


Abstract
Brain metastases outnumber the incidence of brain tumors by a factor of ten. Patients with brain metastases have a dismal prognosis and current treatment modalities achieve only a modest clinical benefit. We discuss the process of brain metastasis with respect to mechanisms and involved targets to outline options for therapeutic intervention and focus on breast and lung cancer, as well as melanoma. We describe the process of penetration of the blood-brain-barrier (BBB) by disseminated tumor cells, establishment of a metastatic niche, colonization and outgrowth in the brain parenchyma. Furthermore, the role of angiogenesis in colonization of the brain parenchyma, interactions of extravasated tumor cells with microglia and astrocytes, as well as their propensity for neuromimicry, is discussed. We outline targets suitable for prevention of metastasis and summarize targets suitable for treatment of established brain metastases. Finally, we highlight the implications of findings revealing druggable mutations in brain metastases that cannot be identified in matching primary tumors.
- Angiogenesis
- astrocytes
- biomarkers for brain metastasis
- blood-brain-barrier
- brain parenchyma
- genetics of brain metastasis
- microglia
- neuromimicry
- transmigration
- review
In the U.S., 200,000 cancer patients with brain metastasis are diagnosed each year (1). The incidence of primary brain tumors is outnumbered at least ten-fold by the number of patients with brain metastases (2). Like primary brain tumors, brain metastases deteriorate neuronal functions, displace and destruct normal brain tissue and induce cerebral edema, resulting in neurocognitive impairment (3). As a rule, multiple metastatic lesions are detected at the time of diagnosis (4). By the time brain metastasis has been diagnosed, the median survival time of these patients is in the range of five weeks and can be extended to 3-18 months by multi-modality therapy (4). Several different types of tumors display a pronounced tropism of metastasis to the brain. The incidence varies with the source of information and ranges between 40-50% for lung cancer, 20-30% for breast cancer, 20-25% for melanoma, 10-20% for renal carcinoma and 4-6% for gastrointestinal tumors (5-8). Different compartments and microenvironments of the brain can be colonized. For example, in patients with malignant melanoma, 49% of the metastases are of intraparenchymal, 22% of leptomingeal and 29% of dural location (9). In this review, we focus on parenchymal brain metastases originating from the tumor types as described above. We outline experimental systems for investigating brain metastases and describe the process and targets involved in metastasis, homing, colonization and, finally, their outgrowth in the brain parenchyma. There are two categories of animal models for studying brain metastases (10). The first one makes use of cell lines that preferentially metastasize to the brain after injection into the arterial circulation and the second one is based on stereotactic injection of cell lines into the brain. We discuss issues, such as transmigration of tumor cells through brain capillaries, dormancy, establishment of a neuronal niche as a prerequisite of metastasis by brain-metastasis initiating cells (BMIC), interactions of tumor cells with endothelial, glial cells and astrocytes, as well as neuronal mimicry, as an adaption strategy for brain colonization. We also outline biomarkers and gene signatures predictive for brain metastasis, as well as their genetics, all in the context of translational applications. The issue of drug delivery into brain metastases was covered by us in a previous review (11). The targets discussed in this review are summarized in Figure 1.
Blood-Brain-Barrier (BBB)
Since there are no lymphatic vessels in the brain, circulating tumor cells (CTCs) reach the brain via the hematogenous route and have to surpass the BBB as a prerequisite for homing, colonization and outgrowth in the brain parenchyma. The BBB is a neurovascular unit acting as a gatekeeper and restricts the entrance of macromolecules into the brain (12, 13). It is composed of endothelial cells, pericytes and astrocytes. The cerebral vasculature exhibits unique features, such as non-fenestration, continuous tight junctions and low pinocytic activity at the capillary level, all contributing to its gatekeeper function (14). At the interface between blood and brain, endothelial cells are stitched together by tight junctions, which are composed of transmembrane proteins, such as occludin, claudins and junctional adhesion molecules (JAMs) (15). These transmembrane proteins are anchored to the cytoskeleton of endothelial cells by an intracellular protein complex, which includes ZO-1 and other associated proteins (15). Endothelial cells are covered by a basement membrane, pericytes, which regulate BBB permeability, and end-feet of astrocytes that form a perivascular sheath (16). Astrocytic end-feet express and regulate expression of several molecules on tumor cells, such as P-glycoprotein, aquaporin 4 (AQP4), glucose-transporter (GLUT1) and connexin 43, which are involved in transport of various compounds across the BBB (17).
While this ensures tight control of vascular permeability under normal circumstances, increased permeability of tumor-associated vessels has been observed in tumor patients and in experimental systems in the mouse (18, 19). Leakiness of the BBB has been found in xenografts of mice with brain metastases that are larger than 0.5 mm due to production of vascular endothelial growth factor (VEGF) by metastatic cells (18).
Another feature of the BBB is its heterogeneity across different regions of the brain with respect to morphology, histochemistry and functionality (19, 20). High heterogeneity of the permeability of tumor-associated blood vessels has been observed by imaging studies tracking isotope-labeled chemotherapeutic drugs (20). Increase of permeability of the BBB has also been observed in ischemic regions of the brain (21). Mediators and mechanisms of penetration of the BBB by CTCs will be described in the next chapter.
Transmigration
In order to colonize the brain parenchyma, CTCs have to traverse brain endothelial cells, a process referred to as transendothelial migration. Arrest of CTCs at branch points of capillaries and postcapillary venules as a first step of the transmigration process has been observed (22, 23). Two pathways for transmigration have been discussed: the paracellular pathway through inter-endothelial junctions and the transcellular pathway through endothelial cells (24). Whereas the process of leukocyte diapedesis can use both routes, for transmigration of CTCs into the brain, the paracellular pathway is clearly the preferred option (24). Real-time imaging of i.v. injected tumor cells reach an extraluminal position in two days, in case of lung cancer cells, and in seven days, in case of breast cancer cells, after their injection (22). The targets promoting transmigration are at least partially organ- and tumor type-specific (17, 25-27). Targets and pathways involved in transmigration are comprehensively summarized in previous reviews (24, 25).
We will now focus on pre-clinically validated targets with potential for translation into agents preventing brain metastasis at the level of transmigration. Gene expression analysis of breast cancer cell lines with high propensity for metastasis into the brain, followed by analysis of their function and their expression in clinical samples, has revealed several gene products involved in transmigration into the brain (26). Some of the mediators of transmigration are shared by both lung and brain tropism of metastasis models, for example cyclooxygenase COX2 (PTSG2), angiopoietin-like 4 (ANGPTL4) and EGFR ligand heparin-binding EGF (HB-EGF). COX2 activity creates prostaglandin, which increases BBB permeability (27), ANGPTL4 is induced by tumor cell derived transforming growth factor-β (TGFβ) and disrupts endothelial junctions (28) and autocrine HB-EGF induces enhanced formation of invadopodia (29). In the breast cancer system, as mentioned above, αN-acetylgalactosaminide α2,6 sialyltransferase 5 (ST6GALNAC5) was identified as a facilitator of tumor cell/brain endothelial cell adhesion. Sialyltransferases are enzymes adding sialic acid to gangliosides and glycoproteins (30). The special impact of this finding is that ST6GALNAC5, a brain-specific enzyme, can be co-opted by cancer cells for penetration of the BBB. Since sialyltransferases can be inhibited by small molecules, identification of ST6GALNAC5 as a mediator of transmigration may have translational impact for the prevention of brain metastases (31). An additional important mediator of transmigration of breast cancer cells was identified as cathepsin S (CTSS) (32, 33). CTSS is a lysosomal cysteine protease, which can be secreted by a process termed lysosomal exocytosis. It is involved in extracellular matrix (ECM) degradation and major histocompatibility complex (MHC) II peptide presentation (34). Disruption of the junctions by degradation of JAMs, especially JAM-B, by tumor cell-secreted CTSS plays an important role in cancer cell extravasation at the BBB (32, 33). Importantly, transmigration could be inhibited with CTSS-specific inhibitor VBY-999 (ViroBay, Menlo Park, CA, USA), emphasizing the druggability of CTSS as a target (35). Another transmigration-promoting system between breast cancer cells and human brain endothelial cells was identified as the interaction between SDF-1α (CXCL12) and its receptor CXCR4, a seven transmembrane G-protein coupled receptor (36). This signaling pathway induces blood vessel instability through increased vascular permeability based on activation of phosphatidylinositol-3-kinase (PI3K), protein kinase B (PI3K, AKT) and focal adhesion kinase (FAK) (36). It should be stressed that the SDF-1α/CXCR4 interaction is druggable since plerixafor, a small molecule inhibiting this interaction, has been approved for stem cell mobilization in patients with lymphoma and multiple myeloma after autologous transplantation (37).

Schematic representation of brain tropism of metastasis-related targets discussed in this review. Targets associated with the primary tumor, disseminated tumor cells (DTC), the blood-brain-barrier (BBB), brain metastases, astrocytes, neutrophils and exosomes are shown. ADAM8, a disintegrin and metalloproteinase domain-containing protein 8; ANGPTL4, angiopoietin-like 4; alpha v beta 3, integrin αvβ3; BCL2L1, BCL2 Like 1; CCL2, CC chemokine ligand 2; COX2, cyclooxygenase 2; CPT1C, carnitine palmitoyl-CoA transferase 1C; CTSS, cathepsin S; CXCL12, CXC chemokine ligand 12; CXCR4, CXC chemokine receptor 4; EGF, epidermal growth factor; FASL, FAS ligand; GABA-R, γ-aminobutyric acid receptor, GLUT-R, glutamate receptor; GSTA5, glutathione S-transferase; HB-EGF, heparin-binding EGF; IL1, interleukin 1; IL6, interleukin 6; L1CAM, L1 cell adhesion molecule; miR-19, micro RNA 19; MMP1, matrix metalloproteinase 1; MTF, melanotransferrin; NCAM, neural cell adhesion molecule; NO, nitric oxide; NS, neuroserpin; PD-L1, programmed death 1 ligand; PLEKHA5, pleckstrin homology domain-containing family A member 5; PLN, plasmin; PTEN, phosphatase and tensin homolog; PTSG2, prostaglandin-endoperoxide synthase 2; STGALNAC5, α-N-acetylgalactosaminide α2,6-sialyltransferase 5; TNFα, tumor necrosis factor α; TWIST1, Twist family BHLH transcription factor 1; t-PA, tissue-type plasminogen activator; u-PA, urokinase plasminogen activator; VEGFA, vascular endothelial growth factor isoform A.
Many investigations have studied transmigration of melanoma cells through brain endothelial cells (38, 39). Several potential targets were revealed; we focus here on a possible key target. It was shown that melanotransferrin (MTF) plays a crucial role in this process (40, 41). MTF is a glycosyl-phosphatidyl-inositol anchored protein and member of the transferrin family of iron (Fe) binding proteins (40). Plasmin formation, through interaction of MTF with plasminogen, might be the underlying mechanism for promotion of transmigration (42). For transmigration of non-small cell lung cancer (NSCLC) cells, N-cadherin and neural cell adhesion molecule (NCAM) are possible transmigration-enhancing targets; however, these targets have to be validated in more detail (43, 44). Transmigration promoting activity has also been assigned to pleckstrin homology domain-containing family A member 5 (PLEKHA5) for melanoma cells (45, 46), as well as α-cyrstallin and matrix metalloproteinase 1 (MMP1) for breast cancer cells (47, 48). These targets will be described in more detail in the section “biomarkers prognostic for brain metastases”.
Angiogenesis
After passage of the BBB, CTCs reach the brain parenchyma. A prerequisite for successful colonization of the parenchyma is the establishment of a metastatic niche based on interactions with several types of cells as outlined in the following chapters. Interactions of extravasated tumor cells (TCs) with endothelial cells (ECs) are crucial for their survival and colonization of the brain parenchyma. In experimental mouse models, intravascularly injected tumor cells colonize the brain at day 7-10 post-injection (22, 23). After extravasation, TCs have been found to be associated with microvessels or sprouting existing vessels (angiogenesis) in case of lung carcinoma cells or with non-proliferating existing blood vessels (co-option) in case of melanoma cells (22, 23). Association of extravasated TCs with brain endothelial cells also has been described by other groups (49-51). Adhesion of TCs to the vascular basement membrane (52) and integrin αvβ3-mediated adhesion of tumor cells to endothelial cells, resulting in production of VEGF, have been observed (53). TCs in the parenchyma can activate angiogenic programs in the adjacent microvascular ECs, increasing the probability of further dissemination (54, 55). L1 cell adhesion molecule (L1-CAM) has been identified as a mediator of co-option between TCs and blood vessels in the brain (56). An interesting observation is that stromal cells associated with the primary tumor can be involved in metastatic nodules in the brain, demonstrating that tumor cells can bring their own “soil” into the brain parenchyma (57). Overall, it is argued that post-extravasation growth is the major rate-limiting step in brain metastasis (22, 58). Tumor vessels associated with metastasizing brain TCs have a lower microvascular density than surrounding normal brain parenchyma and contain dilated blood vessels with large volume (59). It is generally agreed that the growth of metastatic brain tumors is dependent on adequate blood supply (60, 61) and that VEGF is involved in colonization and outgrowth of brain metastases (62). The role of VEGF in this context is underlined by the following observations. VEGF has been shown to increase adhesion of MDA-MB 231 cells to brain endothelial monolayers (63). VEGF expression increases in a metastatic variant of this cell line and inhibition reduced brain metastatic burden after intracarotic injection (64). Six different human cancer cell lines proven to produce brain metastases were investigated with respect to VEGF as a contributor to metastasis (65). The results support investigation of VEGF inhibition in patients with brain metastases. Bevacizumab, a humanized monoclonal antibody (mAb) directed against VEGF-A, in combination with paclitaxel-carboplatin therapy, significantly increased progression-free survival (PFS) and overall survival (OS) in patients with advanced NSCLC (66, 67) recommending further exploration of bevacizumab and small molecule inhibitors of the VEGF/VEGFR signaling system in metastatic brain disease.
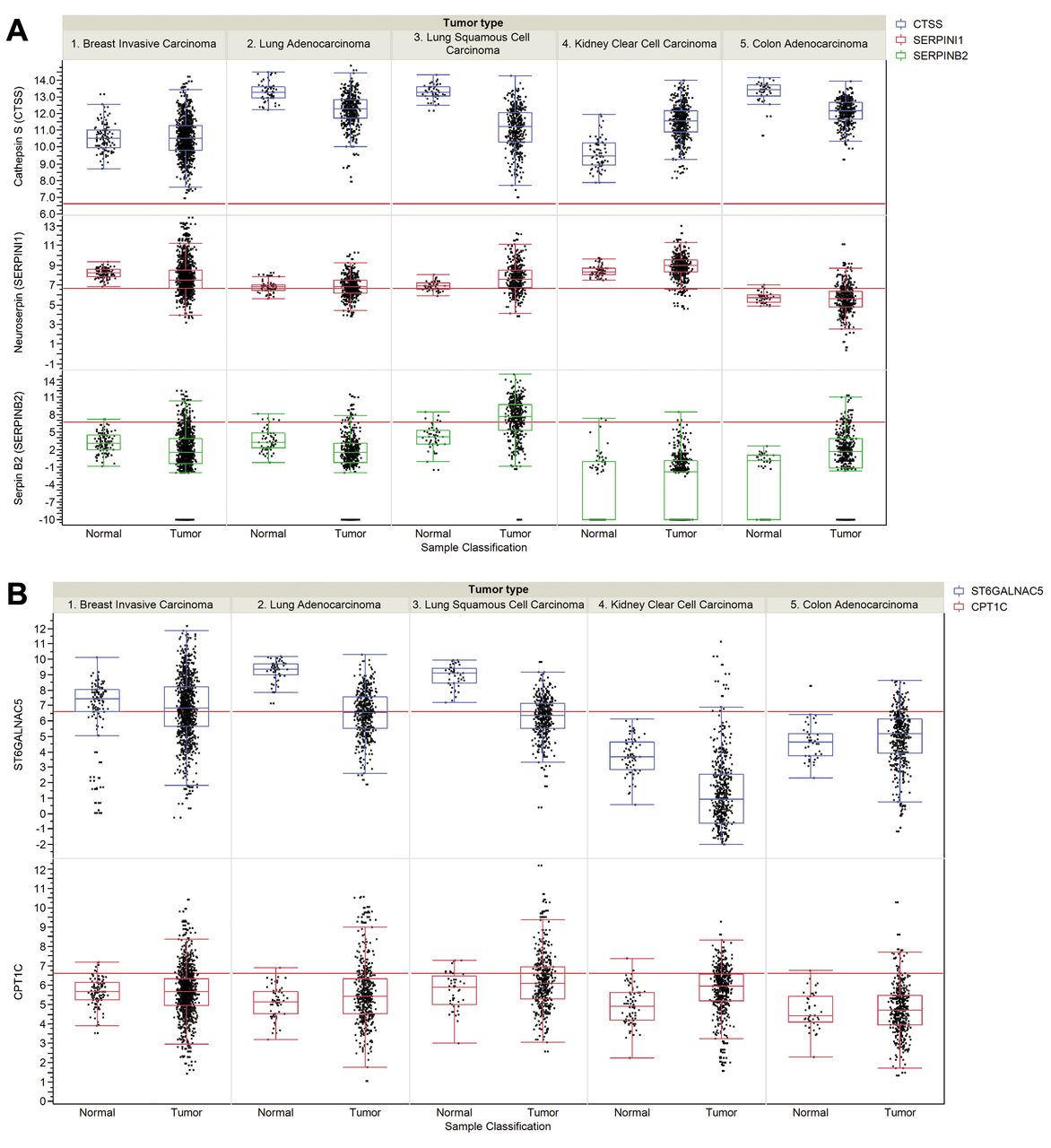
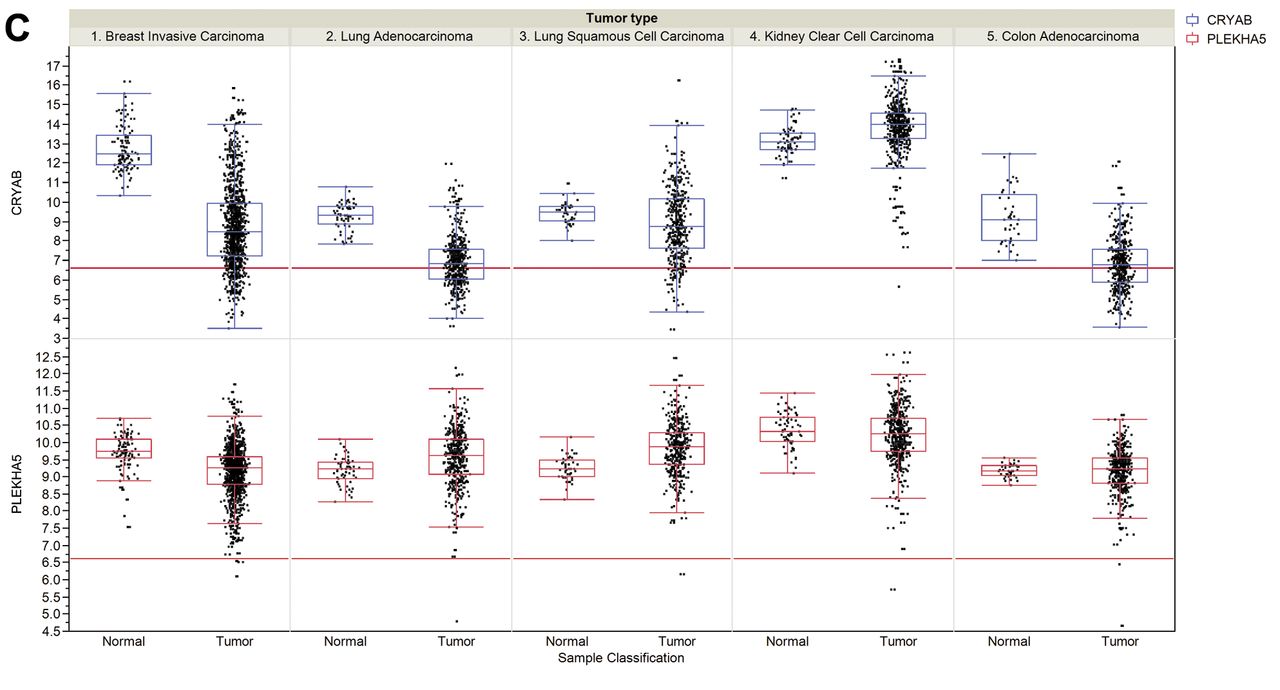
Steady-state RNA levels of selected brain metastasis-related genes in tumors and corresponding normal tissues. A: Protease-related genes. Cathepsin S (CTSS), Neuroserpin (NS) and Serpin B2 (SERPINB2). B: Brain mimicry-related genes. αN-acetylgalactosaminide α2,6 sialyltransferase 5 (ST6GALNAC5), carnitine O-palmitoyl 1 brain isoform (CPT1C). C: Brain metastasis biomarker related genes. Crystallin B (CRYAB), human pleckstrin homology domain-containing family A member 5 (PLEKHA5). Data for RNA expression of selected genes were collected from The Cancer Genome Atlas (TCGA) for breast invasive carcinoma, lung adenocarcinoma, lung squamous cell carcinoma, kidney clear cell carcinoma, colon adenocarcinoma and their corresponding normal tissues. In more detail the cohorts have the following numbers of data points: breast invasive carcinoma (111 matched normal and 1,046 tumor samples), lung adenocarcinoma (58 matched normal and 466 tumor samples), lung squamous cell carcinoma (45 matched normal, 419 tumor samples), kidney clear cell carcinoma (72 matched normal and 519 tumor samples) and colon adenocarcinoma (41 matched normal and 439 tumor samples). Expression values are given as log2 normalized read counts for the TCGA data. The red lines (set at 100 normalized reads) discriminate lower from higher expression. Expression data are shown as box plots where the line in the box represents the data median, whereas the rectangle shows the upper and lower 25% quartile. Therefore, 50% of all data points are included in the rectangle. All other data points, except for outliers, are located within the upper and lower whiskers.
Process of Colonization of the Brain Parenchyma – Interactions of Tumor Cells with the Microenvironment
After passage of the BBB, a dynamic interaction between tumor cells and the new microenvironment takes place, resulting in creation of a neural niche (68). Since tumor cells and cells of the metastatic niche originate from disparate germ-line lineages, an adaption process for tumor cells is required in order to survive in the new microenvironment. Consistently, a phase of dormancy in which tumor cell proliferation is inhibited without induction of cell death mediated by host stress signal-induced activation of the p38 kinase pathway is observed (69, 70). Their reactivation and transition into brain metastasis-initiating cells (BMIC) requires further investigation (71, 72). Dormancy and BMIC are not in the focus of this review. We next outline the interaction of disseminated tumor cells with microglia and astrocytes as pivotal processes for the colonization of the brain parenchyma. Also, the acquisition of novel traits by advantageous mutations (73, 74) may play an important role in the process of neural selection (39, 75), as will be outlined in one of the following chapters in more detail.
Interactions of Tumor Cells with the Microglia
Microglia are brain-type macrophages and considered to be the most important immune effector cells of the brain. Under pathological conditions, they acquire the ability to divide and phagocytose materials. Microglia can activate specific immune responses including B- and T-cells (76). Moreover, microglia-mediated immune-repressive functions, such as expression of programmed death-ligand 1 (PD-L1), the ligand of the inhibitory T-cell receptor PD1, have been identified (77). Activated microglia surrounding areas of tumor burden have been observed (78, 79). In line with anti-tumoral functions of microglia is the observation of a tumoricidal effect of glial cells and macrophages based on nitric oxide (NO)-induced cell death. Inhibition of inducible NO synthase activity in either microglia or macrophages abrogated the tumoricidal effects (80, 81). On the other hand, microglia exert pro-tumoral effects by secretion of multiple cytokines, growth factors and enzymes. Examples are IL1 and IL6, angiogenesis promoter VEGF, tumor proliferation mediator EGF and invasion-promoting metalloproteinases (82). The involvement of the Wnt-pathway in colonization of brain parenchyma with tumor cells mediated by resident macrophages is another example of pro-tumoral activity of these cells. The effect could be antagonized by inactivation of microglia, as well as by Wnt-inhibitor dickkopf-2 (83).
The Role of Astrocytes in Colonization of the Brain Parenchyma by Tumor Cells
Astrocytes maintain homeostasis of the brain micro-environment and protect neurons from injury-induced apoptosis (84, 85). Especially, their survival-promoting role is adapted by tumor cells. Astrocytes release inflammatory cytokines promoting tumor cell proliferation in vitro, such as IL1, tumor necrosis factor α (TNFα) and IL6 (62, 86, 87). An example for harnessing the neuroprotective effects of reactive astrocytes is the protection of melanoma from chemotherapy-induced apoptosis by sequestering intracellular calcium through gap junction channels (88). Calcium acts as a second messenger and plays a crucial role in cell death (89). In line with these findings is the observation that cell contact-dependent induction of survival genes, such as glutathione-S-transferase α5 (GSTA5), BCL2-Like 1 (BCL2L1) and Twist family BHLH transcription factor 1 (TWIST1), are induced in tumor cells after co-culture of human breast and lung cancer cells with murine astrocytes, but not with fibroblasts (90). GSTA5 catalyzes conjugation of glutathione to proteins via sulfhydryl groups and prevents apoptosis by inhibition of c-jun phosphorylation by jun N-terminal kinase (JNK) (91). BCL2-L1 is the longer isoform of BCL-xL and exerts an anti-apoptotic function (92). TWIST1 is a helix-loop-helix transcription factor mediating an anti-apoptotic function by inhibition of the JNK/mitochondrial pathway (93).
Another strategy for survival of tumor cells in the brain parenchyma is to overcome the tumor growth-inhibiting effect of neural stem cells (NSCs). NSCs have been observed in adjacent brain tissue of breast cancer metastases and tumor cell-expressed bone morphogenetic protein 2 (BMP2) has the ability to differentiate NSCs into astrocytes (94). On the other hand, an anti-tumoral function of astrocytes has been observed. Astrocytes secrete tissue-type plasminogen activator (t-PA) and urokinase plasminogen activator (u-PA), both of them being involved in conversion of enzymatically inactive plasminogen to its active form plasmin, which has serine protease activity (95). The latter has been shown to be toxic for tumor cells, thus suppressing brain metastases (96). The lethal effect of plasmin is mediated by a paracrine FAS/FASL-mediated death signal induced by shedding of transmembrane FASL by plasmin on astrocytes (97, 98), as shown in an experimental mouse model with brain-metastasizing breast cancer cells (97, 98). Furthermore, plasmin inactivates cell adhesion molecule L1CAM. This transmembrane receptor is expressed on neurons where it mediates axonal guidance through the growth cone. In the context of brain metastasis, L1CAM has been identified as a mediator of co-option between tumor cells and brain endothelial cells enabling spread of tumor cells on the abluminal surface of brain capillaries (99). In the brain metastasis model as outlined above, expression of serpins by the tumor cells circumvented the lethal effect of plasmin and, thus, promoted colonization of the brain parenchyma (97). Thirty-six serpins inhibiting 18 proteases have been identified (100). Four of these, the brain-specific neuroserpin (NS) and the more broadly expressed serpins B2, E1 and E2 are selective inhibitors of plasmin (101). The most frequently up-regulated serpins in brain-seeking metastasis models were identified as NS and serpin B2, which were found to be expressed in a high percentage of brain metastases from breast and lung cancer patients (97).
An additional remarkable interaction between astrocytes and tumor cells has been noted. Exosomes, derived from astrocytes containing miR-19a, were shown to reversibly down-regulate phosphatase and tensin homolog (PTEN) in tumor cells, resulting in secretion of chemokine (C-C motif) ligand 2 (CCL2), which is able to recruit brain metastasis-promoting myeloid cells (102). This is an example of microenvironment-induced loss of PTEN as a priming event for outgrowth of breast cancer cells in the brain parenchyma due to breast cancer cell-induced gliosis (102). In addition to its transmigration-promoting function as outlined previously, tumor cell-derived and macrophage-derived CTSS supports survival of tumor cells to form micrometastases successfully. In animal models, only combined inhibition of mouse and human CTSS impairs both metastatic seeding and outgrowth (32).
Neuromimicry
Colonization of the brain with tumor cells is the result of reciprocal interactions with the tumor microenvironment. One of the consistent features observed in this context is the acquisition of a neuronal phenotype based on transcriptional reprograming, as outlined below. On the other hand, it has been observed that breast cancer cells derived from primary tumors already express proteins that are essential for outgrowth as brain metastases. Examples are α-crystallin (47, 103), a disintegrin and metalloproteinase domain-containing protein 8 (ADAM8) (104), and metalloproteinases (105). The concept of neuronal mimicry of tumor cells for survival and colonization of the brain parenchyma has been demonstrated with three experimental approaches. The first one is based on comparative transcriptional profiling of triple-negative, as well as HER2-positive breast tumors with brain metastases and representative cell lines (94, 106, 107). The second one is based on transcriptome analysis of four tumor cell lines representing breast, lung, colon cancer and melanoma xenografted into brain, skin and orthotopic sites (108). The third one follows the transcriptional profile of metastases of the brain, bones and lungs after left ventricular injection of two human melanoma cell lines into immunosuppressed rats (109). The common observation in these systems is the emergence of a brain-to-brain transition, adaption to the brain microenvironment and acquisition of characteristics of neuronal lineage cells. The identified molecular candidates are involved in organizing the metastatic niche and promotion of the brain-adaptive phenotype. The derived brain-specific signatures involve genes with neuron-specific functions involved in neurotransmission, neuron excitation, neurogenesis, synaptic plasticity and neuro-inflammation. The first study mentioned above revealed the up-regulation of neurotransmitter γ-aminobutyric acid (GABA)-related components, such as GABA-A receptors, GABA transporters and GABA-transaminase, in brain metastases of breast cancer (94, 110). The neuronal-like GABA metabolism provides a metabolic advantage to breast cancer cells in the brain microenvironment through generation of additional energy due to synthesis of nicotinamide adenine dinucleotide (NAD). GABA-related intervention might emerge as an upcoming treatment for brain metastases. It should be kept in mind that several GABA inhibitors are approved for treatment of epilepsy (110). Components of the glutamate receptor (GLUT-R) signaling pathway, as possible therapeutic targets based on up-regulation of its receptors, were identified in the melanoma-based experimental system as described above (109). GLUT-R activation allows the influx of Ca2+ and activation of Ca2+-dependent effector functions, such as activating transcription factors CREB, EGR1, EGR2, NFAT and NF-κB that regulate genes involved in survival, synaptic development and neuronal plasticity (111). GLUT-R overactivation is involved in exotoxicity and, therefore, corresponding antagonists are presently evaluated in diseases, such as traumatic brain injury, stroke and neurodegenerative diseases (112). In the second experimental system as described above (108), a strong correlation between epigenetic changes and transcriptomic reprogramming was observed, indicating that methylation, at least partly, might be involved in generation of the observed transcriptional changes. However, the key events triggering transcriptome reprograming remain to be elucidated.
Another intriguing observation that a brain-specific enzyme, carnitine palmitoyltransferase 1C (CPT1C) was co-opted by breast cancer cells before metastasis, possibly facilitating metastasis and their outgrowth in the brain parenchyma (113). CPT1C confers proliferation and survival signals to cancer cells. CPT1C catalyzes the conversion of acyl-CoA to carnitine-CoA, facilitating lipid transport through mitochondrial membranes and subsequent β-oxidation of fatty acids and resulting production of ATP (114, 115). Data correlating expression of CPT1C in different types of cancer and corresponding normal tissues and correlation with brain metastasis would be helpful for further elucidation of the function of CPT1C in brain metastasis.
Genetic Aspects of Brain Metastases
Genetics-based information on the relationship between primary tumors and their brain metastases is crucial for understanding generation and evolution of brain metastases and to design individualized, precision medicine-based therapeutic approaches. Whole-exome sequencing has paved the way for high throughput sequencing of matching primary tumors and their brain metastases in comparison to corresponding normal tissues. Comparison of nucleotide sequences of 79 stage IV squamous cell lung cancers (SQCLC) and selected paired brain metastases revealed that brain metastases are genetically highly different from their primary tumors (116). Two subtypes of SQCLC have been identified; one with FGFR1 amplification and another one with alterations in the PI3K pathway, such as loss of function of PTEN and gain of function of PIK3CA mutations (116). The latter patients revealed a higher incidence of brain metastases, higher metastatic burden and strikingly shorter OS compared to patients without these alterations. PTEN loss or PIK3CA activation mutations were maintained in all brain metastases analyzed, despite their genetic heterogeneity. Another recent study is based on whole-exome sequencing of 86 matched primary tumors and brain metastases predominantly from patients with breast, lung and renal cancer (117). An important finding of this study is that 53% of brain metastases had clinically informative alterations not detected in the corresponding primary tumors. Clinically actionable alterations in brain metastases resulting in responsiveness to HER2/EGFR inhibitors (118), MAPK pathway inhibitors (119), CDK inhibitors (120) or inhibition of PI3K/AKT/mTOR signaling (121) were shared in all brain metastases of a patient. In 26 cases, there were 32 alterations predicting sensitivity to HER2/EGFR signaling, 20 mutations were found in both primary and secondary tumors, 2 of them were only in primary tumors and 10 of them were only in brain metastases. Regarding sensitization to MAPK inhibitors based on 29 cases, 36 alterations were found with a distribution of 24/6/6 as described above. Mutations predicting sensitivity to CDK inhibitors are based on 71 alterations with a distribution of 44/7/20. Similarly, 43 alterations in 37 cases were identified, predicting sensitivity to inhibition of the PI3K/AKT/mTOR signaling with a distribution of 24/5/14 (117). Taken together, primary tumors and metastases share mutations; however, unique mutations are discovered in metastases and to a lesser frequency in primary tumors. The mutations found in brain metastases are not shared by lymph node or extracranial metastases, implicating that these metastases are not reliable surrogates for oncogenic mutations of brain metastasis. Analysis confirmed heterogeneity of multiple anatomically and temporally distinct brain metastases and suggests divergent evolution at metastatic sites. The findings, as outlined clearly, indicate that nucleotide sequencing of brain metastases may result in an important translational impact.
Another study has assessed epigenetic differences in primary breast cancers and their brain metastases by bioinformatic screening of genome-wide breast cancer methylation data available at The Cancer Genome Atlas (TGCA) and assessment of the methylation status of eleven pairs of fixed paraffin-embedded primary breast tumors and their corresponding brain metastases (122). The promoters of three genes, GALNT9, CCDC8 and BNC1, were found to be frequently methylated in brain metastases (55%, 73% and 71%) and infrequently or not all methylated in primary tumors. GALNT9 is an initiator of O-glycosylation by linking α-N-acetylglucosamine covalently to Ser and Thr residues (123). CCDC8 encodes a gene product involved in maintaining microtubule dynamics (124). BNC1 (basonuclin 1) encodes a zinc transcription factor involved in expression of many different genes (125). As shown with RNAi and matrigel-coated invasion chambers, reduced expression of either of these genes increased the invasive potential of breast cancer cell lines. It can be speculated that deregulation of these three genes may be more than just a marker for breast cancer brain metastases.
Biomarkers for Brain Metastases
Reliable biomarkers with an expression profile that correlates with brain metastasis would allow treatment of patients with agents preventing brain metastasis. As will be outlined herewith, brain tropism-predicting factors are tumor type-specific and sharing of corresponding markers between different types of tumors occurs rarely.
For breast cancer, several metastasis-predicting markers that are partly validated with respect to their functional contribution to brain metastasis have been identified. In this context, expression of 23 metalloproteinases was evaluated in brain metastasis-free breast cancer patients. Only matrix metalloproteinase 1 (MMP1) was significantly correlated with brain metastasis (48). As previously described, MMP1 degrades claudin and occludin, two key components of the BBB. Interestingly, MMP1 expression was promoted by prostaglandins, the products of metastasis-associated COX2. Another protease, CTSS, a mediator of transmigration of tumor cells through endothelial cells by cleavage of JAM-B, was identified as an additional marker for prediction of metastasis of breast cancer to the brain (32, 33). High CTSS expression at the primary site correlated with decreased metastases-free survival in breast cancer patients (32, 33). Also, α-crystallin was identified as a marker to predict high-risk breast cancer patients for metastasis to the brain (47, 103). α-crystallin is a chaperone member of the small heat-shock protein family that inhibits ECM detachment-induced apoptosis and enhances penetration through an endothelial cell/astrocyte co-culture system of the BBB in vitro. Furthermore, it was shown that α-crystallin acts as a cell death antagonist by inhibition of caspase 3-activation induced by oxidative stress (126) and promotes lung and brain metastasis in an orthotopic model of triple-negative breast cancer (TNBC) in immunodeficient mice (103, 127). A 17-gene signature derived by profiling of brain-seeking breast cancer cells in comparison to their less metastasizing counterparts showed an association with brain relapses in two independent breast cancer datasets (26). Six of the genes involved in brain metastasis were shared with an 18-gene lung metastasis signature that is associated with relapse to the lungs, but not to bones, liver or lymph nodes (128). Genes shared with the lung metastasis signature include cyclooxygenase (COX2), matrix metalloproteinase 1 (MMP1), angiopoietin-like 4 (ANGPTL4), latent TGFβ-binding protein (LTBP1) and fascin 1 (FSCN1). Additionally, STGALNAC5, a brain-specific enzyme promoting transmigration of tumor cells through endothelial cells (26) and heparin-binding EGF (HB-EGF) is also part of the brain-metastasis signature. However, the 17-gene brain metastasis signature remains to be further validated by independent groups since other approaches to identify reproducible genes' signatures for central nervous system (CNS) relapse have failed.
Several efforts have been addressed to identify a gene signature predictive for brain metastasis of lung cancer (129, 130). Three candidate genes, CDH2, KIF1C and FALZ (BPTF) were identified to be associated with brain metastasis of NSCLC by transcriptional profiling techniques (129). N-cadherin (CDH2) is a transmembrane, cell adhesion-mediating protein expressed in multiple tissues (131). KIF1C is a member of the kinesin family of microtubule binding proteins, which slides along and cross-links microtubules (132). FALZ (BPTF) is a histone binding component of the nucleosome-remodeling factor (NURF), a complex that catalyzes ATP-dependent nucleosome binding and transcription of chromatin (133). Bioinformatic analysis of human lung adenocarcinomas resulted in a T-cell factor 4 (TCF-4)-based signature prognostic for metastasis to bone and brain (130). Three gene products, LEF1, HOXB9 and BMP4 were highly correlated with metastatic development. Lymphoid binding factor 1 (LEF1) (134) and homeobox DNA-binding protein HOXB9 (135) are transcription factors, while bone morphogenetic protein 4 (BMP4) stimulates expression of genes related to Smad- and MAPK-based signal transduction pathways (136).
Few biomarkers predictive for brain metastases are shared by different tumor entities. An example is neuroserpin and serpin B2 whose expressions in lung adenocarcinoma and breast cancer are associated with relapse (32).
A brain relapse-predicting factor for malignant melanoma has been described. Expression of PLEKHA5 in primary tumors was associated with a shorter time to development of brain metastases for both cerebral and extracerebral metastases (45, 46). PLEKHA5 was identified by expression profiling of A375P melanoma cells and its cerebrotropic variant A375Br (45, 46). Silencing of PLEKHA5 decreased cell viability and the in vitro potential for crossing the BBB. PLEKHA5 is member of a family of seven proteins, PLEKHA1-7, with pleckstrin (PH) and Trp-Trp WW domains (137). PLEKHA5 expression is associated with cell membranes and microtubules playing a role in cell migration and cell-cell interaction (138). PH domains, as found in PLEKHA5, are known for phosphoinositide binding properties and, therefore, PLEKHA5 might interact with the PI3K/AKT pathway. In melanoma, the PI3K/AKT pathway can be activated through mutations in NRAS (139), whereas loss of PTEN correlates with BRAF activation and earlier development of brain metastases (140).
Expression of Brain Metastasis-related Genes in Tumors and Corresponding Normal Tissues
Steady-state RNA levels for breast-invasive carcinoma, lung adenocarcinoma, lung squamous cell carcinoma, kidney clear cell carcinoma, colon adenocarcinoma and corresponding normal tissues were derived from The Cancer Genome Atlas (TCGA). Colon adenocarcinoma was included as a tumor type that does not exhibit brain tropism of metastasis. Melanoma was not included due to lack of data covering corresponding normal tissues in TCGA. We have focused on protease-related genes, such as cathepsin S (CTSS), neuroserpin (NS) and serpin B2 (SERPINB2), brain mimicry-related genes such as ST6GALNAC5 and CPT1C and brain metastasis biomarker-related genes, such as crystallin α (CRYAB) and PLEKHA5. In many of the pairs analyzed, the data median were identical or even lower in corresponding tumor tissue (Figure 2A, B, C). We observed up-regulation of NS, SERPINB2 and PLEKHA5 in lung squamous cell carcinoma, PLEKHA5 in lung adenocarcinoma and CTSS, NS, CPT1C and CRYAB in kidney clear cell carcinoma. These correlations have to be investigated further by analysis of occurrence of brain tumors and expression levels of the corresponding genes in patients. In invasive breast carcinoma no median up-regulation of CTSS, NS, serpin B2, ST6GALNAC5 and CPT1C was observed; however, outliers representing increased expression in comparison to normal tissue were consistently noted. It cannot be excluded that they are associated with the increased sample numbers in tumor tissue in comparison to normal tissues. Surprisingly, steady-state levels of α-crystallin were found to be down-regulated in invasive breast carcinoma in comparison to normal tissue and no outliers with increased expression were observed. Regarding the discrepancy with respect to α-crystallin, as a biomarker for brain metastasis, it should be kept in mind that steady-state RNA levels do not necessarily correlate with protein steady-state levels in all cases.
Concluding Remarks
The therapeutic benefit of current treatment modalities for brain metastases, such as surgery, whole-brain radiation therapy, stereotactic radiosurgery, chemotherapy and growth factor inhibitors, is limited (4, 61). Medium survival time of 5 weeks for untreated patients can be extended to several months with multimodality therapy by combination treatments as mentioned above (61, 62). Also, prevention of brain metastasis with therapeutic agents is an important medical challenge. We have outlined biomarkers in the primary tumors' predictive metastases to the brain, such as MMP1, α-crystallin, PLEKHA5, cathepsin S, neuroserpin and serpin B2, that still need further pre-clinical validation with respect to their relevance as targets for prevention of brain metastases. Targets involved in transmigration of tumor cells through the BBB, such as CXCL12/CXCR4, ST6GALNAC5, CTSS, MMP1 and α-crystallin can be identified by profiling of the primary tumors. Inhibition of colonization of the brain parenchyma and outgrowth of metastases is another issue. Emerging targets in this context are serpins, such as neuroserpin and serpin B2, as well as neuromimicry-based targets, such as GABA and glutamate receptors. A possible breakthrough was achieved by whole-exome sequencing of brain metastases and their matching primary tumors. These efforts have revealed that temporally and locally distinct metastases from the same patient share druggable driver mutations that are not present in the matching primary tumors. These findings may result in new treatment modalities for brain metastases as personalized medicines based on genetic information of the metastases of individual patients.
- Received February 25, 2016.
- Revision received April 21, 2016.
- Accepted April 25, 2016.
- Copyright© 2016, International Institute of Anticancer Research (Dr. John G. Delinasios), All rights reserved
References
In this issue




{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- Blood-Brain-Barrier (BBB)
- Transmigration
- Angiogenesis
- Process of Colonization of the Brain Parenchyma – Interactions of Tumor Cells with the Microenvironment
- Interactions of Tumor Cells with the Microglia
- The Role of Astrocytes in Colonization of the Brain Parenchyma by Tumor Cells
- Neuromimicry
- Genetic Aspects of Brain Metastases
- Biomarkers for Brain Metastases
- Expression of Brain Metastasis-related Genes in Tumors and Corresponding Normal Tissues
- Concluding Remarks
- References
- Figures & Data
- Info & Metrics