


Abstract
Background: Lymph node metastasis is an important clinicopathological parameter for breast cancer prognostication and treatment. Although the development of metastasis is common in axillary lymph nodes, the mechanisms underlying the locoregional spread are yet poorly understood. In the present study, we outline the involvement of proteins in tumor invasion by comparing the proteome profile of primary breast tumors (PBT) against that of lymph node metastasis (LNM). Patients and Methods: The comparative proteome analyses of seven paired samples were performed using two-dimensional gel electrophoresis (2DE) and mass spectrometry (MS). Results: Recurrent proteins were differentially expressed in PBT and LNM across patients. Higher levels of 1433G, 1433T, K2C8, PSME2, SNAA, TPM4, TRFE and VIME were observed in primary tumors compared to the metastatic site. On the other hand, higher levels of ALDH2 and GDIR2 were identified in metastasis related to tumors. These proteins provide a new insight on breast cancer research. Conclusion: Our achievements strengthened previous omics-based studies and also support the validation of potential markers of tumor invasion and metastasis.
EMT: Epithelial-mesenchymal transition; ER: oestrogen receptor; IDC: invasive ductal carcinoma; IEF: isoelectric focusing; IPG: immobilized pH gradient; LNM: lymph node metastases; MALDI: matrix-assisted laser desorption/ionization; MS: mass spectrometry; MM: molecular mass; pI: isoelectric point; PAGE: polyacrylamide gel electrophoresis; PMF: peptide mass fingerprinting; PR: progesterone receptor; PBT: primary breast tumour; PTM: post translational modification; TOF: time of flight.
Breast cancer is a heterogeneous disease with a wide range of molecular abnormalities leading to cell growth and proliferation, vessel invasion and metastasis (1). Distinct patterns of metastasis can be discerned within breast cancer. However, the dissemination through the body is not a random process. Commonly, regional lymph nodes are the primary sites of metastasis, with patterns of lymphatic spread occurring via pre-existing afferent vessels and/or newly formed capillaries (2-4). Distant metastasis, on the other hand, involves intravasation/extravasation of the circulatory system to other parts of the body, such as bone, lungs, liver and brain (5). The mechanisms underlying regional and distant spread, nevertheless, remain incompletely characterized (4).
Metastases are the major causes of treatment failure and death of patients diagnosed with breast cancer (6, 7). Although numerous clinical and pathological variables (tumor grade and size, as well as hormone status) are used to predict patient outcome; the investigation of locoregional spread is the most important prognostic indicator in breast cancer (8, 9). The sentinel lymph node biopsy has a major role in disease staging. It guides decision-making regarding completion of lymphadenectomy to achieve regional control (10, 11). Furthermore, the number of lymph nodes affected is an important criterion for administering adjuvant therapy (12, 13). In this context, the molecular evaluation of candidate markers in PBT for predicting LNM and/or delineating treatment is a potentially promising strategy for disease management (14).
In the last decade, researchers have attempted to identify differential molecular signatures of the genome, transcriptome and proteome to determine and/or explain intrinsic properties of the disease and its metastatic phenotype (14-17). In a genome-based analysis, Santos et al. (17) reported DNA copy number changes per chromosome across PBT and the sentinel LNM. Following transcriptome approach, Feng et al. (16) and Suzuki and Tarin (15) described a list of differentially expressed genes that may predict clinical outcome of node-positive patients; however, with a small overlap between the two studies. Microarray gene expression analysis performed by Weigelt et al. (8), on the other hand, did not reveal common metastasis-specific gene set within the pairs of matching PBT and LNM.
Patients’ clinicopathological data.
Metastatic-associated proteins have also been investigated and promise new information and knowledge regarding patterns driving tumor progression. In this context, the analysis performed by Li et al. (18) revealed a few significant changes in the quantitative level of individual proteins; however, none common denominator has been identified to distinguish PBT from matched LNM. A similar result has been observed by Gonçalves et al. (1) in comparative analysis of nipple aspirate fluid and LNM in women with breast cancer. Ultimately, differential protein peaks have been demonstrated by Nakagawa et al. (14) with potential ability for predicting presence of LNM, but no particular biomarker has been suggested.
Distinct studies supported the analysis of PBT and LNM with different purposes and conflicting achievements (8, 14, 15, 18-20). Relatively few studies, nevertheless, have been noted for the discovery of remarkable proteins linked to breast cancer metastasis. In this study, we aimed to provide a comprehensive comparative proteome profiling that will contribute to the better understanding of disease progression, assigned by invasion and metastasis. We believe that the differential proteome will bring an insight into the key events that underlie the intrinsic molecular changes. In order to capture the proteome complexity of samples, we used fresh-frozen tissue, as represented in the two stages of invasive ductal carcinomas (IDC). Seven paired PBT and LNM were analyzed using two-dimensional polyacrylamide gel electrophoresis (2D-PAGE) and mass spectrometry (MS). The differential proteins represent potential markers for tumour invasion and also support other studies in the field.
Materials and Methods
Sample collection and clinical evaluation. Matched pairs of sporadic PBT and axillary LNM were obtained from seven patients (females) diagnosed with IDC, within estimated average age of 62±7.64 years (Table I). None of these women had received pre-operative hormonal treatment, radiation or chemotherapy. Samples were collected during surgical intervention at the Hospital Nossa Senhora das Graças in Curitiba, Brazil, and immediately stored at -80°C. Specimen were assembled between February 2008 and December 2009. The project was approved by the Ethics committee from the Hospital Nossa Senhora das Graças (CONEP 7220) and was in accordance with the regulations of our institution (Universidade Federal do Paraná). The participants signed an informed consent to participate in this research.
Protein extraction and quantification. Breast tumour fragments were solubilized in lysis buffer containing 7 M urea, 2M thiourea, 4% CHAPS, 40 mM Tris and 0.2% PMSF and the cells homogenized with an electric tissue disruptor. The total lysate was centrifuged at 12,000 rpm for 5 min to clear debris. The protein concentration was determined using the Bradford method (21). Subsequently, the extract (1,000 μg protein) was solubilised in rehydration buffer (7 M urea, 2 M thiourea, 2% CHAPS, adding 50 mM DTT and 0.5% IPG buffer), and applied to 13 cm linear immobilized pH gradient (IPG) strips (pH 4-7) (GE Healthcare, Little Chalfont, United Kingdom). Rehydration occurred at room temperature for 16 h in the Immobiline DryStrip Reswelling Tray (GE Healthcare).
Two-dimensional gel electrophoresis. The first-dimensional separation was performed with Ettan IPGphor II (GE Healthcare). Strips were placed on a ceramic plate (Manifold/GE Healthcare) and isoelectric focusing (IEF) was performed with stepwise increasing voltage as follows: 500 V for 1 h; 100 V for 1 h; 8,000 V for 2:30 h; 8,000 V for 30 min. After IEF, the strips were equilibrated for 15 min in a solution containing 50 mM Tris-HCl, pH 8.8; 6 M urea; 30% (w/v) glycerol; 2% (w/v) SDS; 50 mM DTT; and traces of bromophenol blue. Free thiol groups were then alkylated by substituting the DTE with 4.5% iodoacetamide in the equilibrating buffer.
Second dimension electrophoresis was performed using Hoefer SE 600 Ruby (GE Healthcare) in running buffer (25 mM Tris, 192 mM glycine and 0.1% SDS) at 11°C for 30 min at 15 mA and 4.5 h at 30 mA. Before staining, gels were placed for 1 h in a fixative solution containing 1.3% orthophosphoric acid (85%) and 20% methanol. Gels were then coloured with 0.01% Coomassie G-250, 1.5% orthophosphoric acid (85%) and 7.7% ammonium sulfate.
Gel replicates (n=3) were produced for each sample using a restricted pH range (from 4 to 7) that focuses on the region with the highest quantities of proteins, as previous tested in our laboratory. No depletion method was used to remove plasma proteins from the samples; however, spots matching albumin in the gel were not considered in the match set.
Image processing. Stained gels were scanned with ImageScanner™ II (GE Healthcare) and analyzed with ImageMaster™ 2D Platinum v6.0 (GE Healthcare). The parameters used to detect spots by the software were: area min - 5; smooth - 3; and saliency - 25. Triplicates were cropped to frame the same cluster of spots across samples and one representative gel was used to create a match-set. In addition, the logarithmic ratios of spots with precise matching were considered for normalization at ImageMaster™. Spots with expression levels above 2 were first filtered for further analysis. The ImageMaster™ software was also used to compute the statistical analysis of the data (Student’s t-test), covering the pre-filtered bands with significant p-value (p<0.05).
Mass spectrometry (MS) and protein identification. The spots of interest were manually excised from the gels and the pieces were destained in 50% acetonitrile and 25 mM ammonium bicarbonate. Dehydration was performed in two rounds of 100 μl of acetonitrile for 5 min. The supernatant was removed and the remaining gels were dried at room temperature. Afterwards, the gel pieces were rehydrated in 15 μl of digestion solution containing 40 mM ammonium bicarbonate, 10% acetonitrile and 15 ng/μl trypsin (Sequencing Grade Modified Trypsin; Promega, Fitchburg, Wisconsin, USA) for 30 min. The digestion was performed at 37 °C for 16-20 h. Tryptic peptide extracts were dissolved (1:1) in a matrix solution (50% acetonitrile, 0.1% trifluoroacetic acid and saturated α-cyano-4-hydroxycinnamic acid) and spotted onto MALDI AnchorChip target (Bruker Daltonics, Billerica, Massachusetts, USA).
MS was performed using the MALDI-TOF/TOF/MS/MS AutoflexII (Bruker Daltonics) and MALDI-Tof/MS micro MX (Waters Corporation, Milford, Massachusetts, USA). The peptide mass fingerprinting (PMF) was compared with theoretical molecular masses (MM) and isoelectric point (pI) from UniProtKB/Swiss-Prot annotation, using the Matrix Science (MASCOT) database. For protein identification, the taxonomic category was restricted to Homo sapiens, maximum 100 ppm of mass tolerance and one missed enzymatic cleavage for trypsin. A number of fixed (carbamidomethylation of cysteine residues) and variable modifications (methionine oxidation) were included as search parameters. Proteins were considered reliable when the score exceeded the threshold value of 56.
Results
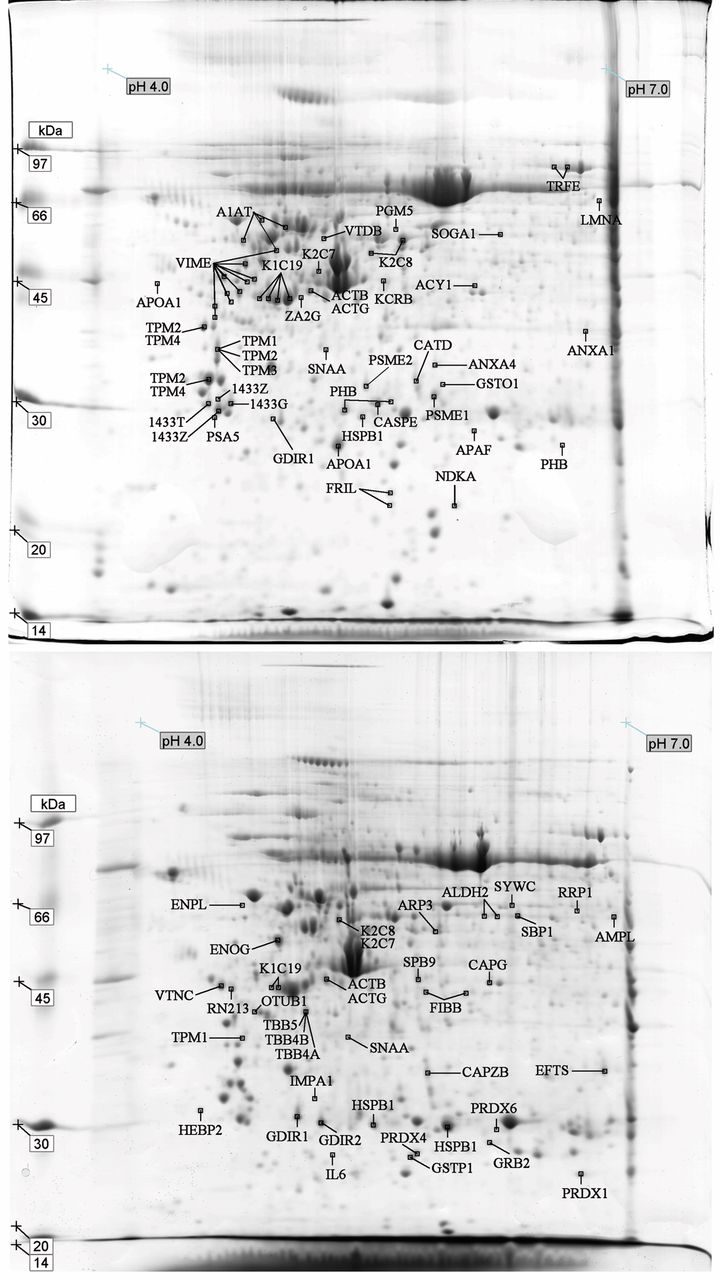
Proteomic profiling of primary breast tumors and matched lymph node metastasis samples. With the purpose of comparing individual protein changes in matched PBT and LNM, seven pairs of surgical samples were obtained from patients diagnosed with IDC. In the present study, the total of 42 gels showed approximately 703 spots detected in PBT and 852 in LNM samples (http://goo.gl/KGEBHM). Figure 1 (a and b) illustrates the most representative gels (CP 645L and CP 645T) of all pairs selected among seven matched proteomic profiles of PBT and LNM.
The differential spots refer to positive protein identification based on master/reference gels from each patient. Spots were labelled according to the protein identity in UniProtKB/Swiss-Prot data base. Overall, 128 spots with differential expression were identified, corresponding to 76 distinct proteins, including isoforms and variants. Ultimately, 119 spots and 67 proteins were selected for further analysis (http://goo.gl/KGEBHM). The proportional values of MM and pI, theoretical and observed, were used in this study based on Dupont et al. (22).
After comparing the seven gels of PBT and the seven of LNM, significant spot overlaps across patients were observed. Hence, there were recurrent spots/proteins identified in the group of primary or secondary site. The results demonstrated nine distinct proteins with increased expression levels in PBT samples (Tables II and III). These proteins are: 14-3-3 protein theta (1433T), 14-3-3 protein gamma (1433G), Keratin, type I cytoskeletal 19 (K1C19), Keratin, type II cytoskeletal 8 (K2C8), Proteasome activator complex subunit 2 (PSME2), Alpha-soluble NSF attachment protein (SNAA), Tropomyosin alpha-4 chain (TPM4), Serotransferrin (TRFE), Vimentin (VIME). In addition, only three common proteins with increased expression were detected in LNM: Aldehyde dehydrogenase, mitochondrial (ALDH2), Rho GDP-dissociation inhibitor 2 (GDIR2) and K1C19.
Certain proteins may have been carried by other peptides or have undergone chemical modifications; all relevant facts that should be exposed. For example, Alpha-1-antitrypsin (A1AT) showed consistent pI in two distinct samples (CP 622T and CP 644T) but, in both cases, the MM parameter was different from the theoretical data. Likewise, two other spots exhibited pI values concordant with the literature and were identified as Fibrinogen beta chain (FIBB) in CP 644L and RING finger protein 213 (RN213) in CP 645L. Although short variances in MM and pI, Cytosol aminopeptidase (AMPL), Ribosomal RNA processing protein 1 homolog A (RRP1), Elongation factor Ts, mitochondrial (EFTS), Endoplasmin (ENPL) and Microtubule-actin cross-linking factor 1, isoforms 1/2/3/5 (MACF1), were also retained for registering. Nine proteins, however, showed unidentifiable values of both MM and pI and have not been considered in our analysis (http://goo.gl/KGEBHM).
Other proteins showed variations of MM/pI. In particular, A1AT and Heat shock protein beta-1 (HSPB1) were identified from different spots mapped in a nearby region. Likewise, VIME and cytokeratins exposed a range of neighbouring spots with similar MM and pI. This may reflect the dynamicity of the cell’s proteome. Major regulations from mRNA transcription to post-translational modifications (PTM) leads to a wide variety of product size, shape and complexity (23).

Proteomic profiling of the master/reference gel in primary breast tumors and lymph node metastasis. Gels were stained with Coomassie blue as described in the Materials and Methods section. The arrows indicate differential spots identified in primary breast tumors and lymph node metastasis across all samples.
Proteins identified with increased expression levels in primary breast tumors.
Proteins identified with increased expression levels in lymph node metastasis.
Grouping proteins into functional categories and cellular location. Proteins were grouped into functional categories based on Pucci-Minafra et al. (24, 25): (i) cytoskeleton and associated proteins (42.64%); (ii) cell growth and proliferation regulators (14.73%); (iii) proteins with extracellular activity (7.75%); (iv) binding proteins (7.75%); (v) proteolysis regulation (7.75%); (vi) metabolic enzymes (4.65%); (vii) detoxification and redox proteins (3.88%); (viii) molecular chaperones/heat shock proteins (3.10%); (ix) membrane-associated proteins with multiple activities (4.65%); and (x) other functions (3.10%) (http://goo.gl/KGEBHM).
Accordingly, Figure 2 (a and b) highlights the percentage of proteins identified with increased expression levels in each group of samples and are labelled according to their respective category. The cytoskeleton and associated proteins are the most abundant in both groups and show a greater coverage in PBT (48.19%) than LNM (32.61%). In contrast, the percentage of metabolic enzymes is higher in LNM (10.87%) when compared to PBT (1.2%). The same occurs for detoxification and redox proteins (8.70 in PBT and 1.2% in LNM). The changes in protein expression levels may support important variations in the cell molecular landscape and functioning.
It is also relevant to understand that the specialized domains of the cellular components determine cell orientation, function and fate. The proteins described here were mainly encountered in the cell membrane, cell junction, cell projection, cytoplasm, cytoskeleton, endosome, endoplasmic reticulum, extracellular matrix, Golgi apparatus, intermediate filament, keratin, lysosome, microtubule, mitochondrion, nucleus, proteasome or secreted (Figure 3, a and b). As regards to the cellular components, PBT and LNM revealed greater numbers of proteins in the cytoplasm, cytoskeleton, intermediate filament and outer cell (secreted) (http://goo.gl/KGEBHM).
Recurrent protein up-regulation in primary breast tumors and lymph node metastasis. 14-3-3 protein gamma (1433G) and 14-3-3 protein theta (1433T) are members of the 14-3-3 family that act as cell growth and proliferation regulators. These proteins exhibit diverse biological activities by mediating signal transduction pathways. They regulate cell division, differentiation, survival and apoptosis (24). It is noteworthy that 1433s are involved in the transition of breast epithelial cells to neoplasia (26). Our study showed that these proteins have higher expression levels in PBT compared to LNM. The protein inactivation and/or down-regulation have also been reported in various cancers and strongly correlated with tumour progression (18, 27-31).
Proteasome activator complex subunit 2 (PSME2) is involved in proteolysis regulation by degrading damaged proteins. PSME2 may participate in the origin and progression of tumours (32). In vitro and in vivo experiments have revealed that up-regulation of PSME2 leads to cell growth, proliferation and tumorigenicity (20, 33). In this study, PSME showed higher levels in PBT when compared to LNM. Contradictory results in the literature involving PSME2, however, emphasize the urgent need for further analyses of this protein. For instance, PSME2 has shown down-regulation in lung cancer (33, 34) but up-regulation in gastric cancer (35) and renal carcinoma (36). Greater levels of PSME2 in metastatic melanoma cell lines have also been observed when compared to primary tumours (28).
Alpha-soluble NSF attachment protein (SNAA) is part of the membrane-associated proteins with multiple activities class. It is a member of the SNAP family and participates as a component of the cell vesicle trafficking machinery. SNAA plays an essential role in anchorage and fusion of vesicles in the membrane-associated SNAP receptor (SNARE) complex. Proteins involved in secretion have been detected in tumour samples, which may suggest a particular cellular activity by transporting substances to regulate the cell environment (37). Higher levels of this protein were found in PBT compared to LNM. Increased levels of SNAA have also been described in colorectal cancers and linked to aggressive phenotype, poor prognosis and high mortality (37). SNAA was also linked to drug resistance, although the mechanisms behind this is are poorly understood (38).
Tropomyosin alpha-4 chain (TPM4) is part of the tropomyosin family and belongs to the cytoskeleton and associated proteins category. Tropomyosins provide stability to intracellular filaments and regulate the access of other actin-binding proteins. This protein has shown a critical role in lamellipodia formation, endocytosis and vesicle trafficking (39). Altered expression of different tropomyosin isoforms has been related to several tumours (25) and appears to be directly responsible for aberrant motile activity and malignant phenotype (40). Our results revealed increased levels of TPM4 in PBT compared to LNM; or decreasing levels in the metastatic node. The protein’s down-regulation has been linked to oncogenic transformation, cell growth and tumour metastasis (41, 42). In a different proteomic approach, TPM4 showed up-regulation in oestrogen receptor (ER) positive breast cancer related to ER-negative carcinomas (43).
Serotransferrin (TRFE) is a glycoprotein classified as a binding protein, which transports iron through the blood (27). It is critical for tumour cells due to their rapid proliferation that require a higher amount of iron than their normal counterparts (44). Dysregulation of proteins involved in iron metabolism plays a critical role in cancer. Quantification of TRFE, therefore, could be valuable to expand the assessment of breast cancer status and improve treatment with targeted chemotherapy. Our study revealed higher levels of TRFE in PBT in comparison to the LNM counterpart. In previous reports, increased levels of TRFE were also demonstrated in proliferative breast cancer (44, 45).
Vimentin (VIME) is a major constituent of the intermediate filament family and is classified within the category of cytoskeleton and associated proteins. VIME shows a varied role in cell architecture, cytoplasm integrity and cytoskeletal interactions; besides controlling critical proteins involved in attachment, migration and cell signalling. This protein has been identified in both tumour and stroma in various epithelial cancers; however, its role in cancer progression remains obscure (46). Our analysis showed up-regulated VIMEs in PBT in a range of neighbouring spots. The VIME’s up-regulation is interpreted as a sign of epithelial-mesenchymal transition (EMT), which is generally associated with tumour cell dedifferentiation, growth, invasion and metastasis (47, 48). Furthermore, the protein levels positively correlates with the migratory capacity of the breast cancer cells (49).

Keratin, type I cytoskeletal 19 (K1C19) and Keratin, type II cytoskeletal 8 (K2C8) are members of the keratin family type I (acidic) and II (basic), respectively, part of the cytoskeleton and associated proteins group. K2C7, K2C8 and K1C19 are normally expressed in the ductal epithelium (50). Keratins are intermediate filament proteins involved in the structural integrity of epithelial cells. Functions performed by keratins include complex regulations of cell signalling, stress response and apoptosis (51). Silencing of K1C19, for instance, has resulted in increased cell proliferation, migration and invasion and is associated with poor patient prognosis and reduced survival (52-54). In breast cancer, this protein has been confirmed as a marker of dissemination to lymph nodes, peripheral blood and bone marrow (55). It has been speculated that K1C19 is also biologically relevant in early metastatic spread (51). In addition, the expression of K2C8 and K1C19 showed correlation with positive receptors ER and PR, HER2 over-expression and low/negative Ki-67, EGFR and c-Kit markers (41, 50). In this report, eight spots showed greater levels of K1C19 in PBT, against two spots in LNM cases. The spots identified in PBT, however, were different from the ones identified in LNM. It may reflect internal regulations that delineate key PTMs in these proteins according to the cell state. K2C8 were found up-regulated in PBT when compared to LNM.

Proteins identified with increased expression levels in (a) primary breast tumours and (b) lymph node metastasis according to cell components. The pie chart illustrates the percentage of proteins identified according to the cellular components in the UniProt database.
Rho GDP-dissociation inhibitor 2 (GDIR2) is a member of the family GDP-dissociation inhibitors and is classified as a cell growth and proliferation regulator. It is critical for controlling small GTPases, such as Ras1, Cdc42 and RhoA (56). These proteins further regulate many cellular functions, including cell polarity, proliferation, apoptosis and migration (57). LNM showed higher levels of this protein when compared to PBT. In the literature, GDIR2 has been correlated with breast cancer metastasis and associated with aggressive phenotypes in gene expression analysis of tumor cell lines (58). Our achievements confirmed previous studies (19) in which GDIR2’s over-expression was reported in breast cancer LNM. Other techniques also confirmed increasing levels of GDIR2 in cell lines (56) and drug resistant tumours (56, 59, 60).
Aldehyde dehydrogenase, mitochondrial (ALDH2) belongs to the aldehyde dehydrogenase (ALDH) family and is categorized as a metabolic enzyme. It is involved in the maintenance of cellular homeostasis by processing both endogenous and exogenous reactive compounds. ALDHs play a functional role in cell proliferation, differentiation and survival (61). Moreover, they exhibit additional non-enzymatic functions, such as the ability to bind hormones and other small molecules. This protein showed higher levels in LNM related to PBT in our study. Other studies have reported ALDH2’s decreased levels in invasive carcinomas when compared to ductal carcinoma in situ and fibroadenoma (62) and normal breast tissue (63). Despite the well-established biochemical properties of ALDHs, it is poorly understood how these proteins are regulated under pathological conditions (64).
Discussion
In this study, we highlighted the significant changes in protein expression levels across primary and metastatic breast tissues. The nine proteins (1433G, 1433T, K1C19, K2C8, PSME2, SNAA, TPM4, TRFE and VIME) were up-regulated among PBT and three proteins (ALDH2, GDIR2 and K1C19) among LNM were first described in the context of PBT and LNM. The achievements corroborate other proteomics analysis of human breast tumors (20, 24, 25, 45, 47) and cell lines (31, 62, 65, 66) in distinct perspectives, thereby, supporting a larger disease characterization. Although some of the proteins (or genes) were previously linked to breast cancer or cancer in general, the comparative proteome analysis of primary and metastatic counterparts brings a novel approach to the field. The reported proteins are, therefore, potential markers of disease progression and metastasis.
Although we emphasized the recurrent up-regulated proteins, other markers portray variances across tumors and patients. In a highly dynamic system, changes from mRNA transcription to protein PTMs are directly associated with the wide variety of the products across individuals (67). Protein trafficking and compartmentalization may also adopt different dynamics in response to physiological changes (68). It has been shown that the proteome varies due to the constant transformation influenced by several factors. These factors include cell interactions, microenvironment and specific hormonal changes that alter proliferation, survival, polarity, differentiation and the ability to invade other tissues (69, 70). In this context, our achievements also endorse previous evidence showing that functional protein components poorly correlate with gene expression data, but support other protein analyses (15, 16, 71, 72).
The functional categories and cellular location of proteins may also bring new insight on breast tumor invasion. In particular, proteins from cytoskeleton and associated proteins and cell growth and proliferation regulators showed the highest changes. Accordingly, a greater number of proteins were found in the cytoplasm, cytoskeleton, intermediate filament and outer cell (secreted), which agreed with the major functional categories reported above. Those proteins are crucial for controlling cell movement and proliferation and may be of value for directing treatment.
Our approach ultimately reiterate the importance of performing proteomics studies (73) in order to describe significant differences in protein abundance accompanying breast cancer invasion and metastasis (19). A number of advanced methods and technologies have improved the existing number of features in proteomics in the last decades (74, 75). Despite its limitations on the coverage, the 2D-PAGE method, has the potential to capture real changes of proteins in comparative analysis. MALDI-TOF/MS studies have also supported the identification of biomarkers that impact the understanding of protein profiles (76) and have also addressed ambiguities arising from other techniques (71).
Conclusion
It is noteworthy that tumor cells have a transitory gene-protein expression control. In this study, we outlined proteins differentially expressed in primary breast tumors and metastatic lymph nodes that highlight molecular changes involved in tumor invasion. However, the selected peptides do not reflect the entire biological heterogeneity of breast cancer. Still, taken in conjunction with other studies, our work strengthens the validation of potential biomarkers. At this point, we encourage further analysis of proteomics and immunohistochemistry to complement the understanding of these different phenotypes.
The description and annotation of human proteomes and integrated maps remain urgent and promising in the field. Proteome analysis is, therefore, essential to understand the significant molecular changes through tumour evolution and metastasis. It is imperative to interpret the biology behind breast cancer heterogeneity. Importantly, further investigation is required for qualitative and quantitative differential ‘omics’ strategies with the purpose of identifying potential biomarkers for diagnosis and prognosis, as well as for delineating tailored therapy.
Acknowledgments
The Authors would like to acknowledge Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) and Fundação Araucária for the financial support. They also recognize the Universidade Federal do Paraná, Hospital Nossa Senhora das Graças and Hospital de Clínicas, Curitiba/BR, for providing structure and professional assistance.
The Authors also acknowledge Chloe Warren for reviewing the manuscript.
- Received January 22, 2015.
- Revision received February 2, 2015.
- Accepted February 4, 2015.
- Copyright© 2015, International Institute of Anticancer Research (Dr. John G. Delinasios), All rights reserved




{kind=link}
{kind=link}
{kind=link}