


Abstract
Background: The genome association databases provide valuable clues to identify novel targets for cancer diagnosis and therapy. Genes harboring phenotype-associated polymorphisms for neoplasm traits can be identified using diverse bioinformatics tools. The recent availability of various protein expression datasets from normal human tissues, including the body fluids, enables for baseline expression profiling of the cancer secretome. Chemoinformatics approaches can help identify drug-like compounds from the protein 3D structures. Materials and Methods: The National Center for Biotechnology Information (NCBI) Phenome Genome Integrator (PheGenI) tool was enriched for neoplasm-associated traits. The neoplasm genes were characterized using diverse bioinformatics tools for pathways, gene ontology, genome-wide association, protein expression and functional class. Chemogenomics analysis was performed using the canSAR protein annotation tool. Results: The neoplasm-associated traits segregated into 1,305 genes harboring 2,837 single nucleotide polymorphisms (SNPs). Also identified were 65 open reading frames (ORFs) encompassing 137 SNPs. The neoplasm genes and the associated SNPs were classified into distinct tumor types. Protein expression in the secretome was seen for 913 of the neoplasm-associated genes, including 17 novel uncharacterized ORFs. Druggable proteins, including enzymes, transporters, channel proteins and receptors, were detected. Thirty-four novel druggable lead genes emerged from these studies, including seven cancer lead targets. Chemogenomics analysis using the canSAR protein annotation tool identified 168 active compounds (<1 μM) for the neoplasm genes in the body fluids. Among these, 7 most active lead compounds with drug-like properties (1-600 nM) were identified for the cancer lead targets, encompassing enzymes and receptors. Conclusion: Over seventy percent of the neoplasm trait-associated genes were detected in the body fluids, such as ascites, blood, tear, milk, semen, urine, etc. Ligand-based druggabililty analysis helped establish lead prioritization. The association of these proteins with diverse cancer types and other diseases provides a framework to develop novel diagnosis and therapy targets.
- Biomarkers
- body fluids
- chemoinformatics
- chemogenomics
- clinical variations
- druggable targets
- expression quantitative loci
- gene ontology
- genome-wide association studies
- leukemia and lymphoma
- open reading frames
- phenome-genome association
- secretome
- rule of five
- tumors
A vast amount of genome-wide association studies (GWAS)-based datasets is becoming available for mining the genome for disease association (1-10). Discovery of novel molecular targets for diverse diseases is greatly aided by the Phenome to Genome analysis tools. The National Center for Biotechnology Information (NCBI) Phenome Genome Integrator bioinformatics tool (PheGenI) offers an effective approach to decipher a gene’s polymorphic association with a disease phenotype (11). The association evidence, together with the Expression Quantitative Trait Loci (eQTL) analysis (12), allows us to prioritize molecular targets for rational drug discovery.
The recent availability of numerous protein expression analysis tools has expanded our capability to monitor the protein levels in diverse normal and tumor tissues (13-19). Tools, such as the human protein reference database (HPRD), Multi-omics profiling expression database (MOPED) and Proteomics database (Proteomics DB) encompass protein expression datasets from a large number of body fluids. Discovery of molecular targets that can be detected in the body fluids (the cancer secretome) offers an advantage for biomarker lead prioritization efforts (20, 21). Changes in the target gene expression level can be readily monitored in response to cancer progression or therapy in a non-invasive manner.
Further, novel diagnostic and response-to-therapy indicator proteins may emerge from a database of neoplasm-associated genes that can be readily detected in the body fluids.
Major druggable class of proteins for small molecular weight compounds in the human genome includes cluster of differentiation (CD) markers, enzymes, ion channel proteins, G-protein-coupled receptors, nuclear receptors and transporters (16, 22). Currently, only 640/22,000 proteins present in the human genome are targeted by the Federal Drug Administration (FDA)-approved drugs. Additional targets are clearly needed to provide a basis for cancer drug discovery. The 3D structures of the human proteins can be readily mined for ligand-based druggability prediction using chemogenomics approaches (23, 24). A cancer-oriented protein annotation tool, canSAR, provides tools to mine the cancer proteome for druggableness with links to the chEMBL repository of compounds (25-28). By utilizing such an approach, recently novel molecular targets were identified for diverse diseases, including diabetes (29), Ebola virus disease (30), neurodegenerative diseases (31) and pancreatic cancer (32).
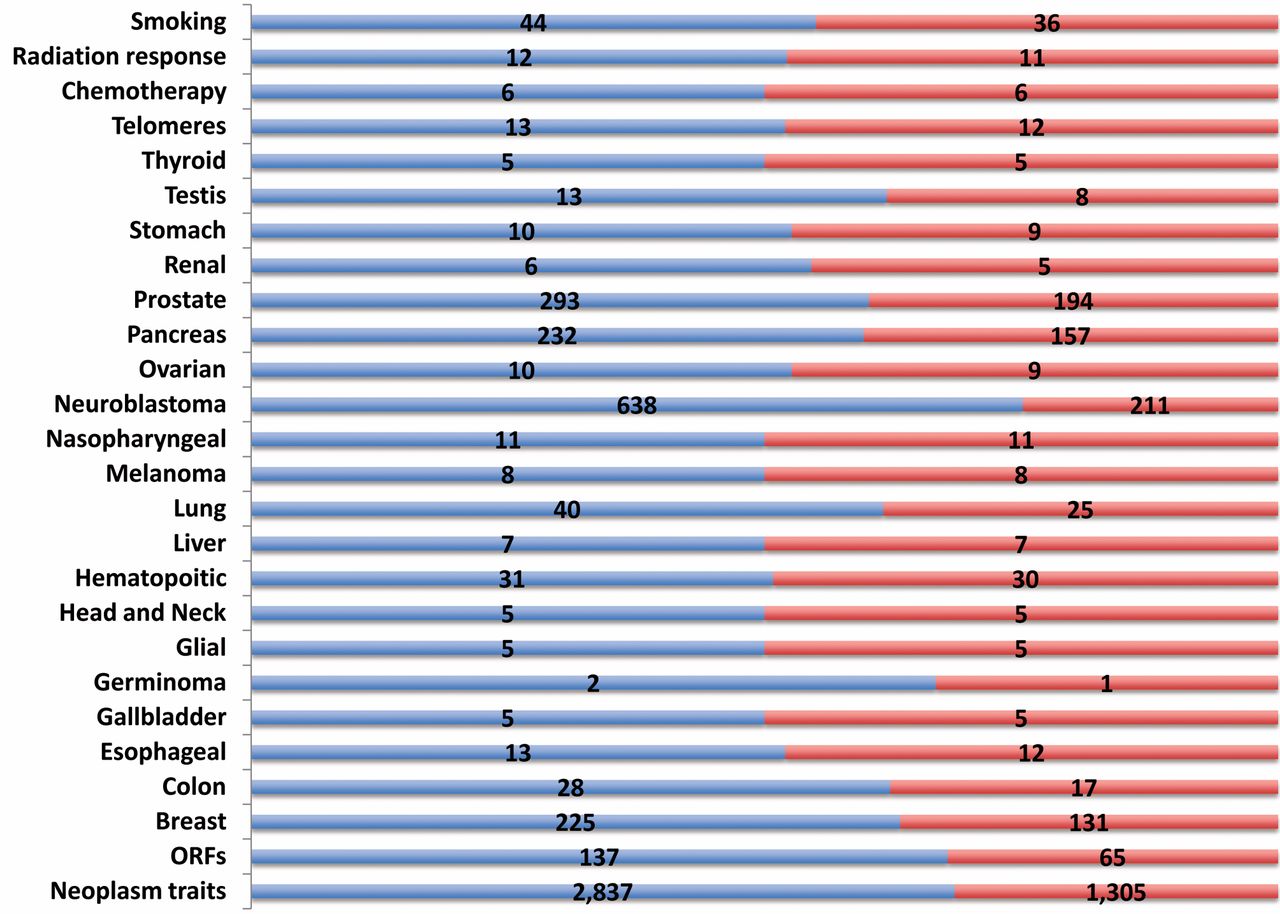
Neoplasm-associated traits in the human genome. The NCBI Phenome-Genome Integrator was used to enrich neoplasm-associated traits. For the indicated traits, the number of genes (red) and the number of associated SNPs (blue) are shown. p-Values <1×10-5. The SNP class included the exon, intron, near gene and untranslated region.
To aid in the discovery of novel cancer-related targets and facilitate drug discovery efforts, a database of genes related to the neoplasm-associated traits was established. Protein expression profile of these genes in diverse body fluids was established. Druggable class of proteins from the secretome was analyzed using chemogenomics approaches. Active lead compounds (<1 μM) with drug-like property were identified. These results open up novel opportunities for cancer drug discovery efforts, as well as for the development of new biomarkers.
Materials and Methods
The bioinformatics and proteomics tools used in the study have been described previously (32-34). The protein annotation and chemical structure-based mining was performed using the canSAR integrated knowledgebase 2.0 (26, 35). The browse canSAR section was used and the neoplasm-associated proteins were batch-analyzed for protein annotations, 3D structures, compounds and bioactivity details. The canSAR compounds link for genes has diverse filters, such as activity and assay types, concentrations, molecular weight, rule of five (RO5) violations, prediction of oral bioavailability and toxicophores. The protein 3D structure information was obtained from the Swiss Protein Database (36). The chemical structures were obtained from the chEMBL (28). Comprehensive gene annotation for the neoplasm-associated genes was established using the GeneCards (37), the DAVID functional annotation tool (38) and the UniProt (39) databases. Protein expression was verified using the HPRD (13), the human protein map (HPM) (14), Proteomics DB (17), the MOPED (18-19) and the human protein atlas (HPA) (16, 40).

Cancer proteome expression in diverse human body fluids. The protein expression in diverse body fluids was inferred from the Multi-Omics Protein Expression Database, the Proteomics DB and the Human Protein reference database. The numbers indicate the number of genes detected for each of the body fluids.
Putative drug hits were filtered from the canSAR datasets for the neoplasm-associated genes using the Lipinski’s rule of five (also known as Pfizer’s rule of five), RO5. The RO5 is a rule of thumb to evaluate druggableness or to determine whether a compound with a certain pharmacological or biological activity possesses properties that would make it a likely orally active drug in humans (41-42). Highest stringency was chosen for the RO5 violation (value=0). Drugs with half-maximal inhibitory concentration (IC50) values, inhibitory activities and inhibitory constant (Ki) values are chosen for the CanSAR output. The chemical structures were verified using the chEMBL tool (27). Toxicophore negative was chosen to filter the hits for toxicity associated compound structures (43). FDA approved listing of drugs were obtained from the DrugBank database (22).
Results
Cancer polymorphic traits in the human genome. The NCBI PheGenI genetic association studies tool was used to establish an initial database of neoplasm-associated traits. Among the diverse association evidence in the GWAS database, neoplasm-associated genes and the single nucleotide polymorphisms (SNPs) were enriched (Figure 1). The human genome segregated into 1,305 neoplasm-associated genes encompassing 2,837 polymorphic SNPs. Sixty five previously uncharacterized open reading frames (ORFs) were also identified in these studies. These novel ORFs, part of the “Dark Matter” proteome have been recently characterized in the context of cancer and other diseases (34, 44). The neoplasm traits were further classified into individual tumor types, risk factor and response to therapy. The largest number of associated genes and SNPs was seen in neuroblastoma, prostate, pancreatic and breast carcinomas. Association evidence was also seen in telomere function, smoking behavior and response to chemotherapy and radiation. This database of cancer subtype-related association provided a framework for detailed bioinformatics and proteomics characterization.

Pathway mapping of the cancer proteome from the body fluids. The neoplasm-associated proteins were analyzed for gene pathways using the GeneALaCart and the DAVID functional annotation tools. The numbers indicate the number of genes associated with the indicated pathways.
Enrichment of cancer proteome in the body fluids. The majority of the neoplasm-associated genes (n=1,305) are well-characterized proteins. However, the data for these proteins exist in diverse databases making the lead gene prioritization for cancer drug discovery often difficult. It was reasoned that the protein biomarkers detectable in diverse body fluids (the cancer secretome) might offer a diagnostic and response to therapy indicator potential. Hence, the protein expression databases, the HPRD, the MOPED and the Proteomics DB, which have proteome expression datasets from normal and cancer patient-derived body fluids, were batch-analyzed for the neoplasm-associated genes (Figure 2). A large number of the neoplasm-associated proteins (913/1,305) were detected in diverse body fluids. Among these, 192 proteins had signal peptide sequence predicted by the Signal P program (45-46). The remaining proteins (n=721) belong to the non-classical secretary pathways. Blood plasma, pancreatic juice and the ascitic fluid showed the maximum number of proteins. In addition, neoplasm proteins were detected in cerumen (earwax), milk, saliva, tear and urine.
Pathway analysis. In order to develop a rationale for novel cancer therapeutic targets discovery, a comprehensive pathway mapping was undertaken for the neoplasm-associated proteins. The GeneALaCart and the Database for Annotation, Visualization and Integrated Discovery (DAVID) functional annotation tools were used and batch-analyzed to cluster the pathways implicated with the cancer proteome (Figure 3). The pathway data was merged from the output from the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway, the Interactome pathway, the Biosystem pathways, the Tocris pathway, the Thomson Reuters pathway and the PharmGKB pathway tools. The pathways segregated into cell growth regulation (angiogenesis, apoptosis, cell cycle, DNA damage and degradative), metabolic (biosynthesis, glycolytic, hormonal and purines and pyrimidines), signaling (Akt, cytokines, ErbB, MAPK, Ras/RaP1 and protein), druggable targets (adhesion, ligands, transporters, receptors,and G protein-coupled receptors (GPCRs), pharmacokinetics and pharmacodynamics, chemotherapy and neoplasm-related diseases (inflammation, infection and immune). These results were also verified from the gene ontology analysis from the canSAR and the DAVID functional annotation datasets. Cell localization ontology categorized neoplasm-associated genes into the membrane (n=516), nuclear (n=446), cytosol (n=416), cytoskeletal (n=81), endoplasmic reticulum (n=87), Golgi (n=81), mitochondria (n=77), receptors (n=233), extracellular (n=206), neuron (n=123), axonal (n=81) and vesicle (n=126).

Druggability analysis of the cancer proteome from the body fluids. The canSAR protein annotation tool was used to batch-analyze the neoplasm-associated proteins from the body fluids. A summary of the output based on the druggable 3D structures of the proteins and the ligand-based druggability indication is shown. The numbers indicate the number of genes for the criteria shown. The filter was chosen at >90% homology for the hits. Only the active compounds for the proteins (<1 μM in bioactivity) are included.
Druggable proteins in body fluids: the cancer secretome. In order to develop an understanding over the druggableness of the neoplasm-associated proteins, genes were classified into classes of coding and non-coding genes. A comparison of all neoplasm-associated genes with the genes identified in the body fluids is shown in Table I. The protein coding genes clustered into druggable class, including cell adhesion molecules, ion channel proteins, enzymes, GPCRs and other receptors and transporters. Enzymes were the largest class of proteins detected in the body fluids (n=144). In addition, 17 uncharacterized ORFs, pseudogenes, RNA genes and linc RNAs were also identified in the body fluids. Utilizing a recent dataset from the HPA (16), these body fluid proteins were further investigated. The list was filtered into authentic secreted proteins with Signal P prediction (n=192) and current FDA-approved drug targets (n=23).
Druggablity profile of cancer secretome. To facilitate the discovery of new cancer therapeutics, the canSAR integrated protein annotation tool was batch-analyzed for the cancer proteome from the body fluids (Figure 4). Using 3D structural evidence and ligand-based druggability ranking (>90% confidence), 121/913 body fluid proteins were predicted to be capable of binding to a ligand. Twenty percent of the body fluid proteins (175/913) were predicted to be druggable based on 3D structures. Active small-molecular-weight compounds were identified (<1 μM bioactivity) for 168 neoplasm-associated protein targets.
Protein classes of the neoplasm-associated genes.
Using a high-stringency definition of druggability percent score (>90%); RO5 violation score (zero); bioactivity cut off (<1 μM); and lack of toxicophore groups, thirty-two secretome proteins were identified as putative drug lead targets (Table II). These lead proteins encompass enzymes (alcohol dehydrogenese, aldo-keto reductase, amine oxidase, demethylase, glucokinase, serine/threonine kinases, lipoxygenease and thymidine phosphorylase), receptors (dopamine receptor, insulin-like growth factor receptor, stem cell growth factor receptor, melanocyte-stimulating hormone receptor and purinoceptor), coagulation factor XIII and cytochrome P450 286 protein.
Cancer lead targets and putative compounds. Seven out of these 32 proteins showed strong Phenome-Genome association evidence in diverse neoplasms: a downstream effector of Cdc42 in cytoskeletal reorganization (CDC42BP) with large B-cell diffused lymphoma, (47); Casein kinase I isoform alpha (CSNK1A1) involved in Wnt signaling with esophageal cancer (48); a Serine/threonine-protein kinase (CHEK2) with stomach neoplasms (49); an intronless melanocyte-stimulating hormone G-protein coupled receptor (MC1R), which is a genetic risk factor for melanoma and non-melanoma skin cancer (50, 51) and basal cell carcinoma (52); a Lysine-specific demethylase 4C (KDM4C), which is an indicator for response to radiation (53); a S-methyl-5’-thioadenosine phosphorylase (MTAP), which is co-deleted in diverse tumors along with the tumor suppressor P16 gene (51) and P2Y purinoceptor 12 (P2RY12), a G-protein coupled receptor with neuroblastoma (54).
These seven lead proteins also had active drug-like compounds (nanomolar bioactivity, RO5 violation value of zero, molecular weight <500 and lack of toxicophore structures) in the chEMBL chemical repository (Table III). A compound against the target, protein, check point kinase 2, CHEK2 (canSAR # 404540|CHEMBL574737), is currently a clinical candidate (55).
Mining the cancer-oriented databases, such as the NCBI ClinVar, the catalogue of somatic mutations in cancer (COSMIC) and the cBioPortal provided additional supporting evidence implicating other tumor types for these genes. Pathogenic clinical variations were seen for the lead genes in malignant melanomas (56-57), neoplastic syndromes, hereditary, familial cancer of breast, Li-Fraumeni syndrome 2 (58), diaphyseal medullary stenosis with malignant fibrous histiocytoma (59) and platelet-type bleeding disorder 8 (60). The COSMIC database showed somatic mutations for diverse tumors and the top three tumor types with mutations are shown (Table III). The cBioPortal Meta analysis tool’s mutational assessor was used to identify high impact missense mutations for distinct tumor types.
Discussion
The human proteome consists of over 22,000 proteins and various isoforms associated with these proteins (61). Detailed knowledge of these proteins at the level of variations, polymorphisms, gene ontology, motifs and domains, gene expression at mRNA and protein levels and disease relevance exist across diverse bioinformatics databases. Despite the large number of proteins, only 620 proteins are the basis of mechanism-based FDA approved drugs (DrugBank listing). These target proteins predominantly involve four protein families, such as enzymes, transporters, ion channels and receptors called druggable genes (62-67). The current drugs largely encompass antagonists or agonists for these protein targets. Gene ontology prediction tools indicate that over 70% of the drug targets are membrane-bound or secreted (16).
Neoplasm-associated lead proteins.
The cancer therapeutics, which is moving toward personalized medicine (68), requires additional drug targets. Reasoning that the GWAS databases can provide an attractive starting point for developing a list of druggable cancer targets, mining of the NCBI Phenome-Genome Integrator database was undertaken. The PheGenI tool from the GWAS database identified a total of 1,305 genes associated with distinct neoplasm traits. Largely, these genes encompassed protein-coding genes (983/1,305); however, non-protein coding sequences, including long intergenic RNAs, linc RNAs (n=12), pseudogenes (n=249) and antisense RNAs (n=5) were also part of this list of genes. A significant number of druggable genes (enzymes, receptors, channel proteins, transporters and cell adhesion molecules) were identified in the study (n=404). Furthermore, 192 putative secreted proteins were identified in the neoplasm-associated gene list.
Drug-like cancer lead compounds. Lead neoplasm -associated secretome proteins were identified using the ligand-based druggability percentile rank (>90%) and with active compounds (<1 μM). Most active compounds are shown with canSAR ID # and IC50 values. The chEMBL IDs are shown in parentheses. Clinical candidate is bolded. Neoplasm association is underlined. Pathogenic clinical variations from NCBI ClinVar are shown. COSMIC mutations are shown for top three tumor types. cBioPortal functional impact score using mutational assessor is shown for high impact missense mutations.
Cancer cells secrete numerous proteins by both classical and non-classical secretory pathways (21, 45). The cancer secretome includes the extracellular matrix components, as well as all the proteins that are released from a given type of cancer cells, such as growth factors, cytokines, adhesion molecules, shed receptors and proteases (20, 68). The secreted proteins in diverse body fluids offer novel biomarker and drug therapy targets. From the neoplasm-associated traits, 913 proteins were identified in diverse body fluids encompassing druggable targets. Only 23 out of these proteins are currently FDA-approved targets (16). Thus, it is distinctly possible that additional biomarkers and drug targets can emerge from the database of the cancer secretome generated in this study.
Using chemoinformatics approaches, 33 of the cancer secretome proteins, encompassing enzymes and receptors, were predicted as druggable. Bioactive compounds (<1 μM) targeting these proteins were identified in the canSAR/chEMBL databases. These 33 protein targets provide a drug discovery rationale for cancer. Furthermore, the discovery of drug-like bioactive compounds targeting seven of these lead secretome proteins provides an immediate starting point for novel cancer therapeutics. The involvement of the neoplasm-associated proteins with other diseases, such as nicotine addiction, Alzheimer’s disease, diabetes, cardiac, hematological, metabolic and respiratory diseases, leprosy and schizophrenia opens-up novel biomarker and therapeutic opportunities for these diseases as well.
In summary, the results presented in this study demonstrate the power of chemogenomics approaches for rational cancer drug discovery. Mining the cancer secretome for neoplasm-associated traits is likely to lead to the discovery of new molecular entities for diagnosis and therapy. The lead compounds identified in the study can be rapidly tested in cell culture and pre-clinical models for efficacy.
Acknowledgments
This work was supported in part by the Genomics of Cancer Fund, Florida Atlantic University Foundation. The Authors would like to thank the canSAR gene annotation tool for valuable datasets and Jeanine Narayanan for editorial assistance.
Footnotes
-
Conflicts of Interest
-
None.
-
Data Availability
-
The detailed data of this study as a supplemental Table is available upon request.
- Received February 18, 2015.
- Revision received February 28, 2015.
- Accepted March 3, 2015.
- Copyright© 2015, International Institute of Anticancer Research (Dr. John G. Delinasios), All rights reserved




{kind=link}
{kind=link}
{kind=link}
{kind=link}