


Abstract
Integrins are heterodimeric transmembrane receptors involved in sensing and transmitting informational cues from the extracellular environment to the cell. This study explored sub-proteome changes in response to elimination of the β3 integrin using a knockout murine model. Cleavable isotope-coded affinity tagging (cICAT) in combination with sub-cellular fractionation, multiple dimensions of separation and tandem mass spectrometry (MS/MS) were used to characterize differentially expressed proteins among β3 integrin–/– (β3–/–) mouse embryonic fibroblasts and isogenic wild-type (WT) controls. From a cytosolic protein fraction, 48 proteins were identified, in which expression differed by >1.5-fold. Predominant ontological groups included actin-binding/cytoskeletal proteins and protease/protease inhibitors. Interestingly, β3 integrin expression was inversely correlated with expression of cathepsin B, a lysosomal cysteine protease, as its expression was greater by over 3.5-fold in the β3–/– cells. This inverse correlation was also observed in stable heterologous cells transfected with β3 integrin, where the intracellular expression and activity of cathepsin B was lower compared to control cells. Our data suggests that the composition of the cellular proteome is influenced by integrin expression patterns and reveals a strong functional relationship between β3 integrin and cathepsin B.
- Cathepsin B
- integrin
- isotope-coded affinity tags
- protease
- cancer-proteomics
- fractionation
- GeLC-MS
- HEK293 cells
Metastatic tumors are the primary cause of death due to cancer. Changes in cell adhesion are prominent during metastatic progression and are directly related to interactions of the extracellular matrix (ECM) with adhesion receptors. Integrins are a large family of heterodimeric αβ transmembrane receptors that mediate cell adhesion, cell migration, and bidirectional signaling between the ECM and the cytosol (1, 2). The β3 integrin group consists of αIIbβ3, found on platelets, and the more abundant αvβ3, found on fibroblasts and other cell types (3-5). The αvβ3 heterodimer is clearly implicated in several pathological processes, such as angiogenesis, rheumatoid arthritis, and osteoporosis (5, 6). Overexpression of αvβ3 has been positively correlated with tumor metastasis (7), yet the complete absence of β3 in murine models leads to enhanced tumorgenicity via elevated angiogenesis (8, 9). This paradox prompted a re-evaluation of its function in angiogenesis (10) and was the impetus for our study focusing on the protein expression changes resulting from elimination of the β3 integrin subunit in mouse cells.
Targeted gene disruption in mice has become a gold standard for characterization of disease and mechanistic pathways (11), but unanticipated compensatory or redundant physiological processes may obscure the expected phenotype (12). The sheer complexity of the eukaryotic proteome presents a challenge for pinpointing and characterizing effects due to loss of a single protein. Furthermore, predicting all potentially affected biochemical networks is impractical; fortunately, the persistent changes occurring in knockout, transgenic, and knock-in models should still manifest at the proteome level (13, 14). Recent reports of global proteomic analyses of isolated cells from knockout mice such as rat fibroblasts lacking the myc oncogene, demonstrated extensive changes that functionally correlated with morphological and proliferative differences (15). Other studies performed in a γ-secretase-deficient background found that caveolin-1 was mislocalized (16), while increased carboxylesterase enzymatic activity was found in adipocytes from a lipase-deficient background (17). Another investigation examined knockout of the cystic fibrosis transmembrane conductance regulator (CFTR), where the identification of reduced murine calcium-activated chloride channel 3 (mClCA3) expression from colon crypts was correlated with reduced glycoprotein secretion (14). Despite these studies, another provocative question remains: Does the loss of an integrin subunit significantly affect the proteome? To date, only one other published proteomic report examines this problem in a β1 integrin knockout using Stable Isotope Labeling of Amino acids in cell culture (SILAC) murine system (18).
To evaluate the sub-cellular proteome changes in cells that do not express the β3 integrin subunit (19), we isolated the cytosolic protein fraction in embryonic fibroblasts and analyzed the protein content using the cleavable isotope-coded affinity tag (cICAT) method (20) in combination with polyacrylamide gel electrophoresis (PAGE), chromatographic separation, and tandem mass spectrometry (GeLC-MS/MS) (21). This strategy revealed a novel inverse relationship between β3 integrin and the cysteine protease, cathepsin B (CatB). The up-regulation of CatB has been linked to several types of cancer (22), arthritis (23), and osteoporotic bone loss (23). CatB protein levels are increased in these disease states, exacerbating enzyme redistribution, secretion, and activity. We have discovered a unique functional correlation between these proteins and validated the relationship in a heterologous cell system, thus corroborating emerging data related to the fundamental role for β3 integrin and protease interaction.
Materials and Methods
Cell lines, media and reagents. The human embryonic kidney cell lines, HEK293 and β3/293, have been previously described (24). Mouse embryonic fibroblasts (MEFs) were derived from 14 day embryos of wild-type or β3–/– mice using a protocol previously described (C57BL6/129 background that had been backcrossed four times to BL6) (19, 25). Briefly, heads and internal organs were removed from the embryos; remaining bodies were rinsed with phosphate-buffered saline (PBS) to remove any traces of blood, minced by sterile scalpels, and the cells homogenized by expelling and drawing the clumps through an 18-gauge needle. The embryonic cells were plated into a 150 mm tissue culture dish with 20 ml of Dulbecco’s modified Eagle’s medium (Hyclone, Logan, UT, USA) plus 10% fetal bovine serum (FBS), Non-Essential Amino Acids Solution (Hyclone), penicillin/streptomycin and placed in a 5% CO2 humidified incubator at 37°C. After one week in culture, the cells on one dish were harvested and split into three new 150 mm dishes for cell maintenance and passage. All other reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Cell adhesion and sub-cellular extraction. Both WT and β3–/– fibroblasts (1×107 cells at passage 3) were seeded onto 150 mm dishes coated with fibronectin (BioCoat; BDBiosciences, Bedford, MA, USA) and allowed to adhere for 1 h before washing 3 times in cold PBS with gentle agitation for 5 min. The extraction protocol was followed according to the manufacturer (EMD/Calbiochem, San Diego, CA, USA). Briefly, 4 ml ice-cold extraction buffer I and 20 μl protease inhibitor cocktail were mixed, added to plates without disturbing the monolayer, and was gently agitated for 10 min at 4°C. The supernatant (fraction 1, cytosolic) was transferred to a 5 ml tube. A similar step was performed using extraction buffer II with incubation for 30 min at 4°C. This supernatant (fraction 2, membrane) was transferred into a new 5 ml tube and stored at -80°C. Comparable steps were performed using extraction buffer III (fraction 3, nuclear) and extraction buffer IV (fraction 4, cytoskeletal). Cytosolic protein extracts (fraction 1) were precipitated using acetone/trichloroacetic acid (TCA), resuspended in tris-buffered saline (TBS)/0.1% sodium dodecyl sulfate (SDS), and quantified by measuring absorbance A280 on a Nanodrop ND-1000 instrument (Nanodrop, Wilmington, DE, USA).
Labeling of cytosolic proteins, SDS-PAGE, and affinity purification of ICAT-peptides. Cytosolic proteins from each genotype (100 μg in 80 μl of 50 mM Tris, pH 7.1 and 20 ϱmol of full-length recombinant human vimentin (an internal quality control for ICAT-labeling efficiency and trypsin digestion) were reduced with Tris(2-carboxyethyl)triphosphate (TCEP) in a boiling water-bath for 10 min. After cooling, protein samples from the WT and β3–/– cells were incubated with the light and heavy acid-cleavable ICAT reagents (Applied Biosystems, Foster City, CA, USA), respectively, at 37°C for 2 h. The labeled samples were combined and concentrated in a Speedvac (ThermoSavant, Waltham, MA, USA) for 1 h, mixed with 5× SDS-PAGE sample loading buffer, boiled for 10 min, and run on a 4-20% gradient minigel for 2 h. The gel was rinsed three times in Milli-Q water for 20 min, stained for 10 min in SimplyBlue (Invitrogen, Carlsbad, CA, USA), and destained three times in Milli-Q water for 20 min. Eight roughly equal-sized gel slices were excised, dehydrated, and in-gel digested with trypsin at 37°C overnight. Digested peptides were extracted from gel slices using 50% acetonitrile/0.1% formic acid in water, dried by Speedvac, and purified over an avidin affinity cartridge. Bound peptides were washed, eluted, and cleaved in a 30% Trifluoroacetic acid (TFA) solution at 37°C for 2 h to release the ICAT-labeled peptides from the acid-cleavable linker. The resulting peptides were completely dried using a SpeedVac. The biological replicates were run in duplicate.
Identification and quantitation of proteins by nanoLC-MS/MS. The ICAT-labeled peptides were suspended in 40 μl of 2% acetonitrile/0.2% formic acid in water and the supernatant was used for nanoLC-MS/MS analyses. A FAMOS/Switchos/Ultimate liquid chromatography system (Dionex-LC Packings, Sunnyvale, CA, USA) was used to load, concentrate, and desalt peptide samples and to deliver a gradient with a flow rate of 200 nl/min. The analytical column was 75 μm I.D.×15 cm packed with PepMap C18 (LC Packings). Using mobile phase A (2% acetonitrile/0.1% formic acid in water) and mobile phase B (80% acetonitrile/0.1% formic acid in water), the gradient was 4%-50% B over 100 min, 50%-100% B over 20 min, then 0% B for remaining 30 min. Peptides eluted from the column were analyzed using a Q-TOF API-US mass spectrometer (Waters-Micromass, Milford, MA, USA) equipped with a nano-electrospray ion source (Waters-Micromass). The capillary voltage was 2.8-3.2 kV, and the cone voltage was set at 35 V. The four most intense peptide ions were dynamically selected for fragmentation. The collision energy varied between 16 and 45 eV, depending on the m/z value and charge state of peptides. Automatic switching between MS and MS/MS modes was controlled by MassLynx 4.0 (Waters-Micromass), which was dependent on both signal intensity (≥25 counts/s) and charge states (2, 3 and 4 only) from MS to MS/MS and either signal intensity or time from MS/MS to MS. Initially, the nanoLC-MS/MS raw data obtained were processed with ProteinLynx 2.01 (Waters-Micromass) to generate a .pkl file, which was then used for protein identification by either Mascot 2.2 (Matrix Science, UK; ions/MOWSE score of ≥40 with our search parameters equals a p≥0.05) and the ProteinLynx software to search against the mouse NCBI non-redundant protein database (release date November 2007) using standard variable protein identifications (acetylation, deamidation, oxidized methionine, ICAT light/heavy, and carbamidomethylated cysteines). ProteinLynx also provided an automatic quantitative analysis of ICAT-labeled peptides (cumulative LadderScore >50) as previously described (26). All identification and quantification results were further verified by manual inspection with MassLynx MaxEnt3 and BioLynx software (Waters-Micromass) and accepted rules for peptide fragmentation in a quadrupole-TOF hybrid MS after integration of peaks for both isoforms of each labeled peptide identified on reconstituted chromatograms obtained following extraction of a specific mass (±0.1 Da) from the nanoLC-MS data using MassLynx. Vimentin (54 kDa) was used as an internal control protein since it contains only a single cysteine residue and migrates in the middle region of the SDS-PAGE. The Decoy checkbox was used for all Mascot identifications and only those matches above a homology threshold were accepted for a 5% false discovery rate.
Fluorescence and phase microscopy and immunoblotting. MEFs were seeded on coverslips overnight, fixed in 3.7% paraformaldehyde for 15 min, then permeabilized in 0.1% Triton X-100/PBS for 5 min. Cells were blocked in 5% bovine serum albumen (BSA) for 30 min at 37°C, then double-labeled with Alexa594-phalloidin (1:400 Invitrogen/Molecular Probes, Carlsbad, CA, USA) and an antibody recognizing paxillin (1:200) for 1 h at 37°C. Cells were washed in PBS, incubated with goat anti-mouse secondary (1:400, Molecular Probes) conjugated to the Alexa488 fluorochrome, then mounted with Vectashield/4’,6-diamidino-2-phenylindole (DAPI) and examined using a Zeiss Axiovert 10 microscope under ×600 magnification.
Western blot analysis was performed with protein bound to nitrocellulose membranes and probed using primary rabbit antisera to Muskelin (a generous gift of Dr. Josephine C. Adams, Lerner Research Institute, Cleveland, OH, USA) and CatB (Santa Cruz, Santa Cruz, CA, USA and BD Transduction Labs, San Diego, CA, USA, respectively); goat antisera to fatty acid binding protein 5 (FABP5) (Santa Cruz); mouse antisera to Na+/K+-ATPase (Santa Cruz); mouse antisera to β3 integrin (Santa Cruz and BD Transduction Labs), paxillin (Sigma-Aldrich), and vinculin (Sigma-Aldrich). Immunoreactivity was detected with horseradish peroxidase (HRP) conjugated goat anti rabbit, goat anti mouse, or donkey anti goat IgG (Santa Cruz) and visualized by enhanced chemiluminescence.
Cathepsin activity assay. The protocols were used according to the manufacturer (EMD/Calbiochem). For the intracellular activity measurement of CatB, fresh cell pellets (approximately 1×106 cells) were washed with ice-cold PBS before the addition of 500 μl of cell lysis buffer then incubated on ice for 30 min. Lysates were vortexed for 1 min and centrifuged at 14,000 ×g in a pre-cooled tabletop microcentrifuge. Supernatants were immediately transferred to clean tubes and the protein concentration was determined by A280 on a Nanodrop ND-1000. Equal amounts of protein lysate were used for each cell line (50-100 μg) and diluted to 200 μl with dilution buffer. Activation buffer (25 μl) was pipetted into each well of a black, opaque-bottom 96-well plate with/without inhibitor. Standard, control, or sample (50 μl) was added to each well in triplicate. Plates were briefly pre-incubated at room temperature for 5 min before the addition of 25 μl of specific substrate solution (Z-Arg-Arg-7-amido-4-methylcoumarin hydrochloride (AMC)). Plates were sealed tightly and incubated at 37°C for 1 h. Fluorescence of free AMC was read on a Gemini EM SpectraMax (Global Medical Instrumentation, Inc., Ramsey, MN, USA) fluorescence plate reader (excitation/emission wavelengths at 355/460 nm). Alternatively, for determination of secreted CatB activity, the procedure by Koblinski et al. (27) was followed with minor modifications. Briefly, fibroblasts were grown on fibronectin-coated plates for 24 h in low serum conditions (0.5% FBS). Media were collected and concentrated in spin columns (5 kDa MWCO), then 100 μl of conditioned medium or media alone (control without cells) were incubated for 1 h at 37°C with 25 μl of activation buffer (0.5 M sodium formate, 20 mM EDTA, pH 3.2, and 0.2 mg/ml pepsin). CatB activity was determined by adding 100 μl of substrate solution (200 mM sodium phosphate buffer, pH 6.7, containing 4 mM EDTA, 10 mM dithiothreitol, 0.1% Triton X-100, and 200 μM peptide). Samples and controls were transferred into 96-well plates and the fluorescence intensities determined. Relative fluorescence units were expressed and normalized to either total protein or cell number as picomoles AMC formed (μl or μg)/cells/h.
Results
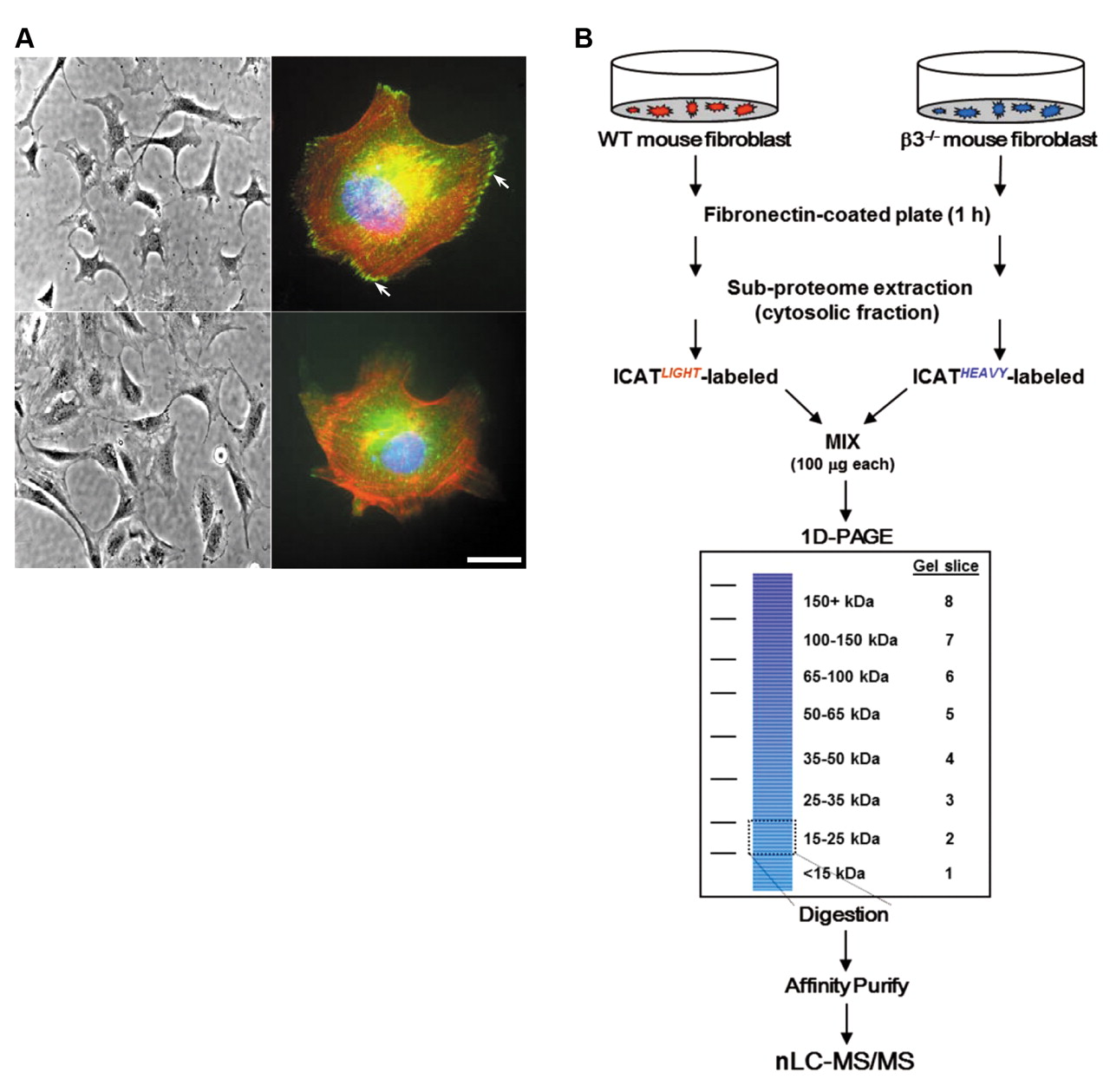
β3 Integrin–/– fibroblasts have an altered pattern of paxillin immunofluorescence. To elucidate the integrin-directed biological events occurring near the cell membrane, we used a simple binary system of MEFs isolated from either the β3–/– or WT isogenic control mice. The initial characterization by phase-contrast microscopy revealed no clear morphological differences between WT (Figure 1A, top left) and β3–/– MEFs (Figure 1A, bottom left). However, immunofluorescent staining of paxillin, a known intracellular binding partner for β3 integrin (28), was localized to canonical adhesive structures in WT (Figure 1A, top right) but not β3–/– fibroblasts (Figure 1A, bottom right). This suggests that the complete elimination of β3 integrin could significantly influence outside-in signaling.
Characterization of differentially expressed membrane proteins from WT and β3–/– fibroblasts by nanoLC-MS/MS. We performed a multidimensional proteomic screening to ascertain changes in expression level of cytosol-associated proteins from WT and β3–/– fibroblasts upon adhesion to and spreading on an extracellular substrate for β3 integrin (Figure 1B). Both WT and β3–/– MEFs were seeded onto fibronectin-coated plates and then subjected to a subcellular extraction. We chose fibronectin rather than vitronectin, another αvβ3 substrate, hypothesizing this would minimize compensation from the other αv-containing heterodimers as has been performed recently (8, 10, 29, 30). Furthermore, we focused the analysis on the cytosolic fraction to facilitate the proteome study towards integrin-associated intracellular interactions. Equal amounts of protein lysate from each cell line were labeled with the cICAT reagent and run on a denaturing gel to provide yet another dimension of fractionation. A known amount of full-length recombinant human vimentin was added to each sample as an internal quality control for cICAT-labeling efficiency, trypsin digestion, and spectra quantitation. Vimentin contains only a single cysteine residue and migrates by SDS-PAGE around 54 kDa mass range.

Analysis of WT and β3–/– embryonic fibroblasts for quantitative proteomics. A: β3 Integrin deficiency does not change morphology but alters immunofluorescence pattern of paxillin (left panels). Phase-contrast images of WT (top) and β3–/– (bottom) fibroblasts under ×20 magnification (right panels). Merged immunofluorescence images of WT (top) and β3–/– (bottom) fibroblasts stained with anti-paxillin-FITC which localizes to focal contacts (white arrows), phalloidin-Alexa594 (actin), and DAPI (chromatin). Scale bar (bottom right) represents 10 μm. B: Outline of quantitative experimental procedure. Mouse embryonic fibroblasts were seeded onto fibronectin-coated plates, harvested and sub-fractionated. One hundred micrograms of cytosolic protein from WT and β3–/– cells were utilized for ICAT labeling and 1D-PAGE. After Coomassie staining, eight equal-sized gel slices (corresponding to mass ranges of <15, 15-25, 25-35, 35-50, 50-65, 65-100, 100-150, and >150 kDa) were excised and trypsin digested. Digested peptides were further purified over an avidin affinity cartridge. Peptides were eluted from the cartridge then acid-cleaved to release the ICAT-labeled peptides from the linker and subjected to nanoLC-MS/MS analysis. Details of the multidimensional procedure and the nanoLC-MS/MS are described in the Materials and Methods.

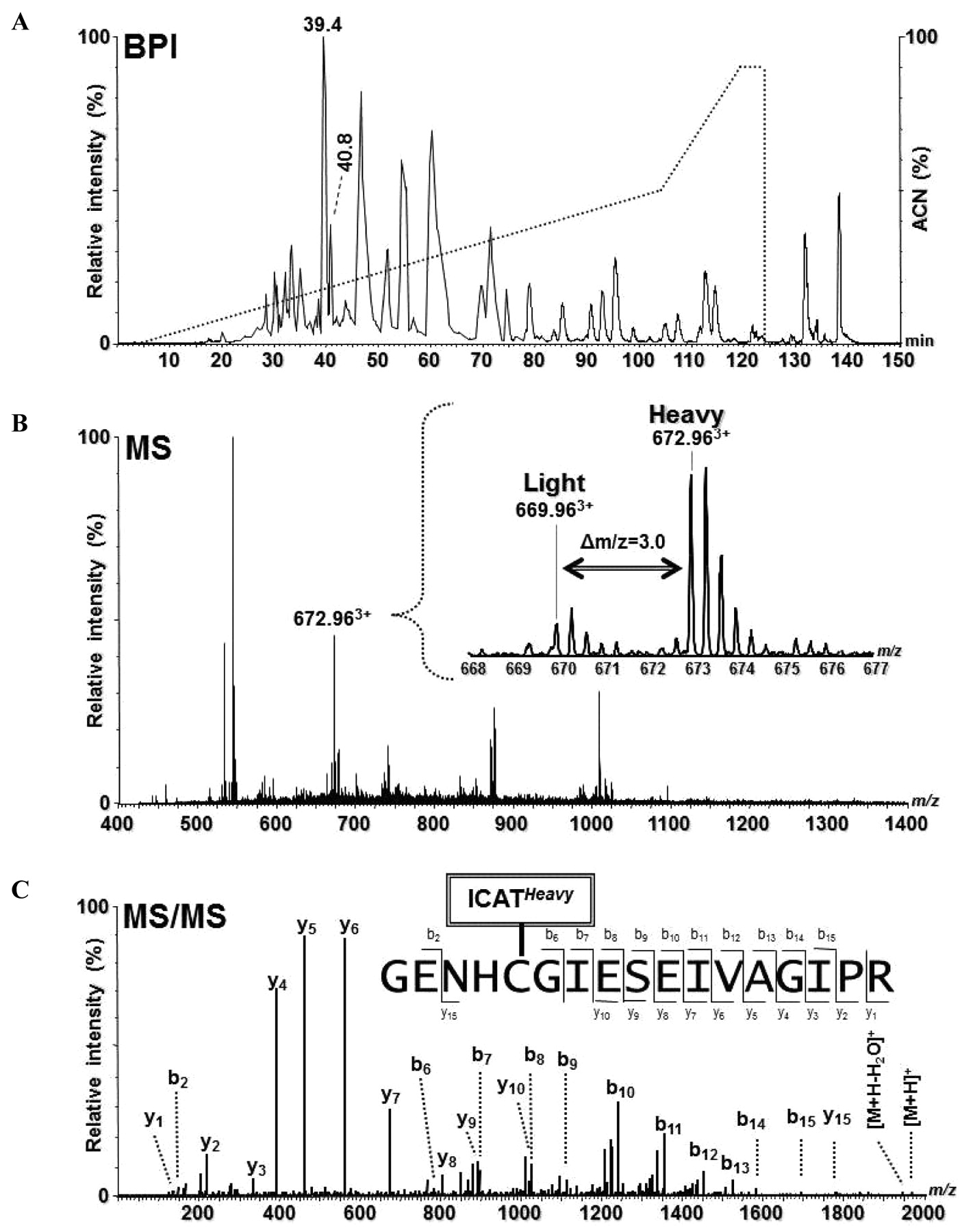
Identification and quantitation of CatB of gel slice 4 using ICAT and GeLC-MS/MS. A: Base peak intensity (BPI) chromatogram for nanoLC-MS/MS of ICAT-labeled peptides (solid line) and the organic elution gradient (dotted line) of the acetonitrile (ACN) solvent. B: MS spectrum of the peptides eluting at 40.25 min (between two peaks at retention time 39.4 and 40.8 min in panel A). Inset: Expanded view of the mass spectrum, in which a triple-charged ICAT-labeled peptide pair were observed at m/z 669.96 (light, L) and 672.96 (heavy, H), respectively. H:L ratio=+3.6. C: MS/MS spectrum of the m/z 672.963+ ion resulting from an ICAT-heavy labeled tryptic peptide of CatB from the β3–/– mouse fibroblasts.
Proteins with expression elevated >1.5-fold in β3–/– mouse embryonic fibroblasts. Proteins displaying up-regulation in β3–/– with respect to WT mouse embryonic fibroblasts from 100 μg each of a membrane fraction labeled with ICATHeavy (13C) and ICATLight (12C) reagent, respectively. Samples were separated by 1D-PAGE, avidin chromatography and analyzed by nanoLC-MS/MS.
Prior to MS analysis, the cICAT-labeled peptides from each of eight equivalent-sized gel slices (see Materials and Methods for specific mass ranges) were further enriched from complex tryptic peptide mixtures through affinity purification. Using an optimized, multi-step 150 min gradient (see Materials and Methods), a better separation and a relatively broader (∼120 min) distribution of peptides retention time (Rt) was achieved. This helped reduce the signal suppression for co-eluted peptides and allowed us to acquire MS/MS spectra of minor components using a relatively low throughput Q-TOF mass spectrometer. For example, the identification and quantification of CatB from gel slice 4 (35-50 kDa mass range) is illustrated in Figure 2. In Figure 2A, a typical base peak intensity (BPI) chromatogram of an enriched, ICAT-labeled peptide mixture is represented. The MS spectrum (Figure 2B) in a single scan obtained at Rt 40.25 min (between two peaks at Rt 39.4 and 40.8 min in Figure 2A) is illustrated. The magnified spectrum (Figure 2B, inset) shows the m/z 669.963+ and 672.963+ ions with a 9 Da mass difference corresponding to a pair of cICAT-light and cICAT–heavy labeled peptides, respectively (H(β3–/–):L(WT)=+3.6). Figure 2C shows an MS/MS spectrum of the 672.963+ ion which was used by an automatic database search engine for protein identification and unambiguously identified as an cICAT–heavy labeled tryptic peptide GENHCGIESEISEIVAGIPR of CatB (100% probability, despite 16 cysteine residues within this protein and a probability of 6-8 cysteine-containing tryptic peptides available) using Mascot or ProteinLynx with stringent filters as described previously (26, 31).
Proteins with expression reduced >1.5-fold in β3–/– mouse embryonic fibroblasts. Proteins displaying down-regulation in β3–/– with respect to WT mouse embryonic fibroblasts from 100 μg of each membrane fraction labeled with ICATHeavy (13C) and ICATLight (12C) reagent, respectively.
From a preliminary list of 123 identified membrane proteins, 48 are differentially expressed between the WT and β3–/– MEFs by more than 1.5-fold, a commonly used threshold for quantitative significance as previously described (see Table I for up-regulated proteins and Table II for down-regulated proteins) (32-36). Examples of the relative quantitation of signal intensities for representative peptide pairs are presented (Figure 3). The observed ratio of H:L=+2.7 for gelsolin (Figure 3A) revealed an increased expression in the β3-null fibroblasts. In contrast, the observed ratio of H:L=–3.8 for galectin-1 (Figure 3B) indicated a decrease in the β3–/– fibroblasts. As shown in Figure 3C, the H:L=1:1 (1:1.02 for two significant figures) for recombinant vimentin (internal standard) is very close to the expected ratio of 1:1 (Figure 3C).
Immunoblot densitometry shows good correlation with MS quantitation. Quantitative MS identifications of the original cytosol fractions were validated independently by western blotting for selected targets (Figure 4A). Western blot analysis was consistent with MS quantitation as determined by band densitometry. For example, MS quantitation indicated CatB is up-regulated in β3–/– cells by over 3.5-fold; densitometric quantitation indicated an increase of 3.1-fold (Figure 4A and 4B). Similarly, proteomic and immunoblot quantitation (1.9 and 1.7) correlated well for the expression of FABP5, the epidermal fatty acid binding protein (Figure 4B). Although, the quantitation was not equal for muskelin or annexin 2A, the trend for their up-regulation in the β3–/– cells was consistent (Figure 4B). Conversely, vinculin was slightly down-regulated in the β3–/– cells compared to WT cells by similar amounts (1.7, which has an inverse value when standardized to 1 of approximately 0.6 for MS and 0.7 for immunoblot quantitations)(Figure 4B). The galectin-1 quantitation was in accordance with the trend for its down-regulation in the β3–/– cells (Figure 4B). The ubiquitously expressed Na+/K+-ATPase α1 unit present on the membrane was used as a loading control (Figure 4B). An overall correlation coefficient for the log-value expression ratios determined by quantitative proteomics and western blot analysis was 0.9 (Figure 4C), which indicates good reliability of our proteomic data (33).
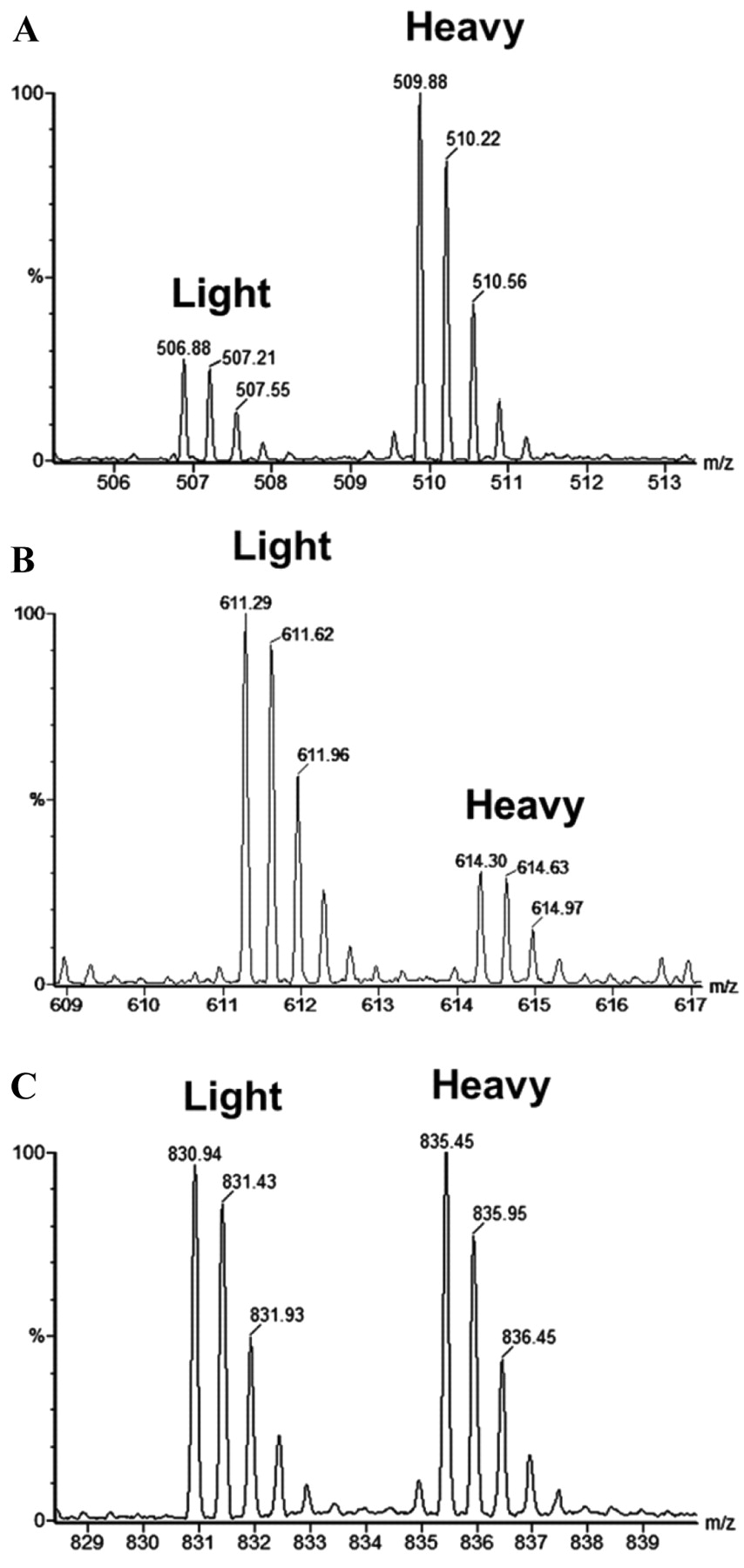
Partial MS spectra showing representative pairs of ICAT-light and ICAT-heavy labeled tryptic peptides. A: SEDCFILDHGR from gelsolin, H:L ratio=+2.7. B: FNAHGDANTIVCNTK from galectin-1, H:L=–3.8. C: QVQSLTCEVDALK from vimentin (control), H:L=+1.0. The H(β3–/–):L(WT) ratios were measured automatically using ProteinLynx software, verified by Mascot, and further validated by manual inspection of the extracted ion chromatograms.
Intracellular CatB is increased in β3 integrin-deficient MEFs. Next, we determined the cell-associated (intracellular) activity of CatB using fluorogenic peptides for an in vitro substrate assay (see Materials and Methods). CatB activity was on average over 1.7-fold higher in the β3–/– cells compared to WT (Figure 5A). The relative difference in activity was not dependent on the manner by which the cells were harvested since both enzymatic (trypsin/EDTA) and mechanical (scraping with a rubber policeman) procedures did not affect the reproducibility. Furthermore, the CatB activity appeared to be linear because the relative difference did not change when either 50 μg or 100 μg of starting lysate was used. In all experiments, activity was completely suppressed with CA-074, a CatB-specific inhibitor, indicating that other proteases were not contributing to the increased activity. To affirm whether increased intracellular CatB activity was due to a deficiency in secretion, latent CatB activity was measured in the media. Pro cathepsin B, the predominant form of CatB secreted from the fibroblasts, requires proteolytic activation (27). Therefore, conditioned media from both WT and β3–/– fibroblasts grown on fibronectin-coated plates was collected, concentrated, and then incubated with pepsin in order to measure the amount of ‘activatable pro-CatB’ being secreted into the media (Figure 5B) (27). Clearly, the amount of pro-CatB exocytosed is significantly lower in β3–/– than in WT cells when grown on fibronectin.

Independent validation experiments of selected markers from nanoLC-MS/MS experiments of the membrane fraction. A: Immunoblotting correlation against muskelin, cathepsin B, fatty acid binding protein 5, annexin 2, vinculin, galectin-1, and Na+/K+-ATPase (control). B: Numerical values of cICAT quantitation and relative densitometry values from immunoblotting for each pair with WT values equal to 1. C: The log10 values of the western blot densitometry for β3–/– expression were plotted versus the log10 values for cICAT ratios from Tables I and II. Both the slope (y) and the correlation coefficient (R2) of 0.903 were calculated from the represented data.

Functional analysis of cathepsin B (CatB) enzymatic activity in cells with reduced or augmented β3 integrin. A fluorometric assay for the determination of CatB proteolytic activity based on cleavage of the synthetic peptide substrate, Z-Arg-Arg-AMC. A: Comparison of cell-associated activity (100 μg protein input) from WT and β3–/– mouse embryonic fibroblast lysates incubated for 1 h with or without the CatB-specific inhibitor, CA-074. B: Activity comparison of secreted pro-CatB from fibroblasts grown on fibronectin in low serum conditions. Media without cells was used as control. C: Comparison of a heterologous cell β3/293 (β3+/+) and HEK293 (β3–/–) human embryonic kidney cell lysates incubated for 1 h with or without the CatB-specific inhibitor, CA-074. Inset: Immunoblot of β3 integrin and CatB protein expression in β3/293 and HEK293. In all experiments, purified recombinant human CatB (10 Units) was used as a control. Values represent the background-subtracted average reading (relative fluorescence units, RFU) from independent experiments repeated at least three times with comparable results.
CatB expression is reduced in β3 integrin-transfected human cells. To confirm the fact that the relationship between CatB and β3 integrin is not limited to fibroblasts, we investigated the human embryonic kidney (HEK293) cell line which normally expresses appreciable amounts of endogenous αv and β1 integrin subunits but negligible levels of β3 and β5 integrin. These cells preferentially form the αvβ1 heterodimer (37); however, transfection with human β3 integrin drives the formation of the αvβ3 heterodimer (24, 38) as developed in the previously described β3/293 stable cell line (24). Whereas HEK293 cells express the αvβ1 integrin and β3/293 cells express the αvβ3 receptor. Both activity (Figure 5C) and expression (Figure 5C inset) of CatB were significantly increased by over two-fold in the HEK293 cells (nominal β3 integrin expression) compared to the β3/293 cells (considerable β3 integrin expression). Taken together, these data demonstrate an inverse correlation between β3 integrin expression levels and cell-associated CatB activity.
Discussion
The goal of this study was to explore the scope and nature of changes to the cytosol-associated proteome in response to integrin knockout. We experimentally influenced the formation of the αvβ3 heterodimer to probe novel proteome relationships in a β3–/– murine model. We quantified the expression of proteins in MEFs by MS/MS analysis and identified a list of 48/123 (39%) unique, non-redundant cytosolic proteins that were differentially expressed by more than 1.5-fold. The expression of CatB, up-regulated by over 3.5-fold, was selected to further validate the potential biological significance of β3 integrin elimination. In β3–/– MEFs, mRNA encoding CatB was unchanged (data not shown), suggesting a post-transcriptional modulation. We determined that the average enzyme activity of CatB increased by nearly two-fold in β3–/– fibroblasts and we confirmed the observations of increased expression and activity in an alternate cell line with high and low β3 integrin expression. Combined, our data establishes an inverse relationship between the expression level of β3 integrin and the cell-associated protease activity of CatB.
Components identified as being functionally related to the protease/proteolytic class were consistently up-regulated in the β3–/– fibroblasts. These included aminopeptidase B, calpain, the endopeptidase inhibitors mug-1 and serpinb6a, and CatB. Interestingly, there are some clear associations with β3 integrin function for some of these proteases. The tripeptide RGD, the primary ligand for αvβ3 integrin is a target of aminopeptidase activity (39). Furthermore, the calcium-dependent protease, calpain, is known to regulate cell migration through cleavage of the β3 cytoplasmic tail (40, 41). Since we found that CatB demonstrated the greatest expression change at 3.6-fold (Table I), by increased activity (Figure 5), we speculate that pools of free CatB increase near cellular membranes. Alternatively, mislocalization, or a reduction in CatB processing (degradation, inhibition, or turnover) may be responsible for the elevated activity.
Increased proteolytic activity is a hallmark of cancer and correlates with metastatic spread, particularly in the context of tumor stromal interaction (42). Recently, observations linking integrin function to cathepsin activity have been reported. Koblinski and colleagues demonstrated that plating human breast fibroblasts on collagen induced α1β1 and α2β1 integrin-dependent pro-CatB secretion (27). This study suggests that i) the secretion of CatB from fibroblasts is modulated by β1 integrin-containing heterodimers when grown on a collagen I matrix, and ii) the inhibition of α1, α2, and β1 subunits with function-blocking antibodies could reduce the secretion of procathepsin B compared to activating antibodies. Klose et al. (43) attained similar results in melanoma cells. They established that function-blocking antibodies against β1 integrin inhibited secretion of pro-CatB upon collagen I binding (43). We propose an analogous mechanism where a lack of β3 integrin-containing heterodimers modulates CatB secretion from fibroblasts plated on fibronectin. Furthermore, results of increased expression of CatB and significant cell-associated enzyme activity were duplicated in HEK293 cells compared to stable transfected cells which express β3 integrin (Figure 5C). Thus, an inverse correlation between β3 integrin loss and intracellular accumulation of CatB is validated in another system that nominally expresses or overexpresses β3 integrin. Corroborating our findings, siRNA-knockdown of CatB and urokinase-type plasminogen activator receptor (uPAR) inhibited glioma cell migration, elicited cytoskeletal condensation, and down-regulated expression of the αvβ3 heterodimer (44). Additionally, inhibition of αvβ3 integrin can reduce metalloprotease activity in melanoma cells (45) and platelets (46). Taken together, these results support the popular mechanism that deregulated integrin function can affect trafficking of proteases to the membrane (47-49).
The αvβ3 integrin is fundamental for establishing the adhesive structure needed to recruit signal and scaffold molecules that initiate reorganization of the actin cytoskeleton and cell migration (28, 50, 51). The majority of proteins in our study grouped under the actin-binding and the cytoskeletal function were up-regulated, including muskelin, L-plastin, gelsolin, cofilin, destrin and capping protein β3. Overexpression of muskelin in mouse myoblasts initiates formation of focal contacts when adherent on thrombospondin, a ligand for αvβ3 (52), and L-plastin peptide induces αvβ3 mediated-adhesion upon actin disassembly (53). Gelsolin’s and associated phosphoinositides’ levels were shown to increase in osteoclast podosomes upon osteopontin binding to αvβ3 while a gelsolin deficiency lead to decreased cell motility signaling (54). Furthermore, adhesion to fibronectin by αvβ3 could promote extensive cytoskeletal reorganization mediated by cofilin and Rho suppression (51). β3 Integrin was not identified as being down-regulated in the β3–/– protein fraction, which is explained by both a lack of β-octylglucoside in the solubilization buffer and the bias towards intracellular components (55). This detergent is required for full saturation and extraction of the membrane. Regardless, the number of identified proteins in the subsets examined and quantified is sufficiently large to suggest substantial changes to the cellular proteome in response to a complete absence of β3 integrin.
By contrast, expression of vinculin and paxillin, both defined components of the classical αvβ3 focal complex (29, 30), was reduced in β3–/– fibroblasts. A consequential reduction in focal adhesions analyzed with vinculin staining has been demonstrated previously in a β3 integrin–/– murine model and also by the common method of antibody blocking for integrin subunits (56, 57). Vinculin and paxillin have also been shown to co-immunoprecipitate with a new subcellular structure, the spreading initiation center (SIC), which seems to exist only during early stages of cell spreading (58). SICs contain focal adhesion markers and an actin sheath as well as ribosomal RNA and RNA binding proteins. We identified several down-regulated nucleic acid-binding proteins (primarily RNA-binding) including heterogeneous nuclear ribonucleoprotein D, in the membrane fraction. Accordingly, the reduction of RNA-binding proteins correlates with the reduction of SIC-related vinculin and paxillin. If the formation of these structures is reduced in β3–/– fibroblasts through a decline of active RNA translation, then this could explain the decrease in vinculin and paxillin expression.
Proteins that share vesicular and secretory functions were another prominent ontological group in our investigation. Integrins internalize within cells, move by vesicular transport to the leading edge for exocytosis, and form new ECM contacts in lymphoid and cervical tumor cells (59, 60), fibroblasts (61), and neutrophils (62). In this study, vesicle amine transport protein (Vat-1 homolog) and esterase D, both membrane-bound vesicle-associated proteins, and members of the annexin family are elevated in β3–/– cells. Annexins comprise a family of type II Ca2+-binding proteins that associate with the cytosolic face of phospholipid-containing cellular membranes. Annexin V can bind the intracellular tail of β5 integrin to pair with the αv subunit (63). Annexin 2 is associated with dynamic actin structures, particularly during times of high cell membrane activity such as phagocytosis, pinocytosis and cell migration (64). The annexin 2 light chain is known to interact with pro-CatB and co-localize to the surface of tumor cells (65), an interaction possibly mediated by β1 integrin (66). This supports a mechanistic link between integrins, annexins, and proteases.
Using a β3–/– murine model, our data suggests that an important subset of proteins influenced by β3 integrin during the initial stages of cell attachment and spreading is actin-related or cytoskeletal in nature. We reveal the first incidence in which β3 integrin influences the post-transcriptional control of CatB, resulting in changes of cell-associated protease activity, potentially related to cytoskeleton and membrane trafficking. These results demonstrate a proteomic change dependent on elimination of one protein. We believe this underscores the caveat that analyses of a single gene knockout can have profound, unexpected biochemical effects that may not be detected without in-depth scrutiny beyond the expected phenotype.
Acknowledgments
We thank Tristan Williams for his excellent technical assistance with MS analysis performed at the Proteomics Facility of the Sanford-Burnham Medical Research Institute. This study was supported by the following grants to J.W.S. (NIH 5U54RR020843-02), to F.P.R. (NIH AR046852, AR048812), and to S.L.T. (NIH AR032788, AR046523, AR048853). Additional support was derived from the Cancer Center grant to the Sanford-Burnham Medical Research Institute (CA30199). J.A.B. was supported by fellowships from the California Breast Cancer Research Program (10FB-0115) and the Canadian Institute of Health Research.
Footnotes
-
↵# Present address: Biology Department, College of Science and Mathematics, California State University-Fresno, 2555 E. Ramon Ave., M/S SB73, Fresno, CA 93740, U.S.A.
- Received October 24, 2011.
- Revision received November 16, 2011.
- Accepted November 17, 2011.
- Copyright© 2012 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.