates (NUNC/Thermo Scientific, Langenselbold, Germany) and luminescence measurements were carried out by automated addition of firefly luciferase substrate buffer (20 mM tricine, 2.7 mM MgCO3, 1 mM MgSO4, 0.1 μM EDTA, 530 μM EDTA, 33.3 μM DTT, 470 mM D-luciferin, 270 μM CoA, pH 7.8) or renilla substrate buffer (0.1 m NaCl, 25 mM Tris, 1 mM CaCl2, 0.9 μM coelenterazine, pH 7.5) using a POLARstar Omega and the Omega software (both from BMG Labtech, Offenburg, Germany).
RNA isolation and analysis. Total RNA was isolated using the RNeasy Kit (Qiagen, Hilden, Germany), according to the manufacturer’s instructions. RNA was resuspended/eluted in Tris-EDTA/water. The quality of total RNA was checked by gel analysis using the total RNA Nano Chip Assay on an Agilent 2100 Bioanalyzer (Agilent Technologies GmbH, Berlin, Germany). RNA concentrations were determined using a NanoDrop spectrophotometer (NanoDrop Technologies, Wilmington, DE). Samples were analyzed in two different ways, firstly by measuring the rRNA integral ratio of 28S to 18S rRNA peaks and by assessing the RNA integrity through a RIN-value. Samples having an rRNA ratio above 2.0 or a RIN-value above 6 were considered for microarray analysis.

Effect of γ-irradiation on cell viability of p53 wild-type (p53+/+) and knockout (p53–/–) HCT116 cells. The data was normalized to the viability of non-radiated cells. The bars indicate SEM values of two independent experiments consisting each of four parallel measurements.
Probe labeling and Illumina Sentrix BeadChip array hybridization. Biotin-labeled cRNA samples were prepared for hybridization on Illumina Human Sentrix-6 or 12 BeadChip arrays (Illumina, Inc), according to Illumina’s recommended sample labeling procedure, based on a modified protocol (41). In brief, 250-500 ng total RNA was used for complementary DNA (cDNA) synthesis, followed by an amplification/labeling step (in vitro transcription) to synthesize biotin-labeled cRNA according to the MessageAmp II aRNA Amplification kit (Ambion, Inc., Austin, TX, USA). Biotin-16-UTP was purchased from Roche Applied Science (Penzberg, Germany). The cRNA was column-purified according to TotalPrep RNA Amplification Kit, and eluted in 60-80 μl of water. Quality of cRNA was assessed using the RNA Nano Chip Assay on an Agilent 2100 Bioanalyzer and spectrophotometrically quantified (NanoDrop Technologies).
Hybridization was performed at 58°C, in GEX-HCB buffer (Illumina Inc.) at a concentration of 100 ng cRNA/μl, unsealed in a wet chamber for 20 h. Spike-in controls for low, medium and highly abundant RNAs were added, as well as mismatch control and biotinylation control oligonucleotides. Microarrays were washed once in High Temp wash buffer (Illumina Inc.) at 55°C and then twice in E1BC buffer (Illumina Inc.) at room temperature for 5 min. Between E1BC washes, microarrays were washed with ethanol at room temperature. After blocking for 5 min in 4 ml of 1% (wt/vol) Blocker Casein in phosphate-buffered saline, Hammarsten grade (Pierce Biotechnology, Inc., Rockford, IL), array signals were developed by a 10-min incubation in 2 ml of 1 μg/ml Cy3-streptavidin (Amersham Biosciences, Buckinghamshire, UK) solution and 1% blocking solution. After a final wash in E1BC, the arrays were dried and scanned.
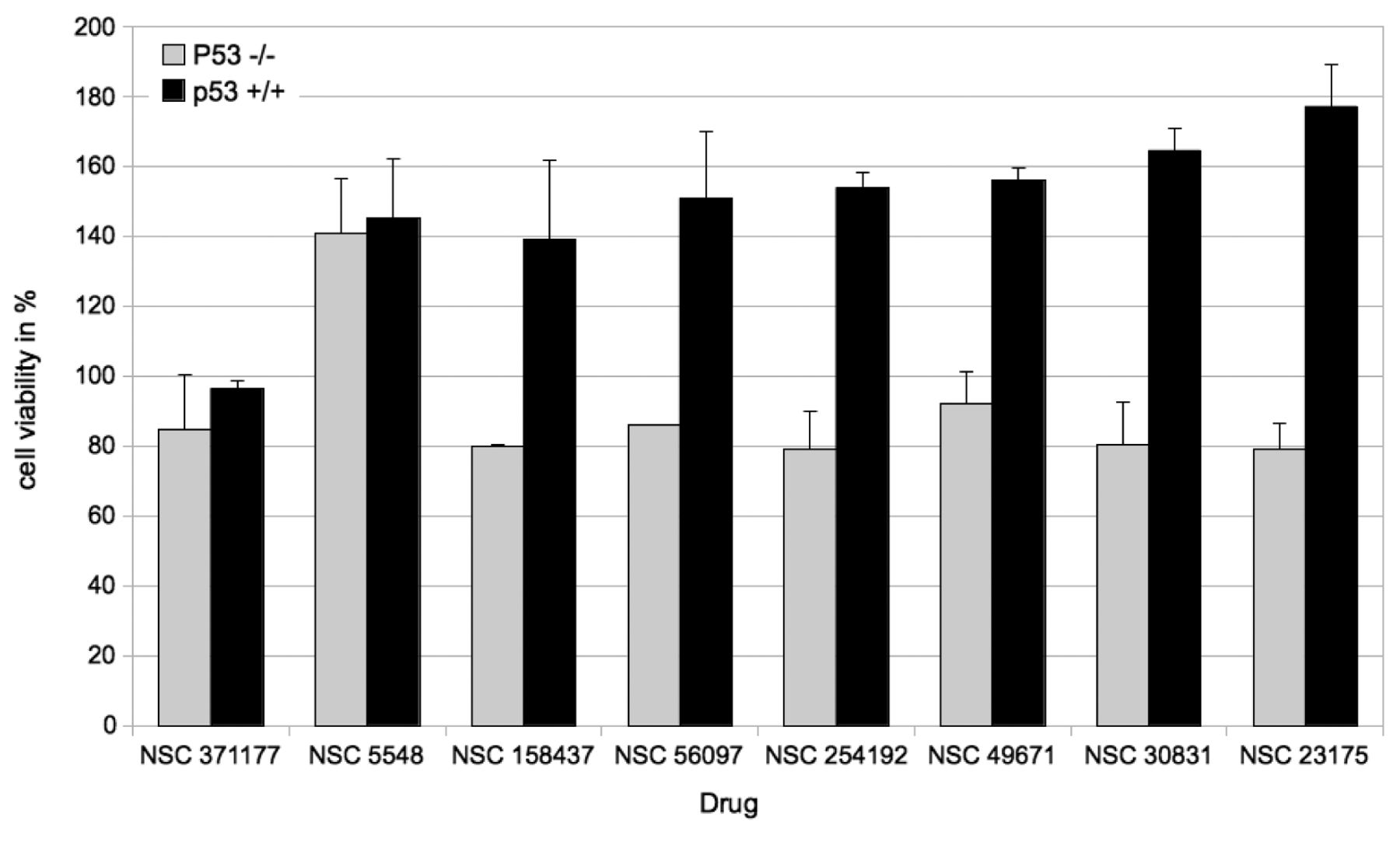
Effect of the candidate compounds (100 μM) on proliferation of p53+/+ and p53–/– cells. The results are expressed as the percentage viability relative to that of the untreated control.
Scanning and data analysis. Microarray scanning was performed using a Beadstation array scanner with settings adjusted to a scaling factor of 1 and PMT settings at 430. Data extraction was carried out for each bead individually, and outliers with values >2.5 median absolute deviation (MAD) were removed. All remaining data points were used for the calculation of the mean average signal for a given probe, and the standard deviation for each probe was calculated.
Data analysis was performed by normalization of the signals using the quantile normalization algorithm without background subtraction, and differentially regulated genes were identified by calculating the standard deviation differences for a given probe in one-by-one comparisons of samples or groups.
Microarray data analysis. Expression data was further analyzed using Chipster (http://chipster.csc.fi/), for filtering genes by varying expression and significance. These steps include filtering of genes to isolate those that were up- or down-regulated by one to three times the standard deviation (depending on number of extremely up- or down-regulated genes). A subsequent assessment of significance using empirical Bayes t-test, further narrowed the pool of genes (12). All genes further considered exhibited a significant difference from the control with a p-value <0.05, or otherwise are noted in the appropriate section. Finally, filtered data were used for Ingenuity pathway analysis for Core analysis, in order to determine networks and pathways influenced by drug treatments (http://www.ingenuity.com/).
Results
In silico screening. Out of approximately 300,000 compounds, eight candidates were chosen after in silico screening using Glide and confirmation by AutoDock (Figure 1). The calculated binding sites of these compounds at the DNA-binding region of p53 are presented in Figure 2.
As a next step, p53+/+ and p53–/– cells were treated with varying doses of γ-irradiation. As shown in Figure 3, p53–/– cells were more sensitive towards 0.5 to 4 Gy than were p53+/+ cells. At higher doses, there was no significant difference between the two cell lines.
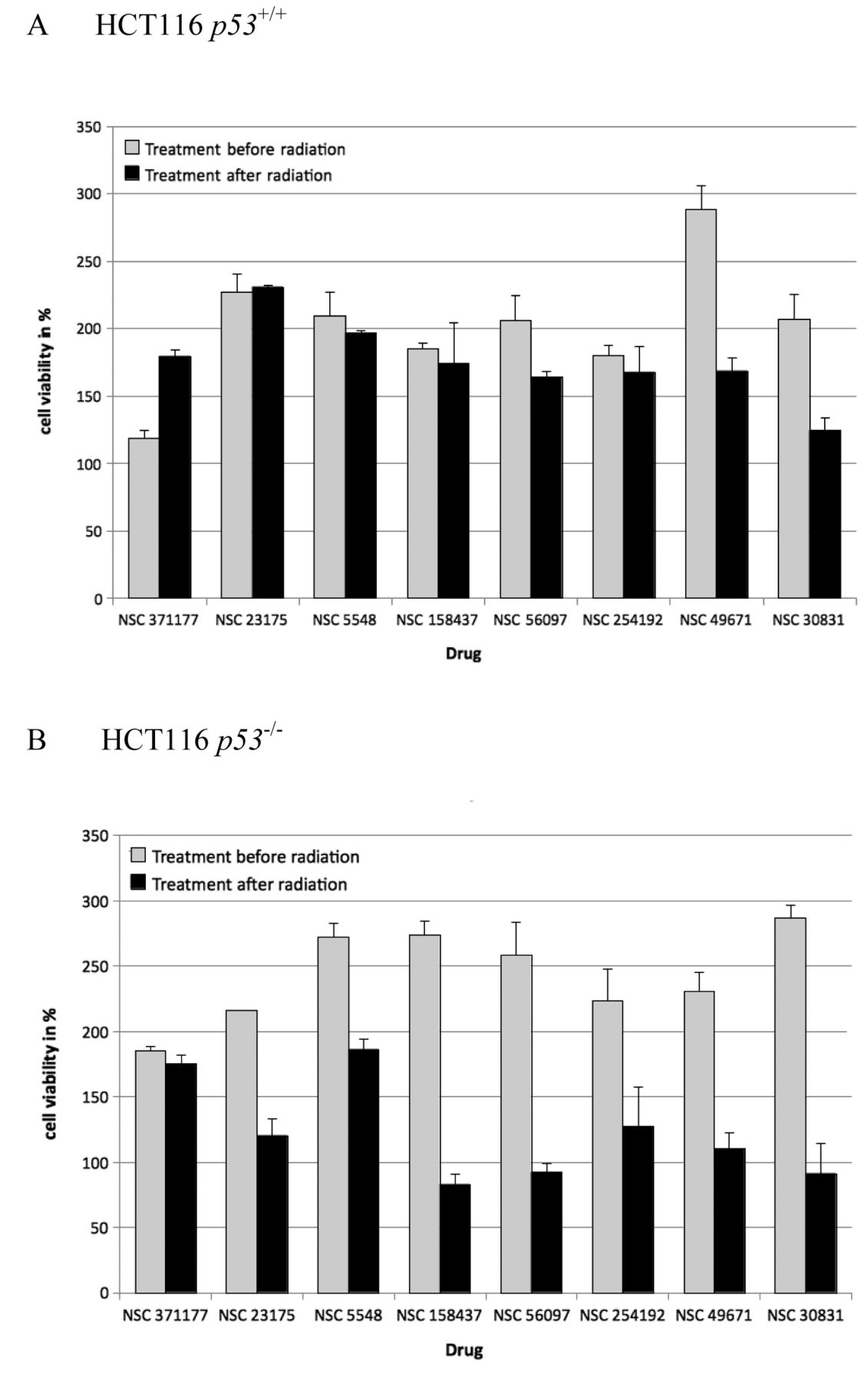
Candidate compounds identified by in silico screening were applied to p53+/+ and p53–/– cells at a concentration of 100 μM in combination with and without γ-irradiation (16 Gy). This radiation dose was chosen for further experiments because it produced similar antiproliferative effects in both cell lines (Figure 3). The effect of each of the eight compounds on cell proliferation is illustrated in Figure 4. The compounds NSC 371177 and NSC 5548 did not produce differing effects on p53+/+ and p53–/– cells. In contrast, all other tested compounds resulted in a significantly higher proliferation rate for p53+/+ cells, as compared to the p53–/– cells. NSC 23175 produced the strongest effect on p53+/+ cells of the compounds tested.
Proliferation assays. The putative p53-interfering compounds were further analyzed in order to investigate the effect on proliferation rates when the compounds were applied before and after irradiation with 16 Gy (Figure 5). The cells were exposed to the compounds either directly before radiation, or immediately after radiation to test for the sensitivity of the compounds to radiation in dependence of the mode of application. Differing effects were observed in p53–/– cells for seven out of the eight compounds, indicating that radiation may cause a functional change of these compounds. The most significant difference in viability for treatment with a substance after irradiation was observed for NSC23175 at 100 μM.
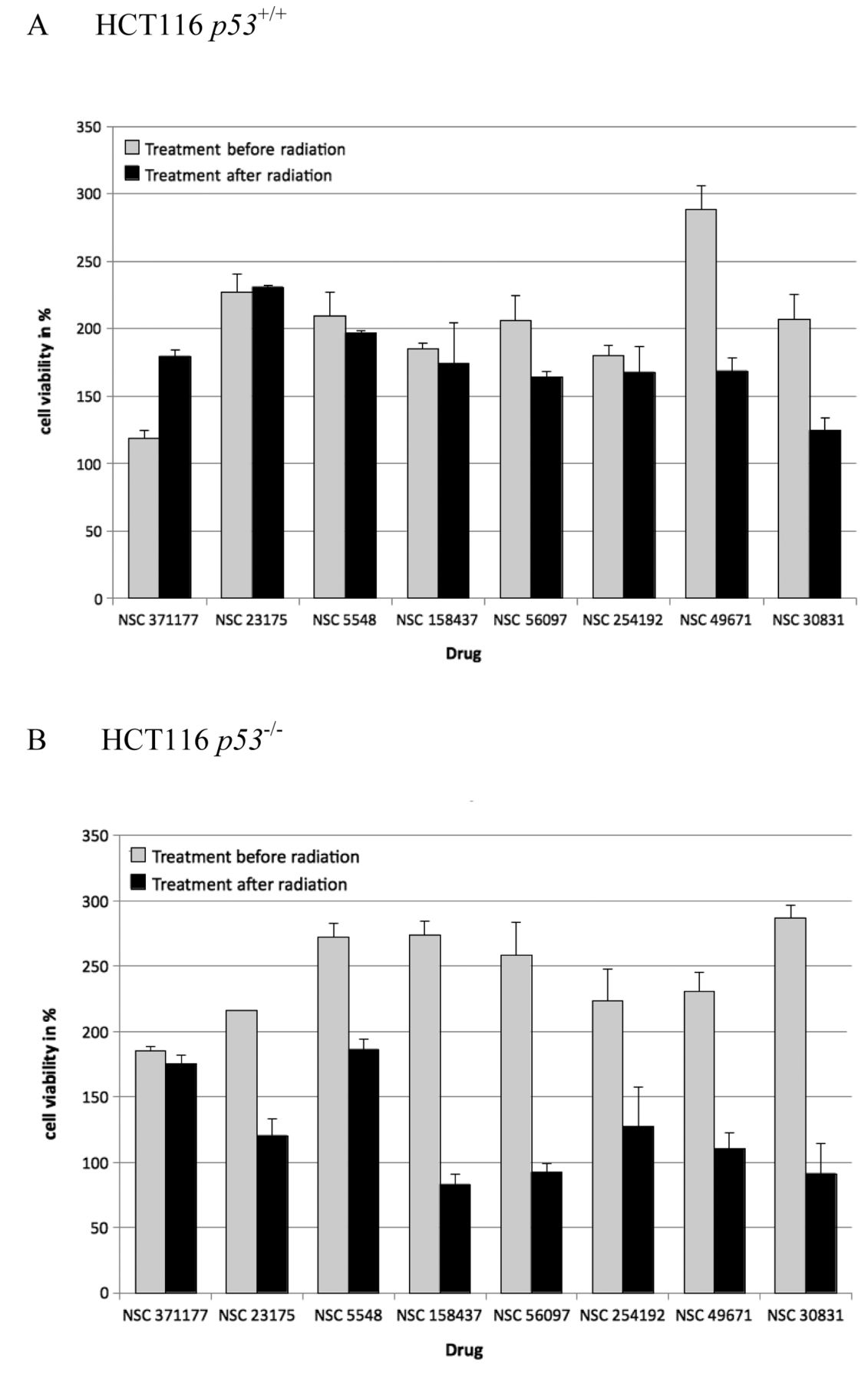
Effects of the candidate compounds on proliferation of p53+/+ (A) and p53–/– cells (B) before and after γ-irradiation (16 Gy). The results are expressed as the percentage viability relative to that of the untreated controls.
NSC 23175 was selected for further analyses, since it presented the strongest growth promoting effects out of all eight compounds in p53+/+ cells, in comparison to p53–/– cells (Figure 5).
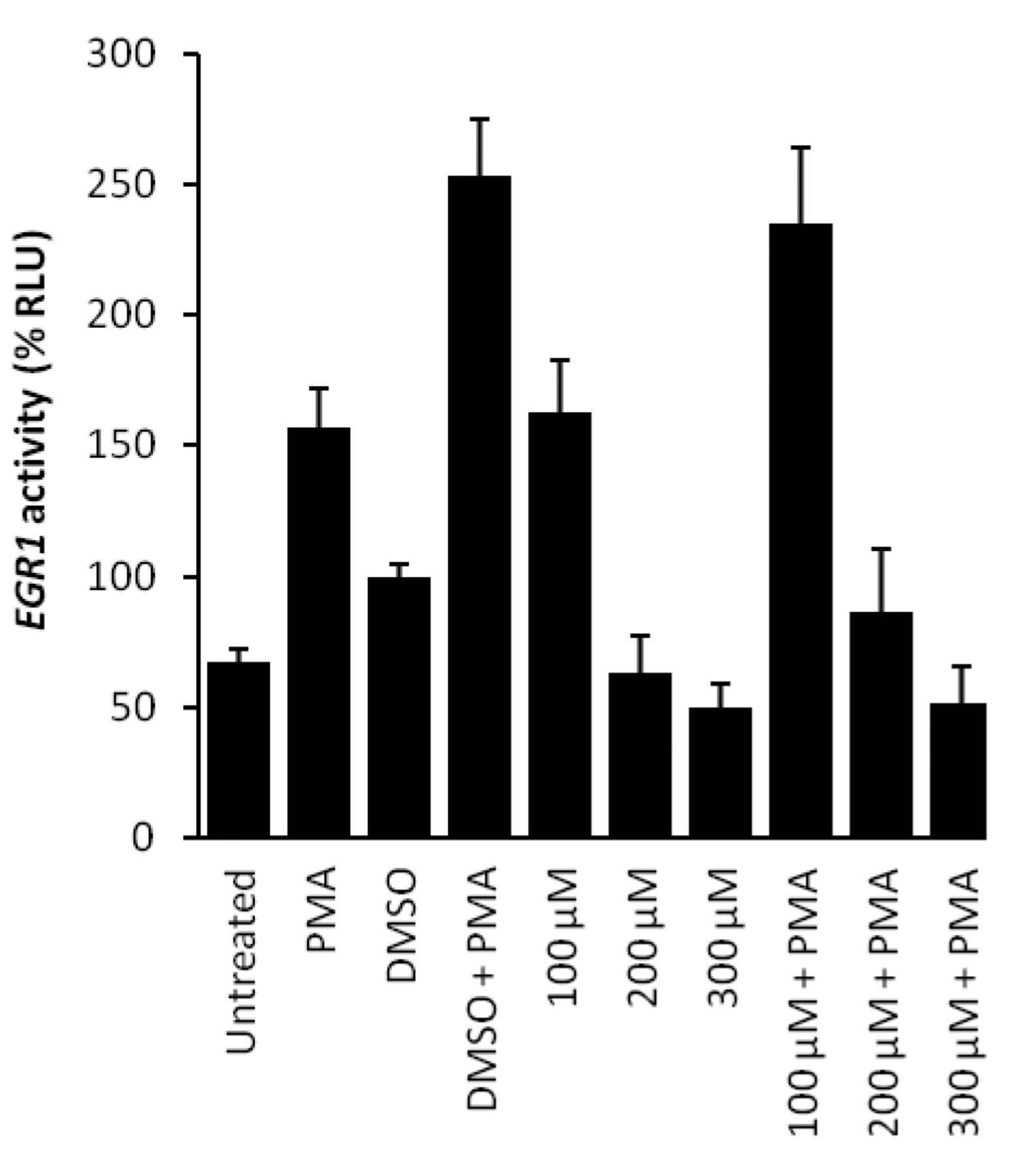
p53 luciferase reporter assay. A reporter assay based on EGR1 as a p53-regulated downstream gene was applied to analyze the activity of NSC 23175. We that hypothesized luciferase luminescence would be inhibited if NSC 23175 inhibits p53 binding to EGR1 promoter sequences. Phorbol 12-myristate 13-acetate (PMA) is a well-known inducer of EGR1 expression (42). Therefore, PMA was used to stimulate EGR1 promoter activity (Figure 6). DMSO used as a solvent for NSC 23175 also induced egr-1 activity, if applied alone. However, the addition of NSC 23175 with or without PMA addition inhibited EGR1 promoter activity in a dose-dependent manner, suggesting that p53 binding to the EGR1 promoter was suppressed by this compound (Figure 6).
Gene expression profiling of HCT116 cells after treatment with NSC 23175. As a next step, cells irradiated with 16 Gy were treated with 100 μM of NSC 23175 to investigate for gene expression profiles of p53+/+ and p53–/– cells. RNA samples of treated and untreated cells from two independent experiments were subjected to microarray hybridization. Using the Chipster filtering tool and Bayes t-test, 213 differentially regulated genes were identified in p53–/– cells and 844 genes in p53+/+ cells. Then, we compiled the genes known to be regulated by p53. Table I shows the p53-downstream genes, which were differentially regulated in p53+/+ and p53–/– genes upon irradiation. Comparing a list of genes downstream of p53 for from the Cold Spring Harbor Laboratory (Cold Spring Harbor, New York, USA) (http://rulai.cshl.edu/TRED/GRN/p53.htm), it was revealed that 34 genes in p53+/+ cells, but only eight genes in p53–/– cells were differentially regulated upon treatment with NSC 23175. As expected the number of genes was much higher in p53+/+ cells than in p53–/– cells, indicating that NSC 23175 may indeed induce p53-dependent effects by binding to p53.
Discussion
The severe side-effects of radio- and chemotherapy prohibit the application of doses high enough to kill all cancer cells, which in turn leads to the development of radio- and chemoresistance. Many strategies have been proposed to improve radiotherapy, most of which are based on two main concepts: Firstly, the sensitization of tumor tissues for radiation, which allows radioresistant tumor populations to be killed and/or radiation doses to be reduced (43). Secondly, the protection of normal tissues from radiation damage by chemical compounds. A novel concept is the specific targeting of the tumor suppressor p53 as a key player of apoptosis to reach radiosensitization of tumors and radioprotection of normal tissues. Small molecules such as PRIMA-1, RITA, and pifitrin-α (PFT), change the sterical conformation of mutated p53 back to the conformation of wild-type p53 (44-47). Hence, p53 is then able to induce apoptosis again in tumor cells and confer re-sensitization to radiation therapy. In the present investigation, we demonstrated that wild-type p53 can also be targeted by small molecules. The inhibition of the function of wild-type p53 in normal tissues may suppress induction of apoptosis in normal tissues and, hence, may prevent radiotherapy-associated damage in healthy tissues of cancer patients (Figure 7).

p53 luciferase assay with NSC 23175. HCT-116 cells were co-transfected with pFW-EBSI4-luc and pRL-TK renilla and pre-treated with different concentrations of NSC 23175, or an equal volume of DMSO as solvent control for 2 h. EGR1 activation was induced by addition of 2 μM PMA for another 5 h and measured by luciferase assay. Luciferase values were normalized to renilla values, and data for DMSO-treated cells were set as 100%. Data shown are the mean±SEM of two independent experiments performed in quadruplicates each.
After virtual screening of approximately 300,000 compounds of the NC1 (http://dtp.nci.nih.gov/repositories.html), eight candidate compounds were identified as interacting with the DNA-binding domain of p53. The compounds identified by Glide were confirmed by molecular docking using AutoDock. In cell proliferation assays, NSC 23175 showed the most potent effect on HCT116 p53+/+ cells, compared with HCT116 p53–/– cells, indicating that this compound likely targeted p53. In combination treatments, NSC 23175 revealed the most considerable effect on p53+/+ cells upon irradiation, but had no effect on p53–/– cells. Compound administration before or after radiation was investigated, since it is important to demonstrate that radiation did not modify the compound resulting in unpredictable cellular effects (e.g. as a radical molecule). Another possibility addressed was the non-specific protection of cells in case the compound itself would absorb radiation after treament. After radiation and subsequent treatment with NSC 23175, no considerable effects on proliferation were observed.
Differentially regulated genes downstream of p53 in HCT116 p53 wild-type and knockout cells after irradiation and treatment with NSC 23175.

Radiosensitivity of tumor cells and radioprotection of normal cells by small molecules binding to wild-type or mutated p53.
Afterwards, p53+/+ and p53–/– cells were radiated, then treated with NSC 23175, and subjected to microarray-based gene expression profiling. Among the differentially regulated genes were genes from diverse functional groups such as cell proliferation (BCL3, BIRC5, CCNA2, CCNB1, CCND1, CDC25C, DGKD, E2F2, EGFR, GDF15, MCM2, MKI67, PLK2, POLD1, S100A4, STAT1, STMN1, TOP2A, NDRG1, TNFRSF10A), DNA repair (BLM, PTTG1, RAD51), apoptosis regulation (EGFR, TP53I3, NDRG1), oxidoreductases (ALDH1A3, PRODH, TP53I3), and others (EZH2, FBLN1, LGALS3, SESN2, TK1). Some of them act as oncogenes (ETS1, FOS), tumor suppressors (FBLN1, PLK2), and/or transcription factors (ATF3, BCL2, BTG1, ETS1, FOS). A synopsis of this set of differentially regulated genes show that NSC 23175 may affect proliferation and apoptosis by p53-mediated regulation. This hypothesis merits further investigation.
Some genes were differentially regulated by NSC 23175 in both p53+/+ and p53–/– cells (ALDH1A3, FBLN1, FOS, GDF15, STMN1 and S100A4). This may be explained by the fact that our virtual compound screening focused only on the DNA-binding domain of p53. Therefore, close relatives to p53 such as p63 or p73 could also be targets of NSC 23175. The DNA-binding domains of p53, p63 and p73 are highly similar (63% similarity between p53 and p73, and 60% similarity between p53 and p63) (48). From a functional point of view, p73 in particular shares a large subset of overlapping downstream genes with p53 and is also involved in response to DNA damage (48). Furthermore, experiments in mouse models have shown that loss of p73 is linked to chronic inflammation (49), since p73-deficient mice reveal inflammatory defects. If NSC 23175 inhibits not only the DNA-binding domain of p53, but also that of p73, it is conceivable that networks related to inflammation may be detected not only in p53+/+ cells but also in p53–/– cells.
Although our data, obtained by microarray hybridization and luciferase reporter assays, provide clear evidence that NSC 23175 may indeed affect binding of p53 to DNA, it should be kept in mind that many differentially expressed genes found in the microarray analysis were not related to p53. This may indicate that NSC 23175 exerts additional modes of action, independently of p53. The further development of NSC 23175 as p53-specific drug would, therefore, necessitate the identification of derivatives with higher specificity for p53 and fewer off-target effects. Nevertheless, NSC 23175 may serve as a valuable lead compound to develop a novel class of compounds inhibiting DNA-binding of wild-type p53.
Another potential issue might be that the protection of normal tissue by NSC 23175 from radiation-induced cell death may lead to the acquisition of persistent irradiation-induced DNA damage and, hence, to carcinogenesis. The therapeutic utility of radioprotective drugs with target specificity to wild-type p53 may, therefore, depend on their plasma half-life. If such a drug has a short half-life, it will be rapidly degraded after radiotherapy. Mechanisms which maintain cell integrity, e.g. DNA repair and apoptosis, can then repair or eliminate damaged cells preventing tumor development. At present, the half-life of NSC 23175 in tumors is unknown. This compound may, however, serve as lead compound for further derivatization, not only to improve binding to p53 and target specificity, but also to obtain compounds with a sufficiently short half-life to prevent carcinogenesis.
In conclusion, NSC 23715 protects cells from the inhibitory effects of γ-irradiation. A bioinformatical approach suggested that NSC 23715 binds to p53, which was supported by microarray-based mRNA expression profiling. The fact that NSC 23715 protects p53+/+ implicates that normal cells with wild-type p53 might also be protected by this compound during radiotherapy of p53-mutated tumors. The role of NSC 23715 as a radioprotecting agent to reduce radiotherapy-related side-effects, merits further investigations.
Acknowledgments
This work was supported by a grant of Mathworks, USA.
- Received December 11, 2012.
- Revision received January 14, 2013.
- Accepted January 14, 2013.
- Copyright© 2013 International Institute of Anticancer Research (Dr. John G. Delinasios), All rights reserved
References
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.