


Abstract
Celecoxib, a selective inhibitor of cyclooxygenase-2 (Cox-2), was efficacious in clinical prevention trials of patients with familial adenomatous polyposis (FAP) and sporadic colorectal cancer. To identify as yet poorly defined molecular determinants of celecoxib efficacy, a multidimensional serum fractionation approach was used coupled with nanospray tandem mass spectrometry to perform label-free global proteomic profiling of serum samples from the FAP/celecoxib prevention trial. Subsequently, the application of an algorithm for large-scale biomarker discovery on comparative serum proteomic profiles of pre- and post-celecoxib treatment samples identified 83 potentially celecoxib-responsive proteins from various cellular compartments, biological processes and molecular functions. Celecoxib modulation of some of these proteins was confirmed in serum samples of FAP patients and colorectal cancer cell lines by Western blotting. Thus, using a shotgun procedure to rapidly identify important celecoxib-modulated proteins, this pilot study has uncovered novel systemic changes some of which are highly relevant for carcinogenesis and vascular biology. Validation of selected markers, especially those involved in key signaling networks and those considered molecular indicators of cardiovascular pathology, in larger celecoxib clinical trials is expected to provide insights into the molecular mechanisms of celecoxib and the efficacy/toxicity issues related to its use as a chemopreventive agent.
Familial adenomatous polyposis (FAP) patients carry a germ-line mutation in the adenomatous polyposis coli gene and develop numerous adenomatous polyps which invariably progress to colorectal cancer. The therapeutic approach directed against the enzyme cyclooxygenase-2 (Cox-2), which is overexpressed in about 50% of adenomatous polyps, has shown success in FAP patients. Treatment of FAP patients with celecoxib, a selective inhibitor of Cox-2, in a randomized clinical prevention study resulted in a significant reduction in the polyp burden (1). Subsequently, celecoxib has shown anticancer potential in clinical trials of sporadic adenomas albeit with an increased risk of adverse cardiovascular events following its long-term use (2, 3). The molecular mechanisms underlying the growth-inhibitory activity or cardiovascular toxicity of celecoxib remain poorly defined. Despite its selectivity for Cox-2, modulation of several non-Cox-2 targets by celecoxib has also been widely recognized.
Recent advances in global proteomic profiling technologies may enable the identification of celecoxib-mediated proteomic changes in the sera of patients treated with celecoxib. Several mass spectrometry-based approaches have been employed for profiling the human plasma/serum proteome that often use front-end depletion of high-abundance proteins or sub-proteome fractionation to overcome the wide dynamic range of serum proteins (4-7). For quantitative protein estimation, labeling with stable isotopes is often the method of choice (8, 9). The combination of depletion and labeling strategies certainly enhances the discovery of novel serological biomarkers, but also introduces unknown variability, loss of information and the high cost of isotope labeling. More recently, alternative label-free approaches for global quantification of differentially expressed proteins in complex proteomes have been reported (10-14).
Previously, we have used a global proteomic profiling technology, two-dimensional differential gel electrophoresis, and identified wide-ranging, celecoxib-mediated Cox-2-independent quantitative changes in a Cox-2-deficient colorectal cancer cell line (15). In this investigation, another previously described tandem liquid chromatography in conjunction with tandem mass spectrometry (LC/LC/MS/MS) method of label-free quantification (16-18) was used and an algorithm was developed to identify large-scale expression changes in the serum proteins of FAP patients following treatment with celecoxib.
Materials and Methods
Serum samples. The FAP/celecoxib clinical trial along with the baseline characteristics of patients and treatment outcome have been described in detail elsewhere (1). The baseline serum samples prior to the start of treatment from four FAP patients enrolled at the MD Anderson Cancer Center and after treatment with 400 mg twice daily dose of celecoxib for six months were used in this analysis. The serum samples were stored in aliquots of 10 μl at -80°C before analysis.
Experimental design. Trypsin-digested serum samples, in duplicate, were subjected to multidimensional LC/LC/MS/MS and the fragmentation spectra were searched for protein identification. Subsequently, cross-comparison of all the pre- and post-treatment samples analyzed resulted in the identification of potential celecoxib-modulated proteins. A few selected putative celecoxib-modulated proteins were validated by Western blotting either in the patients' serum samples or in celecoxib-treated colorectal cancer cell lines.
LC/LC/MS/MS profiling of serum proteome. Separation of the tryptic peptides by two-dimensional liquid chromatography using a strong cation exchange (SCX) column in the first dimension and a reversed-phase (RP) microcapillary column in the second dimension has been described elsewhere (17). Elution of peptides with increasing salt concentrations and MS/MS analysis by an LCQ Deca XP Plus ion trap mass spectrometer (Thermo Fischer Scientific Inc., San Jose, CA, USA) was also as described elsewhere (17). The protein identification was performed using the TurboSequest algorithm in the Bioworks 3.1 software package (Thermo Fischer Scientific Inc., San Jose, CA, USA) and the Swiss-Prot database (Swiss Institute of Bioinformatics, Geneva, Switzerland). The identified peptides were further evaluated using charge state versus cross-correlation number (Xcorr). SEQUEST results were filtered for false-positive identifications; the criteria for positive identification of peptides being Xcorr >1.5 for singly charged ions, Xcorr >2.0 for doubly charged ions, and Xcorr >2.5 for triply charged ions. Only the best matches were considered. The peak areas of all the identified peptides were calculated using Thermo Fischer Scientific Inc. propriety software. The peak intensity of the precursor mass (±2 Da) in close vicinity of the identification (±60 scans = ±30 s) was added. The threshold was set to 106 below which the intensities were not added to the total peak area. The ratios of the calculated peak areas were used to calculate the relative protein concentration in the pre- and post-treatment groups.
Data analysis. The integrated areas from the pre- and post-treatment samples were first scaled to ensure a reasonable comparison. The scaling was based on the assumption that the serum concentrations, and therefore peak areas, of most protein fragments would be unchanged due to celecoxib treatment. Using the first pre-treatment analysis as the base, each time the same peptide ion from the same salt fraction was identified, a scaling factor equal to the ratio of the areas was determined. The resulting list of ratios for common PSC (peptide fragment, salt concentration, charge) species was used to determine the most likely ratio, which was then used to scale the selected analysis relative to the first. Peptide ratios of all PSCs from the same protein were combined and used to calculate an average log-ratio and its standard deviation. Student's t-test was used to determine the probability that the set of log ratios had a mean value of zero (i.e. no change in the protein concentration due to celecoxib). An additional requirement was the presence of at least 10 log ratios needed to obtain a reasonable estimation of the mean and standard deviation needed for the t-test. The proteins without 10 log ratios were considered up- or down-regulated if the ratio of post-to-pre or pre-to-post PSC observations was greater than four, respectively. A more complete description of this analysis is posted on http://www.abcc.ncifcrf.gov/suppmaterial.shtml
Pathway analysis. Classification and functional enrichment analysis of the identified proteins were performed using the in-house WPS software (19) for the biological processes and cellular component of Gene Ontology (http://www.geneontology.org/). The identified proteins were also analyzed by the Ingenuity Pathway Analysis (IPA) tool (Ingenuity Systems, Redwood City, CA, USA) for their functional significance and in the context of biological association networks.
Western blot analysis. Equal quantities of pre- and post-treatment FAP patients' serum samples or the cell lysates from control or celecoxib-treated colorectal cancer cell lines, HCA-7 and HCT-116 (15), were subjected to Western blotting. The following antibodies were used: anti-Apo A-II, anti-PRDX3 (US Biologicals, Swampscot, MA, USA), anti-ApoA-IV, anti-CD72 and anti-ATM (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA), anti-mTOR (Cell Signalling, Danvers, MA, USA), anti-DNA ligase3, anti-SMAD2/3 (BD Biosciences, San Diego, CA, USA) and anti-actin (Amersham, Piscataway, NJ, USA).
Results
Identification of serum proteins in FAP patients. A total number of 7,716 and 8,631 peptides were identified in the pre- and post-treatment samples respectively, resulting in the non-redundant identification of 885 proteins. A large fraction of these proteins (46.4%) was identified by a single peptide observation. Identification of 25% of the proteins was based upon 2-3 peptides, and ~29% of proteins were identified by 4 to >25 peptides. A stringent criterion was set for positive identifications namely, proteins were identified by four unique peptides or by the same peptide in four or more analyses eluting at the same salt step and charge state (PSC) in one or more pre- or post-treatment runs. Using this criterion, the shotgun LC/LC/MS/MS analysis of the serum samples from the four FAP patients before and after treatment with celecoxib resulted in the identification of 252 proteins with at least four PSC observations (Supplemental Table I, http://www.abcc.ncifcrf.gov/supp_material.shtml).
Celecoxib-modulated proteins in FAP serum samples based upon peptide ratios+.

Large-scale proteomic analysis. A, Multiple comparisons of serum samples: duplicate runs of each of the pre-treatment samples were compared with the corresponding post-treatment runs from the same patient and cross compared with the post-treatment runs of three other patients. B, Distribution of the mean and standard deviation of the log-ratios for selected celecoxib-modulated proteins: the ensemble of log-ratios for each protein was from the scaled peak areas of peptide salt charge (PSC) species that were common between pre- and post-treatment samples. There were 12, 10, 12, 17, 42, 398, 58, 38, 11 and 11 log-ratios for Apo A through SN24, respectively.
Large-scale discovery of celecoxib-modulated proteins in FAP serum samples using mass spectrometric data without labeling. For the large-scale proteomic analysis of celecoxib-modulated proteomic markers, PSCs from the pre- and post-treatment runs were cross-compared (Figure 1A). This approach resulted in an increase in the number of peptide ratios that could be used for statistical analysis and included variations in the peak areas between different runs from the same sample and runs from different samples. Application of the bioinformatics algorithm (described in Supplemental Material) to the LC/LC/MS/MS data identified 45 proteins with altered expression levels in the pre- versus post-treatment samples (p>0.05) (Table I): nine of these proteins showed increased expression, whereas 36 were down-regulated following celecoxib treatment. It should be stressed that these were average increases or decreases and it was entirely possible for an individual to have no change or, for example, even to have an opposite change in the serum level of a protein in the post-treatment sample instead of the predicted increase or decrease. Because this analysis cross-compared all the pre- and post-treatment samples, variations between samples in protein concentrations increased the variance in the recorded log-ratios (Figure 1B). However, the t-test identified those proteins whose log-ratios were sufficiently different from zero while including the effect of this inter-sample variance.
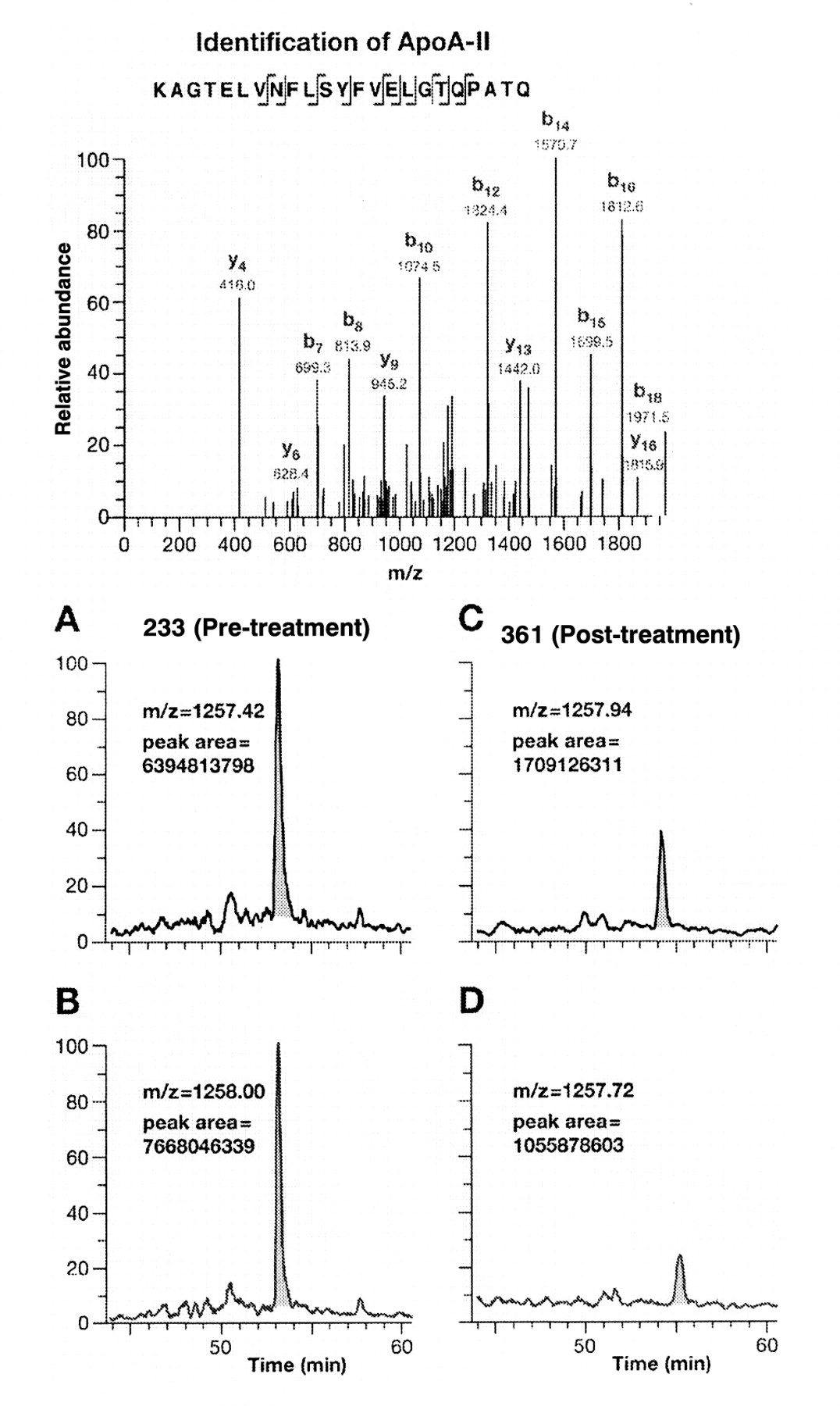
Identification and relative quantification of Apo A-II. A, Tandem mass spectrum derived by collision-induced dissociation of the (M + 2H)2+ precursor ion, m/z 1257.42. Fragment ions in the spectrum mainly represent single-event preferential cleavage of peptide bonds resulting in sequence information recorded from both N (b-ions) and C (y-ions) termini of the peptides simultaneously. SEQUEST searches of the MS/MS data against human database downloaded from National Center for Biotechnology revealed the sequence KAGTELVNFLSYFVELGTQPATQ, which is a tryptic peptide of the C-terminus of human Apo A-II. B, Calculation of the peak area of a selected peptide in all four LC/MS analyses for a single patient (A and B, pre-treatment; C and D, post treatment). The reconstructed ion chromatogram of the peptide KAGTELVNFLSYFVELGTQPATQ from Apo A-II shows the peak area and base peak. Peak areas were calculated using Bioworks 3.1 software.

Validation of proteomic markers by Western blotting. A, Serum samples from FAP patients; B, Colorectal cancer cell lines.
An example of down-regulation, Apo A-II is presented in Figure 2 showing a tryptic peptide of Apo A-II identified through fragmentation information using tandem mass spectrometry and the fully automated SEQUEST search algorithm. A comparison of the peak areas of peptide KAGTELVNFLSYFVELGTQPATQ from Apo A-II before and after treatment of a selected patient is displayed in the panels A - D in Figure 2.
Using another approach to identify candidate celecoxib-modulated proteins, Table II displays celecoxib modulation of another set of 38 proteins based upon the number of identifications in pre-treatment or post-treatment serum samples rather than the differences in the peak areas of common peptides between the two categories. Of these, 14 might have been up-regulated while 24 might have been down-regulated since the ratio of the number of PSC observations in the post-treatment samples was at least four times larger or smaller, respectively than those in the pre-treatment samples.
Validation of celecoxib-modulated proteins. Despite the stringent statistical criteria used for the comparative protein analysis, inaccurate identifications resulting from analytical incompleteness and/or other technical issues inherent to mass spectrometric identifications could not be ruled out. A few selected proteins were therefore validated by Western blot analysis. Figure 3A shows the expression levels of Apo A-IV, Apo A-II, CD 72 and Prx-3 identified as down-regulated proteins by the multiple comparison analysis (Table I). Western blotting showed down-regulation of the proteins in the post-treatment samples in four (Prx-3), three (Apo A-IV), and two (Apo A-II, CD 72) out of the four pairs analyzed. However, as expected and pointed out in the previous section, in some cases, the expression was found to be either unchanged (Apo A-II 435/257 and 387/274: CD 72, 435/247 and 233/361) or even up-regulated (Apo A-IV, 233/361) in the post-treatment versus pretreatment serum samples (Figure 3A).
The attempts to validate many other celecoxib-modulated proteins in the serum by Western blotting using multiple antibodies were not successful. Examples included mTOR, DNA ligase, ATM, and SMAD 2 (Tables I and II). The presence of peptide fragments of these proteins in the serum identified by LC/LC/MS/MS may be a consequence of degradation and subsequent leakage into the serum. The possibility of their modulation by celecoxib was therefore tested in the Cox-2 expresser and non-expresser colorectal cancer cell lines, HCA-7 and HCT-116 respectively. Western blotting showed elevated expression of mTOR and DNA ligase and decreased expression of ATM, especially at the high dose of celecoxib. Thus, the altered expression in colon cancer cell lines was in the same direction as identified by the bioinformatics algorithm in the sera of the FAP patients (Figure 3B and Tables I and II). The expression level of SMAD2 was unaffected by celecoxib in HCT-116 cells, whereas it was increased in the HCA-7 cell line.
Bioinformatics analysis using Gene Ontology and Ingenuity Pathway Analysis Tool. The bioinformatics analysis of the dataset of celecoxib-responsive proteins using Gene Ontology showed representation from diverse cellular compartments including low abundance proteins, such as those involved in signal transduction, transcriptional regulation, and those with antioxidant and transporter activities (Figure 4A). The IPA (www.ingenuity.com) analysis showed that the significant functional categories possibly affected by celecoxib included cardiovascular system development/cardiovascular disease, cell cycle and cell death (Figure 4B). Furthermore, Figure 4C shows network analysis of multiple members on the list of potential celecoxib-modulated proteins (Tables I and II) that are related through direct or indirect biochemical or genetic interactions.
Discussion
In this pilot investigation, the label-free MS-based quantitative profiling of serum samples from the FAP/celecoxib clinical trial identified potential celecoxib-responsive expression changes in many novel proteins, which have not previously been implicated in celecoxib-mediated adenoma regression. A significant finding of this study was the identification of several novel proteins including mTOR, ATM, DNA ligase, and possibly SMAD2 as potential targets for celecoxib modulation. The mammalian target of rapamycin (mTOR), a protein serine-threonine kinase, is a crucial member of complex biological pathways that respond to growth factors and availability of nutrients and are involved in cancer cell metabolism, cell cycle progression, and cellular proliferation (20). The mTOR regulatory network is therefore a target in clinical trials of various carcinomas. The ATM kinase and DNA ligase are involved in DNA damage repair with obvious implications in carcinogenesis (21, 22). Similarly, the signaling of SMAD proteins, downstream of TGF-β superfamily members, is also implicated in carcinogenesis (23). Without the availability of adenoma tissues from FAP patients, one way to unambiguously demonstrate the effect of celecoxib on the expression levels of specific proteins, identified in global proteomic profiles of FAP serum samples, is through celecoxib-modulation of these proteins and/or their transcripts in colorectal cancer cell lines. We have previously shown regulation of mTOR, ATM, DNA ligase, and SMAD 2 by celecoxib at the transcription level (24) and of ATM and DNA ligase at the protein level by isotope-coded affinity tag labeling (unpublished observations). Celecoxib modulation of all four proteins, which are highly relevant for carcinogenesis, in Cox-2 expresser and/or non-expresser colorectal cancer cell lines in the present study (Tables I and II, Figure 3B) strongly suggested that these proteins may be involved in celecoxib-mediated adenoma regression in FAP patients.
Celecoxib-modulated proteins in FAP serum samples based upon number of identifications

Bioinformatics analysis of proteins identified by LC/LC/MS/MS analysis. A, Classification of 252 proteins within selected Gene Ontology (GO) terms for biological processes and cellular components. B, Functional enrichment analysis of celecoxib-modulated proteins by Ingenuity Pathway Analysis Tool. The functional terms 1-12 were identified for their significant enrichment levels using p-values calculated with right-tailed fisher's exact test. The threshold line in the graph corresponds to a p-value of 0. 05 as conventional cut-off. The functional terms above the threshold line would be considered as significantly modulated by celecoxib. 1: cardiovascular system development, 2: hematological system development, 3: cardiovascular disease, 4: hematological disease, 5: cell cycle, 6: cancer, 7: cell death, 8: immune response, 9: immune and lymphatic system development, 10: immunological disease, 11: tumor morphology, 12: inflammatory disease. C, Network view of associated proteins created within Ingenuity Pathway Analysis Tool (modified for clarity) with some of the celecoxib-modulated proteins. Red nodes: proteins up-regulated by celecoxib, green nodes: proteins down-regulated by celecoxib; white nodes: proteins not in celecoxib-modulated list, but with evidence of association with celecoxib-modulated proteins in the network annotated in Ingenuity database.
Celecoxib-modulation of the expression levels of Apo A-II and Apo A-IV, considered to be potential markers of coronary heart disease (CHD) (25, 26), in the post-treatment samples of the FAP patients is noteworthy in the light of safety concerns due to adverse cardiovascular events associated with the long-term use of celecoxib (27-29). It should be emphasized here that the celecoxib treatment of the FAP patients was for six months and in a much younger patient population (median age of FAP patients 39 years vs. 59 years in sporadic adenomas trials) as opposed to three years in the two adenoma recurrence trials (2, 3). A lack of uniform down-regulation of potential markers of cardiac toxicity is not surprising given the biological complexity of individual responses of patients treated with celecoxib for a relatively short duration and therefore can only be considered as suggestive. Analysis of expression levels of Apo A-II, Apo A-IV and other apolipoproteins in patients of clinical trials with long-term treatment with high doses of celecoxib would be useful for exploring if these apolipoproteins might serve as valuable diagnostic markers to monitor cardiovascular toxicity.
A possible association of adverse cardiovascular events with celecoxib treatment was indicated by the IPA analysis (Figure 4 B). Cell death was also one of the significant terms identified that included five proteins, mTOR, ATM, PLG (plasminogen), PTPN13 (Fas-associated protein tyrosine phosphatase) and VTN (vitronectin), apparently modulated by celecoxib. A sub-network of genes (Figure 4C), along with their functional partners, is heavily involved in cardiovascular development/cardiovascular disease, lipid metabolism and molecular transport, processes that may be directly relevant to celecoxib-associated cardiovascular problems. Modulation of the expression levels of three members of this network, Apo A-II, Apo A-IV, and mTOR, was experimentally validated in some of the FAP serum samples or in the colorectal cancer cell lines.
In conclusion, MS-based proteomic profiling of pre- and post-treatment samples from FAP patients allowed multiparametric measurements of serum proteins and, together with the computational analysis, identified a wide range of differentially expressed novel proteomic markers. Our study provides evidence that celecoxib modulates the expression of proteins originating from a wide range of biological classes and molecular functions, some of which may be involved in adenoma pathology and some may serve as indicators of cardiovascular toxicity. Despite the small number of samples in the study, the analysis provides a systems biology perspective of the effects of celecoxib on physiological processes that culminate in adenoma regression and may also lead to cardiovascular toxicity. Future investigations validating and characterizing the novel celecoxib-responsive proteins, identified in this pilot study, would help clarify efficacy/toxicity issues and to better understand the molecular targets of one of the most widely used chemopreventive agents.
Acknowledgements
This project was funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under Contract NO1-CO-12400. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the United States Government.
Footnotes
-
↵* These authors contributed equally to this work.
- Received September 8, 2008.
- Revision received November 14, 2008.
- Accepted December 4, 2008.
- Copyright© 2009 International Institute of Anticaner Research (Dr. John G. Delinassios), All rights reserved




{kind=link}
{kind=link}
{kind=link}
{kind=link}